PeptoGrid—Rescoring Function for AutoDock Vina to Identify New Bioactive Molecules from Short Peptide Libraries

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussions
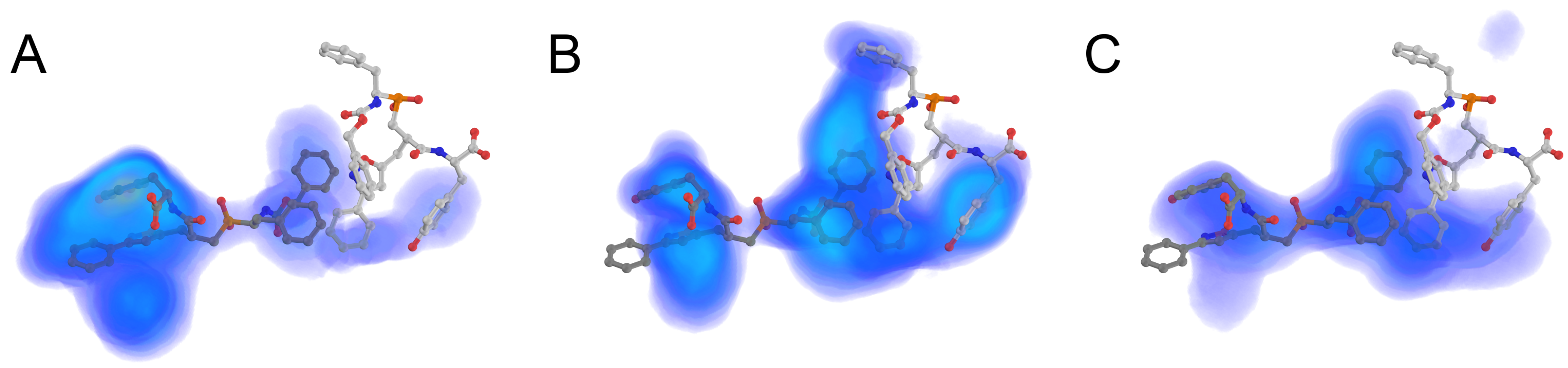
2.1. PeptoGrid Algorithm
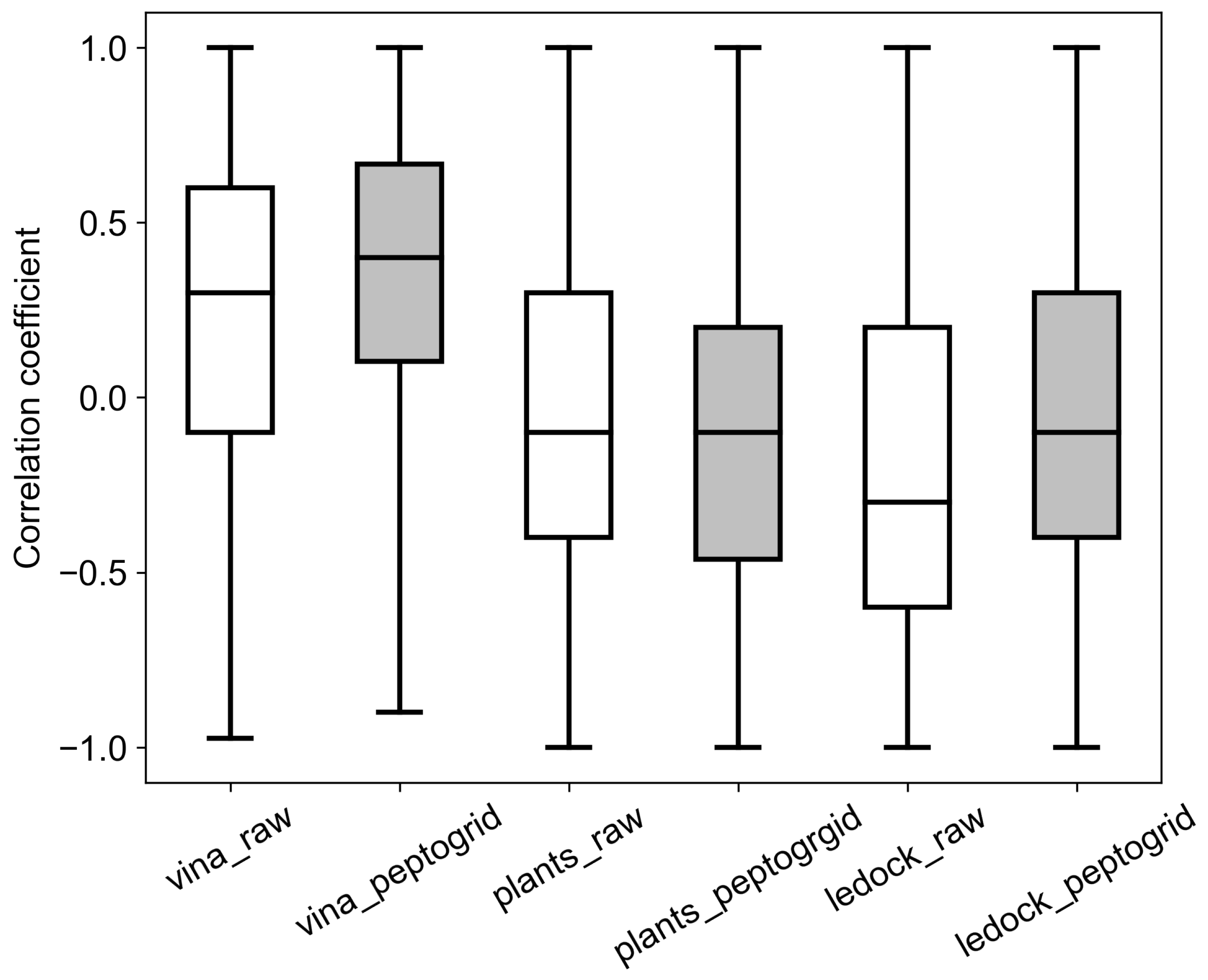
2.2. Benchmark System
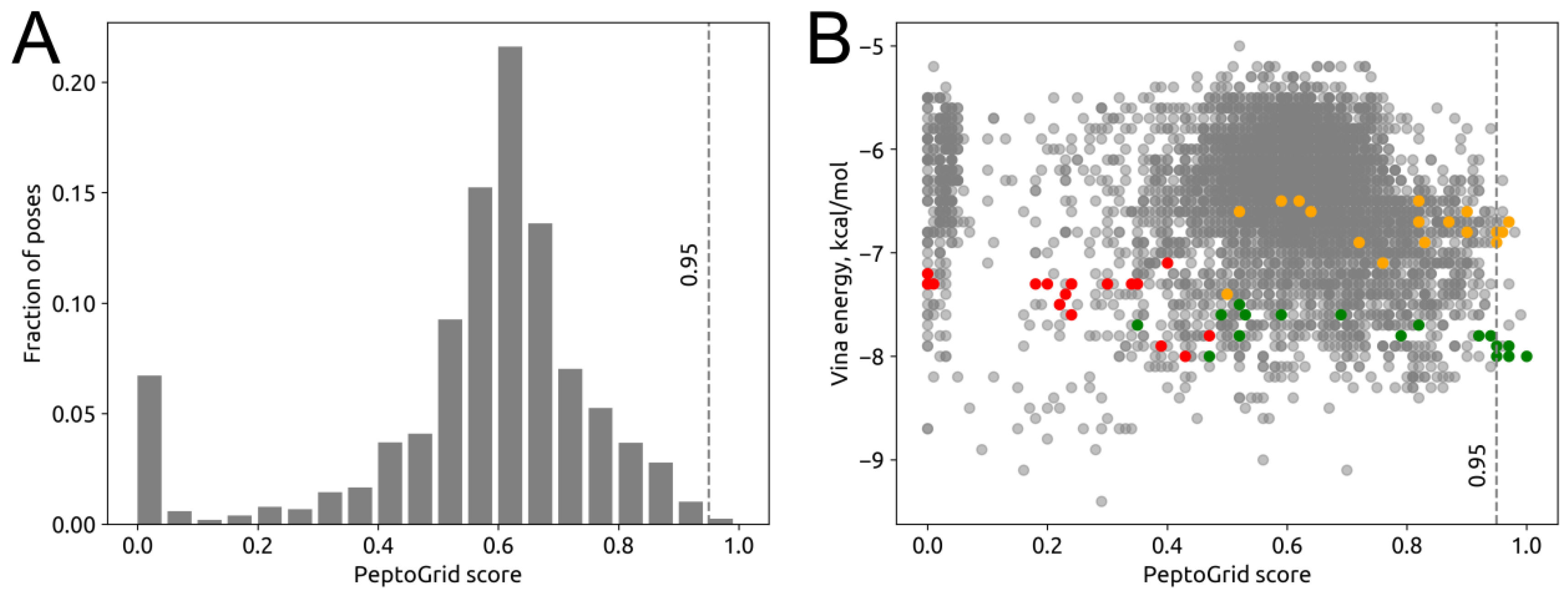
2.3. Screening System
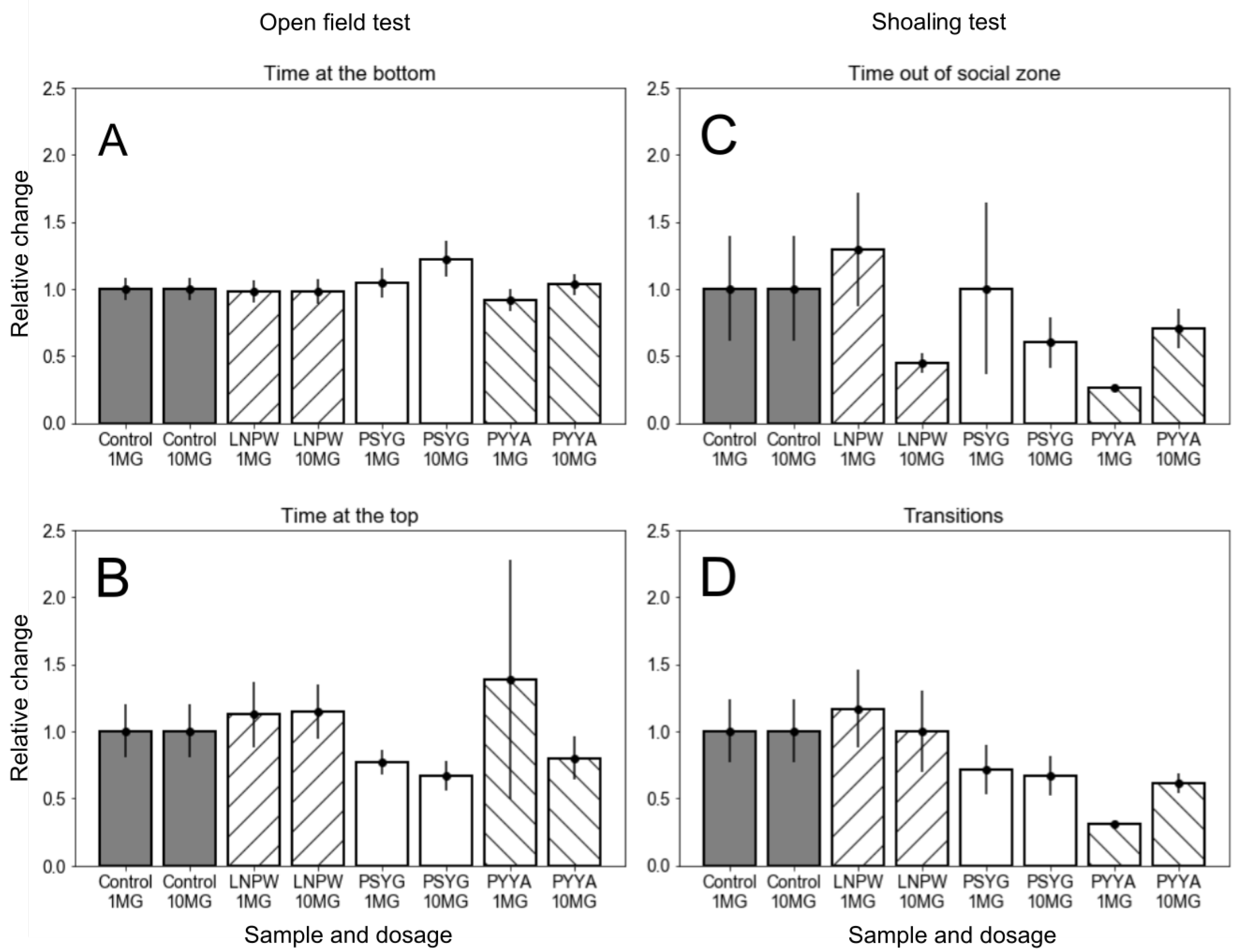
2.4. Experimental Validation of the Virtual Screening Results
- Open field test (OFT). Inactivity or swimming on the periphery of the fish tank is connected with the anxiety-like state. Alternatively, exploring the center of the arena is associated with boldness.
- Shoal cohesion test (SCT). Zebrafish prefer swimming in pods, and this behavioral strategy is thought to be effective against predators. Avoidance of group aggregation (shoal cohesion) is associated with an anxiolytic response.
3. Materials and Methods
3.1. Preparation of Peptide Libraries
3.2. Peptide Structures
3.3. Docking with AutoDock Vina
3.4. Docking with LeDock
3.5. Docking with PLANTS
3.6. Rescoring
3.7. Animals and Housing
3.8. Preparation and Admission of Compounds
3.9. Behavioral Assays
3.10. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.K.; Smith, G.P. Searching for peptide ligands with an epitope library. Science 1990, 249, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Mattheakis, L.C.; Bhatt, R.R.; Dower, W.J. An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc. Natl. Acad. Sci. USA 1994, 91, 9022–9026. [Google Scholar] [CrossRef] [PubMed]
- Kruger, D.M.; Glas, A.; Bier, D.; Pospiech, N.; Wallraven, K.; Dietrich, L.; Ottmann, C.; Koch, O.; Hennig, S.; Grossmann, T.N. Structure-Based Design of Non-natural Macrocyclic Peptides That Inhibit Protein-Protein Interactions. J. Med. Chem. 2017, 60, 8982–8988. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-based virtual screening for drug discovery: A problem-centric review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.; Kurcinski, M.; Kamel, K.; Kolinski, A.; Alam, N.; Schueler-Furman, O.; Kmiecik, S. Protein-peptide docking: opportunities and challenges. Drug Discov. Today 2018, 23, 1530–1537. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Thompson, D.C.; Humblet, C.; Joseph-McCarthy, D. Investigation of MM-PBSA rescoring of docking poses. J. Chem. Inf. Model. 2008, 48, 1081–1091. [Google Scholar] [CrossRef]
- Kurkinen, S.T.; Niinivehmas, S.; Ahinko, M.; Latti, S.; Pentikainen, O.T.; Postila, P.A. Improving Docking Performance Using Negative Image-Based Rescoring. Front. Pharmacol. 2018, 9, 260. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Froestl, W. Chemistry and pharmacology of GABAB receptor ligands. Adv. Pharmacol. 2010, 58, 19–62. [Google Scholar] [PubMed]
- Rentzsch, R.; Renard, B.Y. Docking small peptides remains a great challenge: an assessment using AutoDock Vina. Brief. Bioinform. 2015, 16, 1045–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaput, L.; Mouawad, L. Efficient conformational sampling and weak scoring in docking programs? Strategy of the wisdom of crowds. J. Cheminform. 2017, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Ramsland, P.A.; Edmundson, A.B. Docking of combinatorial peptide libraries into a broadly cross-reactive human IgM. J. Mol. Recognit. 2001, 14, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, I.; Hu, X.; Fumagalli, V.; Contini, A. An Efficient Implementation of the Nwat-MMGBSA Method to Rescore Docking Results in Medium-Throughput Virtual Screenings. Front. Chem. 2018, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Liebeschuetz, J.W.; Cole, J.C. Testing assumptions and hypotheses for rescoring success in protein-ligand docking. J. Chem. Inf. Model. 2009, 49, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leung, K.S.; Wong, M.H.; Ballester, P.J. Improving AutoDock Vina Using Random Forest: The Growing Accuracy of Binding Affinity Prediction by the Effective Exploitation of Larger Data Sets. Mol. Inform. 2015, 34, 115–126. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Kumar, R.; Chaudhary, K.; Sharma, M.; Nagpal, G.; Chauhan, J.S.; Singh, S.; Gautam, A.; Raghava, G.P. AHTPDB: A comprehensive platform for analysis and presentation of antihypertensive peptides. Nucleic Acids Res. 2015, 43, D956–D962. [Google Scholar] [CrossRef]
- Akif, M.; Schwager, S.L.; Anthony, C.S.; Czarny, B.; Beau, F.; Dive, V.; Sturrock, E.D.; Acharya, K.R. Novel mechanism of inhibition of human angiotensin-I-converting enzyme (ACE) by a highly specific phosphinic tripeptide. Biochem. J. 2011, 436, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korb, O.; Stutzle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Natesh, R.; Schwager, S.L.; Sturrock, E.D.; Acharya, K.R. Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature 2003, 421, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Devaurs, D.; Moll, M.; Lizee, G.; Kavraki, L.E. General Prediction of Peptide-MHC Binding Modes Using Incremental Docking: A Proof of Concept. Sci. Rep. 2018, 8, 4327. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhang, W.; Rondard, P.; Pin, J.P.; Liu, J. Complex GABAB receptor complexes: How to generate multiple functionally distinct units from a single receptor. Front. Pharmacol. 2014, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Bettler, B.; Kaupmann, K.; Mosbacher, J.; Gassmann, M. Molecular structure and physiological functions of GABA(B) receptors. Physiol. Rev. 2004, 84, 835–867. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Bush, M.; Mosyak, L.; Wang, F.; Fan, Q.R. Structural mechanism of ligand activation in human GABA(B) receptor. Nature 2013, 504, 254–259. [Google Scholar] [CrossRef]
- Kalueff, A.V.; Gebhardt, M.; Stewart, A.M.; Cachat, J.M.; Brimmer, M.; Chawla, J.S.; Craddock, C.; Kyzar, E.J.; Roth, A.; Landsman, S.; et al. Towards a comprehensive catalog of zebrafish behavior 1.0 and beyond. Zebrafish 2013, 10, 70–86. [Google Scholar] [CrossRef]
- Levin, E.D.; Bencan, Z.; Cerutti, D.T. Anxiolytic effects of nicotine in zebrafish. Physiol. Behav. 2007, 90, 54–58. [Google Scholar] [CrossRef]
- Egan, R.J.; Bergner, C.L.; Hart, P.C.; Cachat, J.M.; Canavello, P.R.; Elegante, M.F.; Elkhayat, S.I.; Bartels, B.K.; Tien, A.K.; Tien, D.H.; et al. Understanding behavioral and physiological phenotypes of stress and anxiety in zebrafish. Behav. Brain Res. 2009, 205, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Stewart, A.; Wu, N.; Cachat, J.; Hart, P.; Gaikwad, S.; Wong, K.; Utterback, E.; Gilder, T.; Kyzar, E.; Newman, A.; et al. Pharmacological modulation of anxiety-like phenotypes in adult zebrafish behavioral models. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Cocco, A.; Ronnberg, A.M.; Jin, Z.; Andre, G.I.; Vossen, L.E.; Bhandage, A.K.; Thornqvist, P.O.; Birnir, B.; Winberg, S. Characterization of the γ-aminobutyric acid signaling system in the zebrafish (Danio rerio Hamilton) central nervous system by reverse transcription-quantitative polymerase chain reaction. Neuroscience 2017, 343, 300–321. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tao, B.; Chen, J.; Jia, S.; Zhu, Z.; Trudeau, V.L.; Hu, W. GABAergic Neurons and Their Modulatory Effects on GnRH3 in Zebrafish. Endocrinology 2017, 158, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Maximino, C.; da Silva, A.W.; Araujo, J.; Lima, M.G.; Miranda, V.; Puty, B.; Benzecry, R.; Picanco-Diniz, D.L.; Gouveia, A.; Oliveira, K.R.; et al. Fingerprinting of psychoactive drugs in zebrafish anxiety-like behaviors. PLoS ONE 2014, 9, e103943. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Tien, M.Z.; Sydykova, D.K.; Meyer, A.G.; Wilke, C.O. PeptideBuilder: A simple Python library to generate model peptides. PeerJ 2013, 1, e80. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Ten Brink, T.; Exner, T.E. pK(a) based protonation states and microspecies for protein-ligand docking. J. Comput. Aided Mol. Des. 2010, 24, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Wojcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A new open-source player in the drug discovery field. J. Cheminform. 2015, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Collette, A. Python and HDF5; O’Reilly: Sebastopol, CA, USA, 2013. [Google Scholar]
- Oliphant, T.E. Guide to NumPy, 2nd ed.; CreateSpace Independent Publishing Platform: North Charleston, SC, USA, 2015. [Google Scholar]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.8. Available online: http://pymol.sourceforge.net/overview/index.htm (accessed on 13 January 2019).
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | IC50, M | Sequence | IC50, M |
|---|---|---|---|
| AutoDock Vina | PeptoGrid | ||
| AWW | 6.5 | GAW | 240.0 |
| VWY | 9.4 | GHG | 122.0 |
| FGG | 82.5 | ||
| GGY * | 1.3 | ||
| GTW | 464.5 | ||
| GVW | 240.0 | ||
| PLANTS | PeptoGrid | ||
| KFY | 45.0 | AGS | 527.9 |
| YKW | 13.3 | VAA | 13.0 |
| RFH | 330.5 | GAP | 9.3 |
| IWH | 3.5 | GTG | 5.5 |
| GHG | 122.0 | ||
| AVV | 66.6 | ||
| LeDock | PeptoGrid | ||
| WYS | 500.0 | AGS | 527.9 |
| KFY | 45.0 | GAP | 9.3 |
| FWN | 18.3 | GPA | 405.0 |
| FNQ | 333.5 | GTG | 5.5 |
| GPV | 4.7 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zalevsky, A.O.; Zlobin, A.S.; Gedzun, V.R.; Reshetnikov, R.V.; Lovat, M.L.; Malyshev, A.V.; Doronin, I.I.; Babkin, G.A.; Golovin, A.V. PeptoGrid—Rescoring Function for AutoDock Vina to Identify New Bioactive Molecules from Short Peptide Libraries. Molecules 2019, 24, 277. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24020277
Zalevsky AO, Zlobin AS, Gedzun VR, Reshetnikov RV, Lovat ML, Malyshev AV, Doronin II, Babkin GA, Golovin AV. PeptoGrid—Rescoring Function for AutoDock Vina to Identify New Bioactive Molecules from Short Peptide Libraries. Molecules. 2019; 24(2):277. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24020277
Chicago/Turabian StyleZalevsky, Arthur O., Alexander S. Zlobin, Vasilina R. Gedzun, Roman V. Reshetnikov, Maxim L. Lovat, Anton V. Malyshev, Igor I. Doronin, Gennady A. Babkin, and Andrey V. Golovin. 2019. "PeptoGrid—Rescoring Function for AutoDock Vina to Identify New Bioactive Molecules from Short Peptide Libraries" Molecules 24, no. 2: 277. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24020277