
Novel C7-Substituted Coumarins as Selective Monoamine Oxidase Inhibitors: Discovery, Synthesis and Theoretical Simulation

 and
and
Abstract
:
1. Introduction
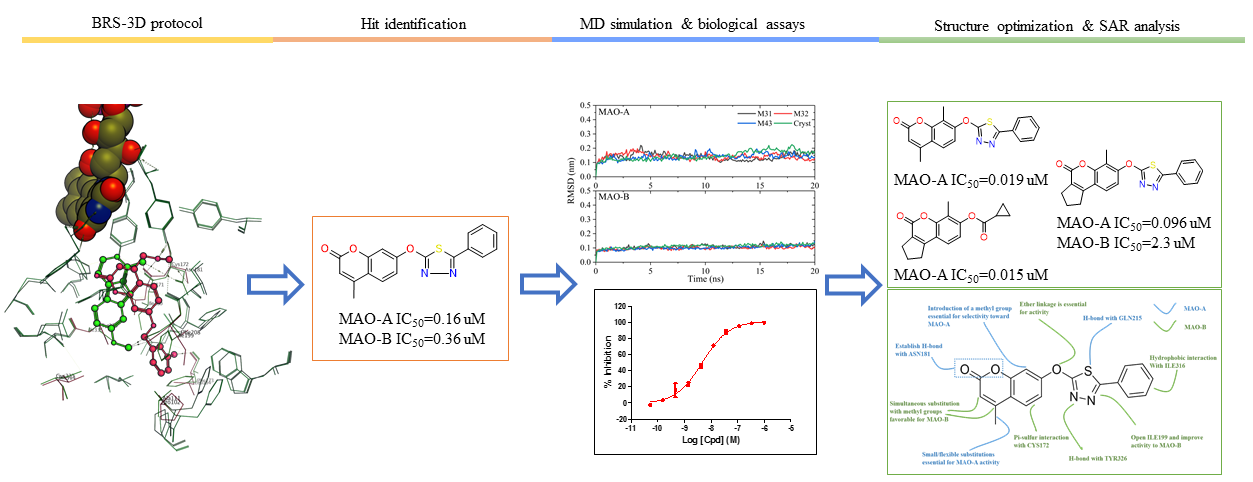
2. Results and Discussion
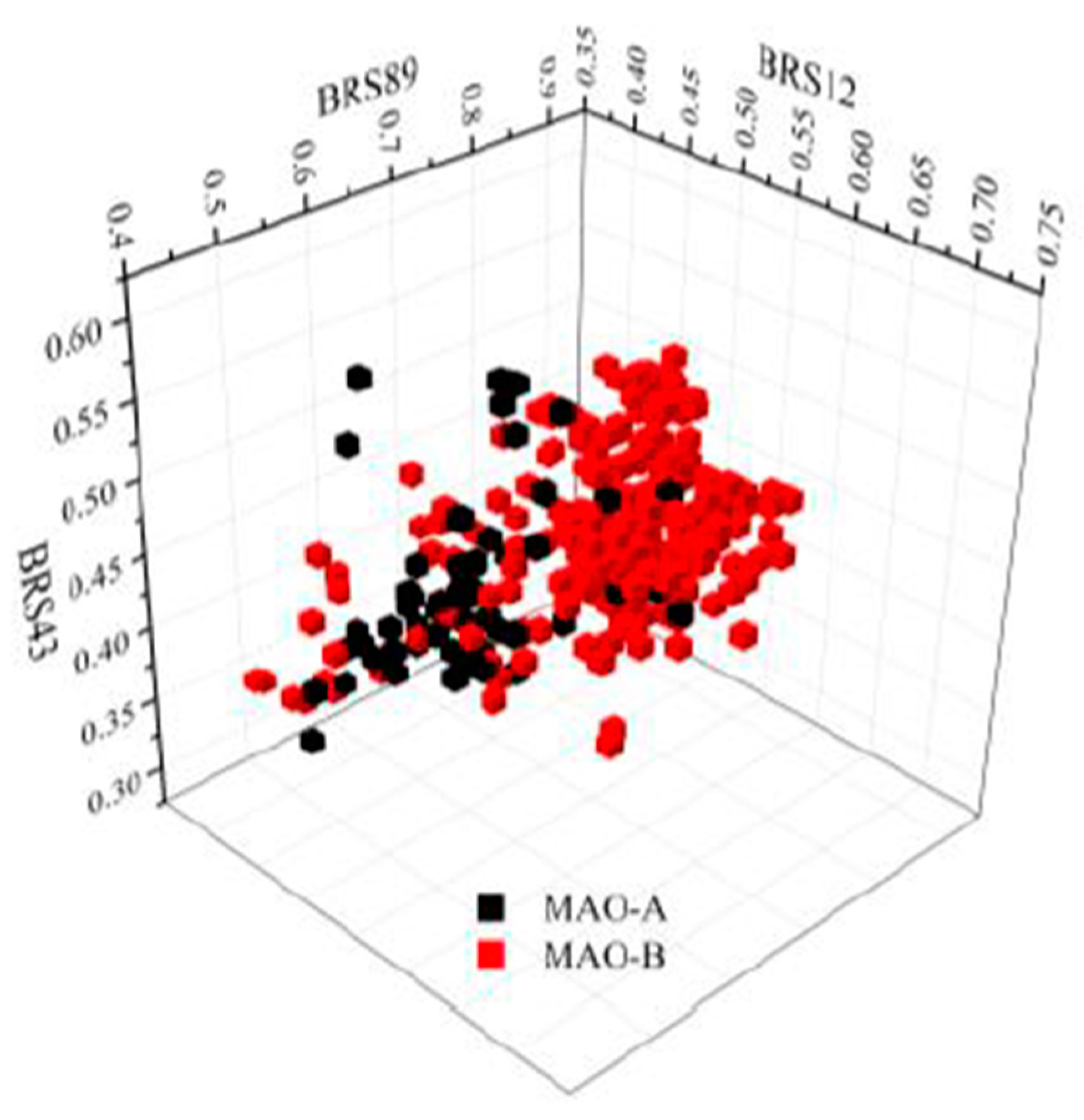
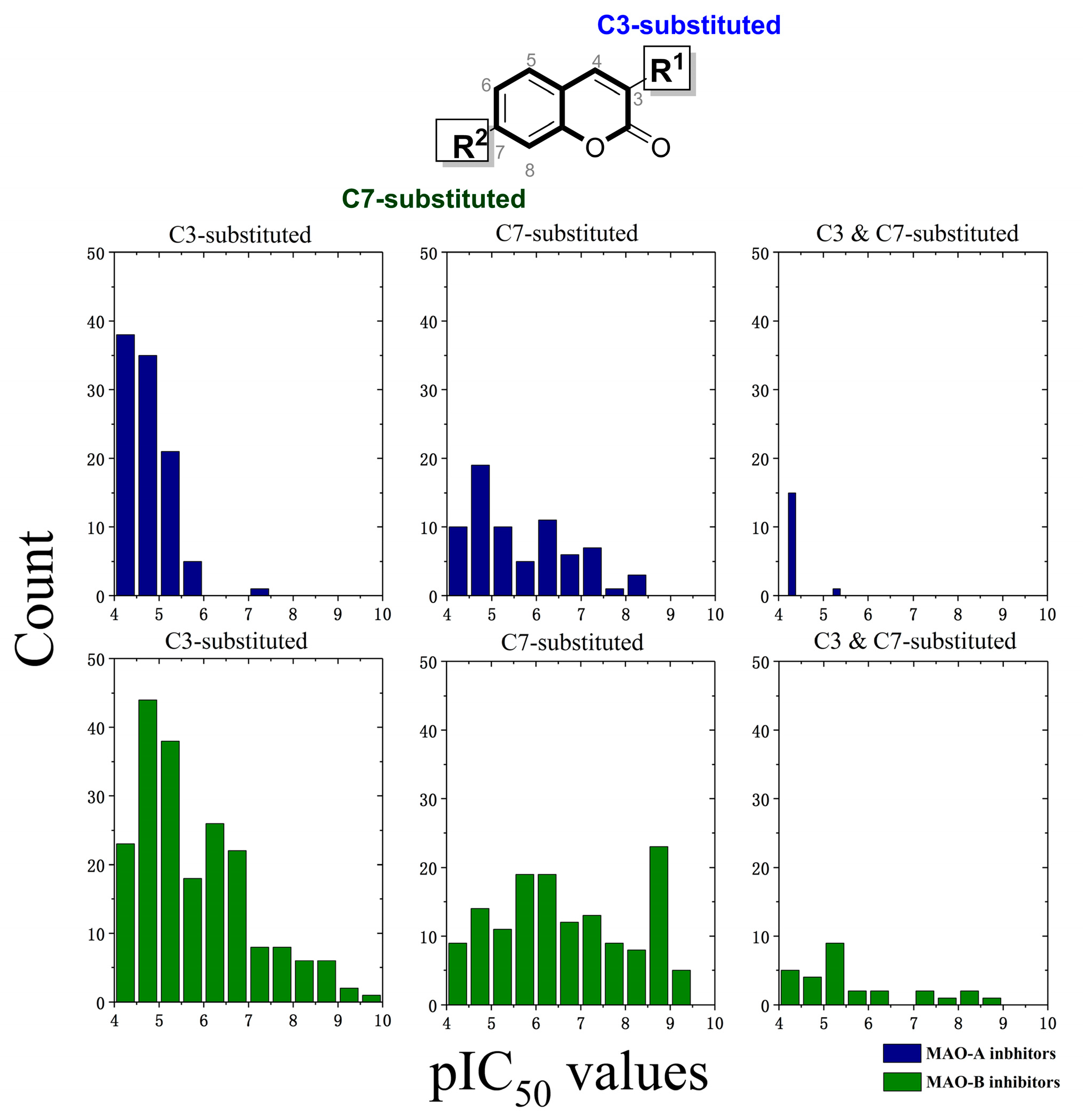
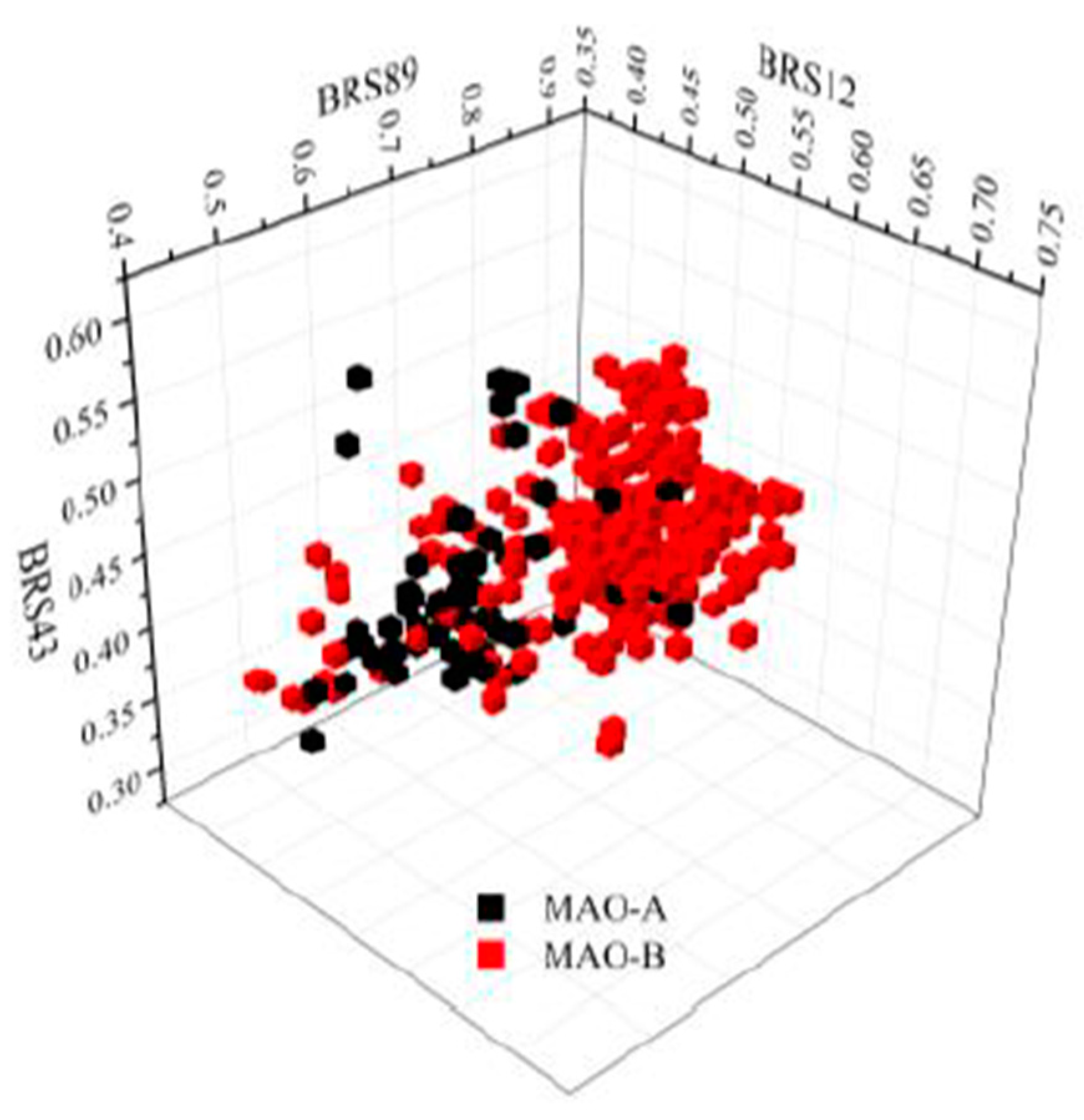
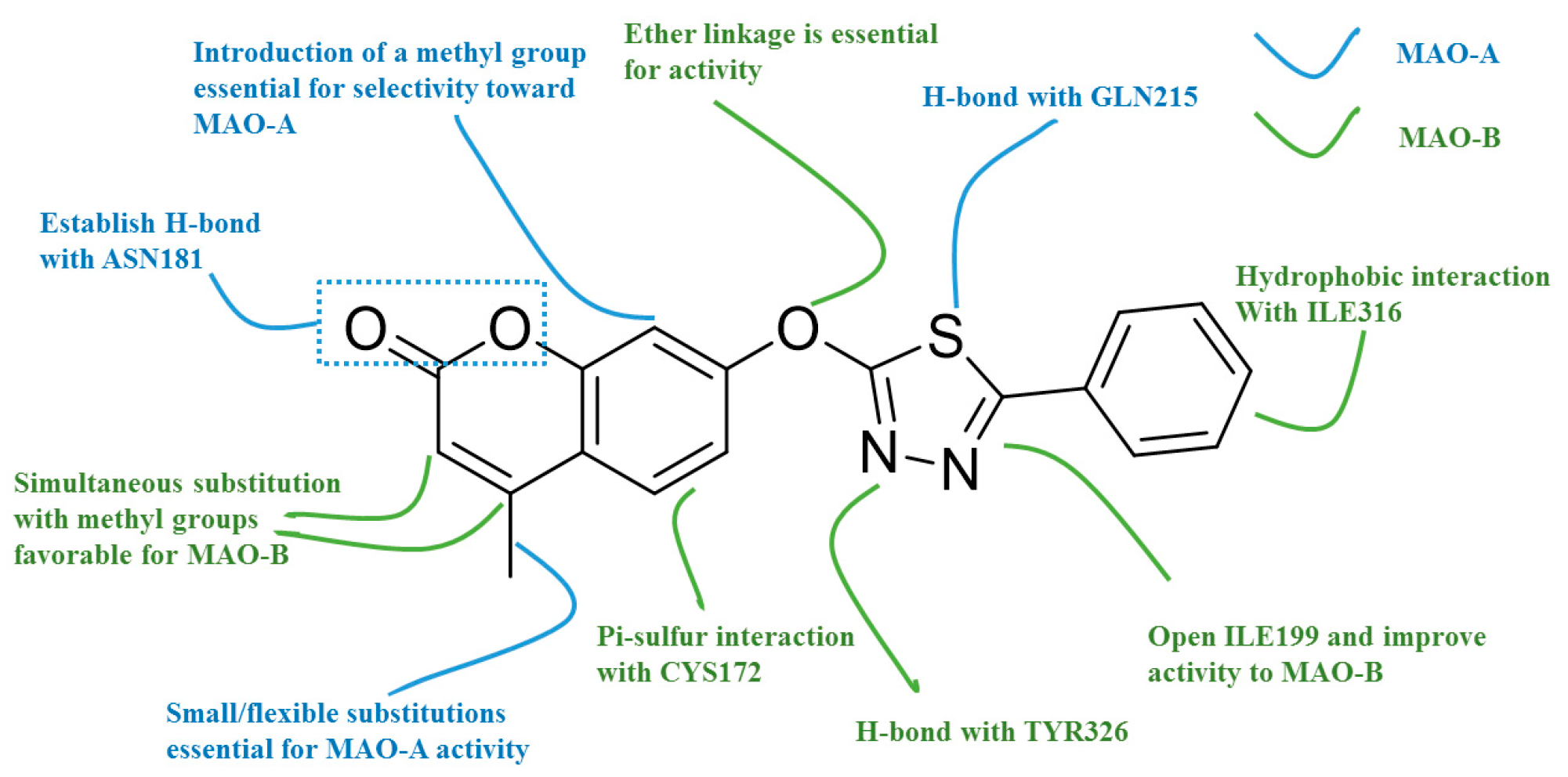
2.1. Qualitative SAR of Previously Identified Hits
2.2. MM/PBSA Binding Energy and Decomposition Analysis
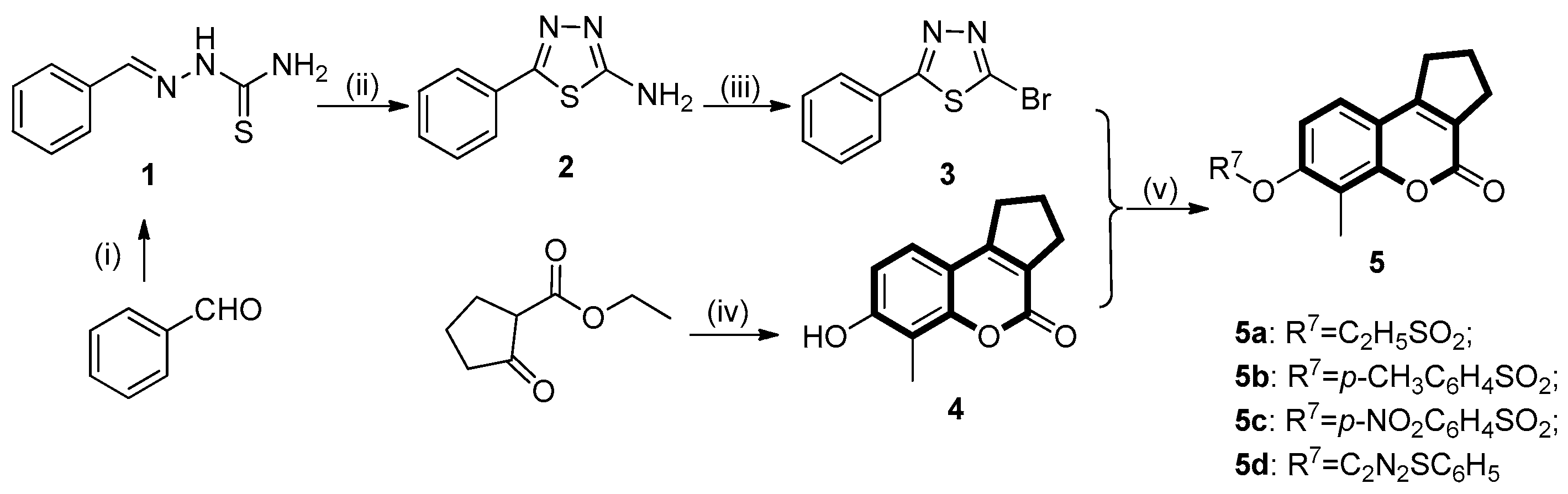
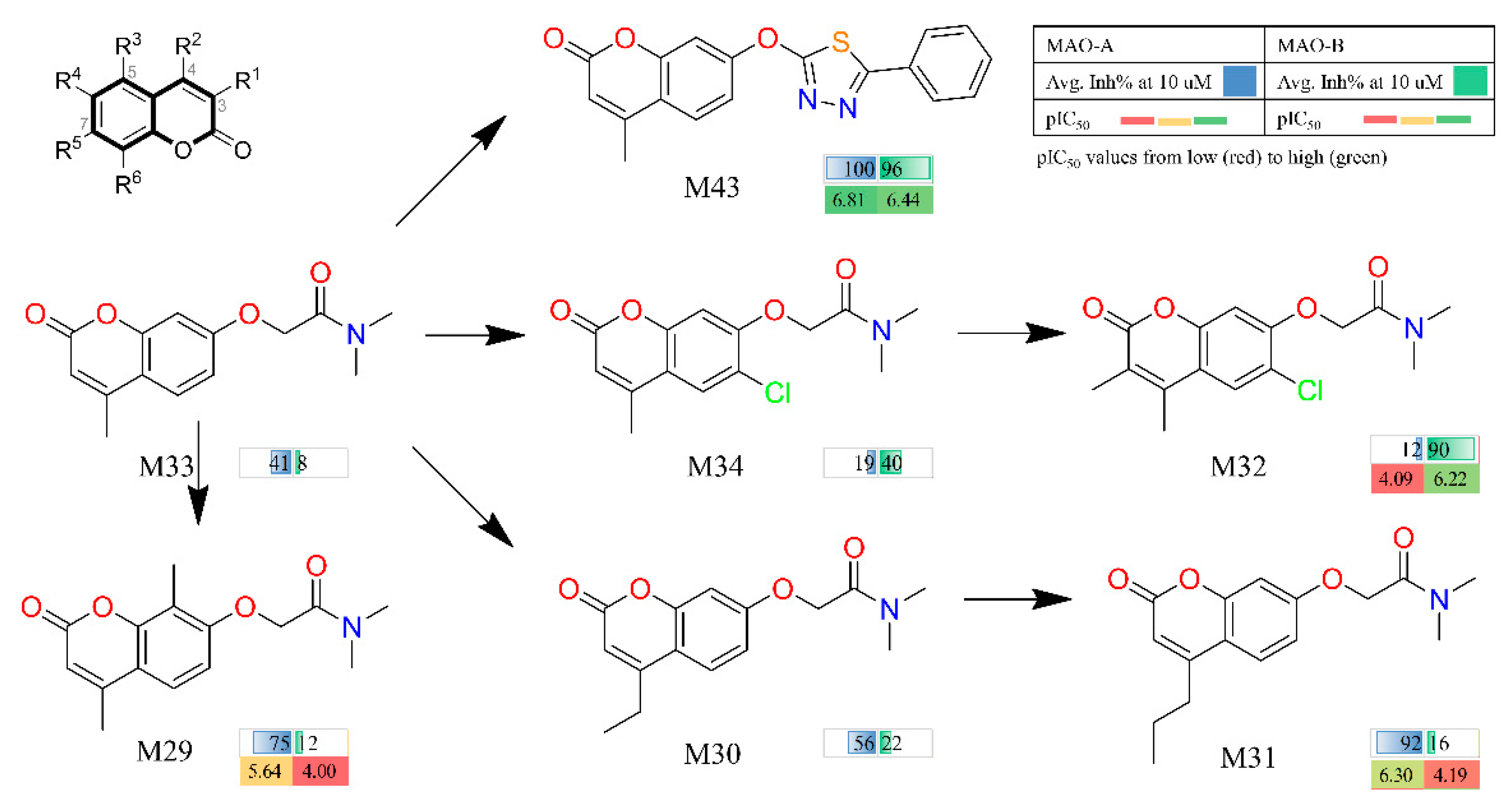
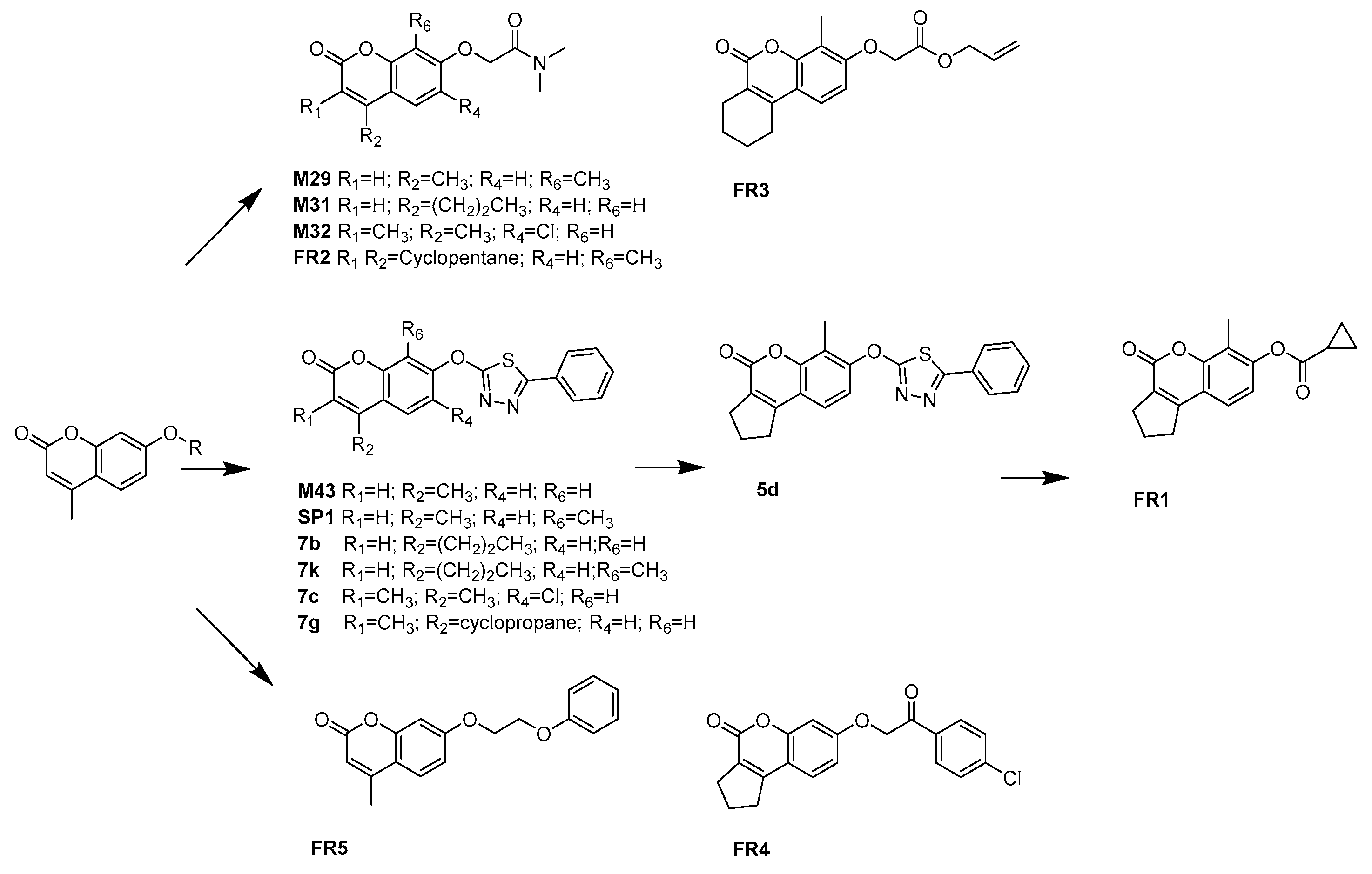
2.3. Discovery, Synthesis and Biological Activity
2.4. In Silico Pharmacokinetic Properties
3. Conclusions
4. Materials and Methods
4.1. Docking Simulations
4.2. Molecular Dynamic Simulations
4.3. MM/PBSA Calculations and Energy Decomposition Analysis
4.4. Chemistry
4.4.1. Synthesis of (E)-2-benzylidenehydrazinecarbothioamide (1)
4.4.2. Synthesis of 5-phenyl-1,3,4-thiadiazol-2-amine (2)
4.4.3. Preparation of 2-bromo-5-phenyl-1,3,4-thiadiazole (3)
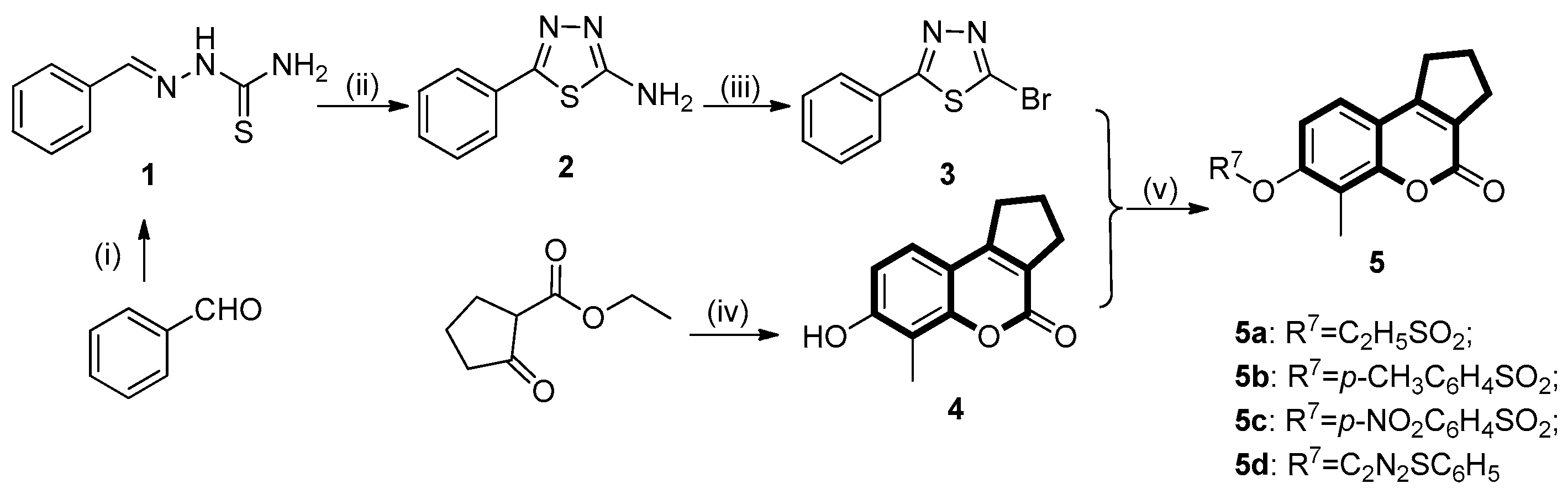
4.4.4. General Procedure for the Synthesis of 7-hydroxy-6-methyl-2,3-dihydrocyclopenta[c]chromen- 4(1H)-one (4)
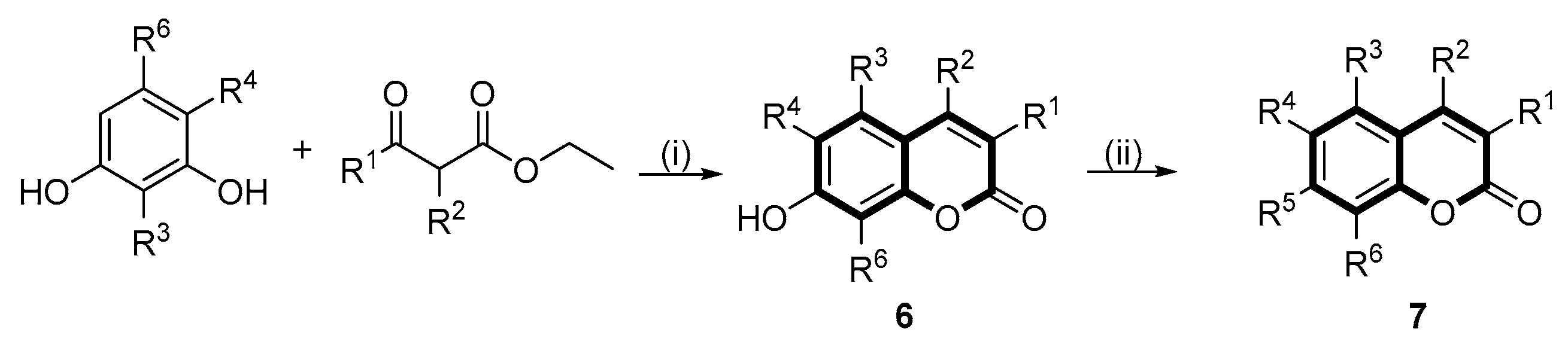
4.4.5. Preparation of 7-hydroxyl Substituted Coumarin Derivatives (6a–e)
4.4.6. General Procedure for the Synthesis of Target Compounds (5d, 7a–c, 7g and 7k)
4.4.7. Synthesis of the Coumarin Sulfonates (5a–c, 7d–f and 7h–j)
4.5. Biological Assays
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tripathi, A.C.; Upadhyay, S.; Paliwal, S.; Saraf, S.K. Privileged scaffolds as MAO inhibitors: Retrospect and prospects. Eur. J. Med. Chem. 2018, 145, 445–497. [Google Scholar] [CrossRef] [PubMed]
- Entzeroth, M.; Ratty, A.K. Monoamine Oxidase Inhibitors—Revisiting a Therapeutic Principle. Open J. Depress. 2017, 6, 31–68. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nature Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Grimsby, J.; Chen, K.; Wang, L.J.; Lan, N.C.; Shih, J.C. Human monoamine oxidase A and B genes exhibit identical exon-intron organization. Proc. Natl. Acad. Sci. USA 1991, 88, 3637–3641. [Google Scholar] [CrossRef]
- Eisner, P.; Klasen, M.; Wolf, D.; Zerres, K.; Eggermann, T.; Eisert, A.; Zvyagintsev, M.; Sarkheil, P.; Mathiak, K.A.; Zepf, F.; et al. Cortico-limbic connectivity in MAOA-L carriers is vulnerable to acute tryptophan depletion. Hum. Brain Mapp. 2017, 38, 1622–1635. [Google Scholar] [CrossRef]
- Edmondson, D.E.; Binda, C.; Wang, J.; Upadhyay, A.K.; Mattevi, A. Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry 2009, 48, 4220–4230. [Google Scholar] [CrossRef]
- Narayanaswami, V.; Drake, L.R.; Brooks, A.F.; Meyer, J.H.; Houle, S.; Kilbourn, M.R.; Scott, P.J.H.; Vasdev, N. Classics in Neuroimaging: Development of PET Tracers for Imaging Monoamine Oxidases. ACS Chem. Neurosci. 2019, 10, 1867–1871. [Google Scholar] [CrossRef] [Green Version]
- Is, Y.S.; Durdagi, S.; Aksoydan, B.; Yurtsever, M. Proposing Novel MAO-B Hit Inhibitors Using Multidimensional Molecular Modeling Approaches and Application of Binary QSAR Models for Prediction of Their Therapeutic Activity, Pharmacokinetic and Toxicity Properties. ACS Chem. Neurosci. 2018, 9, 1768–1782. [Google Scholar] [CrossRef]
- Matos, M.J.; Teran, C.; Perez-Castillo, Y.; Uriarte, E.; Santana, L.; Vina, D. Synthesis and study of a series of 3-arylcoumarins as potent and selective monoamine oxidase B inhibitors. J. Med. Chem. 2011, 54, 7127–7137. [Google Scholar] [CrossRef]
- Blair, H.A.; Dhillon, S. Safinamide: A Review in Parkinson’s Disease. CNS Drugs 2017, 31, 169–176. [Google Scholar] [CrossRef]
- Pisani, L.; Farina, R.; Nicolotti, O.; Gadaleta, D.; Soto-Otero, R.; Catto, M.; Di Braccio, M.; Mendez-Alvarez, E.; Carotti, A. In silico design of novel 2H-chromen-2-one derivatives as potent and selective MAO-B inhibitors. Eur. J. Med. Chem. 2015, 89, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Xie, S.S.; Jiang, N.; Lan, J.S.; Kong, L.Y.; Wang, X.B. Multifunctional coumarin derivatives: Monoamine oxidase B (MAO-B) inhibition, anti-beta-amyloid (Abeta) aggregation and metal chelation properties against Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2015, 25, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Joubert, J.; Foka, G.B.; Repsold, B.P.; Oliver, D.W.; Kapp, E.; Malan, S.F. Synthesis and evaluation of 7-substituted coumarin derivatives as multimodal monoamine oxidase-B and cholinesterase inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 125, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Catto, M.; Nicolotti, O.; Grossi, G.; Di Braccio, M.; Soto-Otero, R.; Mendez-Alvarez, E.; Stefanachi, A.; Gadaleta, D.; Carotti, A. Fine molecular tuning at position 4 of 2H-chromen-2-one derivatives in the search of potent and selective monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2013, 70, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.; Reis, J.; Silva, T.; Matos, M.J.; Bagetta, D.; Ortuso, F.; Alcaro, S.; Uriarte, E.; Borges, F. Coumarin versus Chromone Monoamine Oxidase B Inhibitors: Quo Vadis? J. Med. Chem. 2017, 60, 7206–7212. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Bizzarri, B.; Granese, A.; Carradori, S.; Yanez, M.; Orallo, F.; Ortuso, F.; et al. Synthesis, molecular modeling, and selective inhibitory activity against human monoamine oxidases of 3-carboxamido-7-substituted coumarins. J. Med. Chem. 2009, 52, 1935–1942. [Google Scholar] [CrossRef]
- Secci, D.; Carradori, S.; Bolasco, A.; Chimenti, P.; Yanez, M.; Ortuso, F.; Alcaro, S. Synthesis and selective human monoamine oxidase inhibition of 3-carbonyl, 3-acyl, and 3-carboxyhydrazido coumarin derivatives. Eur. J. Med. Chem. 2011, 46, 4846–4852. [Google Scholar] [CrossRef]
- Matos, M.J.; Vina, D.; Picciau, C.; Orallo, F.; Santana, L.; Uriarte, E. Synthesis and evaluation of 6-methyl-3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 5053–5055. [Google Scholar] [CrossRef]
- Matos, M.J.; Vina, D.; Quezada, E.; Picciau, C.; Delogu, G.; Orallo, F.; Santana, L.; Uriarte, E. A new series of 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3268–3270. [Google Scholar] [CrossRef]
- Matos, M.J.; Vilar, S.; Garcia-Morales, V.; Tatonetti, N.P.; Uriarte, E.; Santana, L.; Vina, D. Insight into the functional and structural properties of 3-arylcoumarin as an interesting scaffold in monoamine oxidase B inhibition. ChemMedChem 2014, 9, 1488–1500. [Google Scholar] [CrossRef]
- Matos, M.J.; Rodriguez-Enriquez, F.; Vilar, S.; Santana, L.; Uriarte, E.; Hripcsak, G.; Estrada, M.; Rodriguez-Franco, M.I.; Vina, D. Potent and selective MAO-B inhibitory activity: Amino- versus nitro-3-arylcoumarin derivatives. Bioorg. Med. Chem. Lett. 2015, 25, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Delogu, G.; Picciau, C.; Ferino, G.; Quezada, E.; Podda, G.; Uriarte, E.; Vina, D. Synthesis, human monoamine oxidase inhibitory activity and molecular docking studies of 3-heteroarylcoumarin derivatives. Eur. J. Med. Chem. 2011, 46, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Costas-Lago, M.C.; Besada, P.; Rodriguez-Enriquez, F.; Vina, D.; Vilar, S.; Uriarte, E.; Borges, F.; Teran, C. Synthesis and structure-activity relationship study of novel 3-heteroarylcoumarins based on pyridazine scaffold as selective MAO-B inhibitors. Eur. J. Med. Chem. 2017, 139, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rendenbach-Müller, B.; Schlecker, R.; Traut, M.; Weifenbach, H. Synthesis of coumarins as subtype-selective inhibitors of monoamine oxidase. Bioorg. Med. Chem. Lett. 1994, 4, 1195–1198. [Google Scholar] [CrossRef]
- Santana, L.; Gonzalez-Diaz, H.; Quezada, E.; Uriarte, E.; Yanez, M.; Vina, D.; Orallo, F. Quantitative structure-activity relationship and complex network approach to monoamine oxidase A and B inhibitors. J. Med. Chem. 2008, 51, 6740–6751. [Google Scholar] [CrossRef]
- Abdelhafez, O.M.; Amin, K.M.; Ali, H.I.; Abdalla, M.M.; Batran, R.Z. Synthesis of new 7-oxycoumarin derivatives as potent and selective monoamine oxidase A inhibitors. J. Med. Chem. 2012, 55, 10424–10436. [Google Scholar] [CrossRef]
- Abdelhafez, O.M.; Amin, K.M.; Ali, H.I.; Abdalla, M.M.; Batran, R.Z. Monoamine oxidase A and B inhibiting effect and molecular modeling of some synthesized coumarin derivatives. Neurochem. Int. 2013, 62, 198–209. [Google Scholar] [CrossRef]
- Gnerre, C.; Catto, M.; Leonetti, F.; Weber, P.; Carrupt, P.A.; Altomare, C.; Carotti, A.; Testa, B. Inhibition of monoamine oxidases by functionalized coumarin derivatives: Biological activities, QSARs, and 3D-QSARs. J. Med. Chem. 2000, 43, 4747–4758. [Google Scholar] [CrossRef]
- Deng, Z.L.; Du, C.X.; Li, X.; Hu, B.; Kuang, Z.K.; Wang, R.; Feng, S.Y.; Zhang, H.Y.; Kong, D.X. Exploring the biologically relevant chemical space for drug discovery. J. Chem. Inf. Model. 2013, 53, 2820–2828. [Google Scholar] [CrossRef]
- Hu, B.; Kuang, Z.K.; Feng, S.Y.; Wang, D.; He, S.B.; Kong, D.X. Three-Dimensional Biologically Relevant Spectrum (BRS-3D): Shape Similarity Profile Based on PDB Ligands as Molecular Descriptors. Molecules 2016, 21, 1554. [Google Scholar] [CrossRef]
- Wang, D.; Li, Z.; Liu, Y.; Chen, M.; Chen, N.; Zuo, Z.; Kong, D.X. Identification of novel monoamine oxidase selective inhibitors employing a hierarchical ligand-based virtual screening strategy. Future Med. Chem. 2019, 11, 801–816. [Google Scholar] [CrossRef] [PubMed]
- He, S.B.; Ben, H.; Kuang, Z.K.; Wang, D.; Kong, D.X. Predicting Subtype Selectivity for Adenosine Receptor Ligands with Three-Dimensional Biologically Relevant Spectrum (BRS-3D). Sci. Rep. 2016, 6, 36595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, Z.K.; Feng, S.Y.; Hu, B.; Wang, D.; He, S.B.; Kong, D.X. Predicting subtype selectivity of dopamine receptor ligands with three-dimensional biologically relevant spectrum. Chem. Biol. Drug Des. 2016, 88, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Catto, M.; Nicolotti, O.; Leonetti, F.; Carotti, A.; Favia, A.D.; Soto-Otero, R.; Mendez-Alvarez, E.; Carotti, A. Structural insights into monoamine oxidase inhibitory potency and selectivity of 7-substituted coumarins from ligand- and target-based approaches. J. Med. Chem. 2006, 49, 4912–4925. [Google Scholar] [CrossRef] [PubMed]
- ACD/Percepta Platform. Advanced Chemistry Development; Advanced Chemistry Development, Inc.: Toronto, ON, Canada. Available online: www.acdlabs.com (accessed on 26 May 2019).
- Shao, C.-Y.; Su, B.-H.; Tu, Y.-S.; Lin, C.; Lin, O.A.; Tseng, Y.J. CypRules: A rule-based P450 inhibition prediction server. Bioinformatics 2015, 31, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Gaussian 09. Revision E. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MAO-A | MAO-B | |||||
|---|---|---|---|---|---|---|
| Cmpd | Inh% | pIC50 | binding free energy | Inh% | pIC50 | binding free energy |
| M29 | 75 | 5.6 | −121.03 ± 0.92 | 12 | 4 | −109.72 ± 1.07 |
| M30 | 56 | ND | −116.90 ± 1.09 | 22 | ND | −102.79 ± 1.27 |
| M31 | 92 | 6.3 | −127.03 ± 1.28 | 16 | 4.2 | −113.63 ± 1.26 |
| M32 | 12 | 4.1 | −112.54 ± 1.12 | 90 | 6.2 | −122.61 ± 1.12 |
| M33 | 41 | ND | −115.25 ± 1.01 | 8 | ND | −105.59 ± 1.08 |
| M34 | 19 | ND | −107.67 ± 0.99 | 40 | ND | −111.18 ± 0.95 |
| M43 (7a) | 102 | 6.8 | −129.34 ± 1.10 | 96 | 6.4 | −135.59 ± 1.11 |
| C18 | - | 4.8 | −96.05 ± 1.31 | - | 7 | −144.57 ± 1.29 |
| Terms | MAO-A vs. M31 | MAO-B vs. M31 | MAO-A vs. M32 | MAO-B vs. M32 |
|---|---|---|---|---|
| ΔGvdW | −211.794 ± 0.990 | −198.535 ± 1.147 | −214.799 ± 0.969 | −198.570 ± 1.187 |
| ΔGele | −38.523 ± 0.817 | −61.460 ± 0.704 | −31.376 ± 0.547 | −1.362 ± 0.727 |
| ΔGpol | 140.734 ± 0.539 | 164.645 ± 1.247 | 151.326 ± 0.787 | 144.800 ± 0.895 |
| ΔGnonpol | −17.431 ± 0.071 | −18.241 ± 0.073 | −17.715 ± 0.066 | −17.418 ± 0.065 |
| ΔG | −127.034 ± 1.281 | −113.631 ± 1.260 | −12.539 ± 1.116 | −122.598 ± 1.144 |
| Residue | MAO-A vs. M31 | MAO-B vs. M31 | MAO-A vs. M32 | MAO-B vs. M32 | |
|---|---|---|---|---|---|
| MAO-A | MAO-B | Total Energy | Total Energy | Total Energy | Total Energy |
| Tyr69 | Tyr60 | −1.4631 ± 0.0443 | −3.5967 ± 0.0614 | −2.7071 ± 0.0511 | −3.0942 ± 0.0708 |
| Ala111 | Pro102 | 0.3332 ± 0.0714 | 0.8449 ± 0.1369 | 0.5828 ± 0.0985 | −0.0127 ± 0.0890 |
| Phe112 | Phe103 | −0.3019 ± 0.0205 | 0.4229 ± 0.0305 | −0.1243 ± 0.0153 | 0.0942 ± 0.0165 |
| Pro113 | Pro104 | −0.0764 ± 0.0091 | −0.0264 ± 0.0079 | 0.0414 ± 0.0055 | −0.0514 ± 0.0163 |
| Trp128 | Trp119 | −0.0145 ± 0.0064 | −0.0097 ± 0.0097 | 0.0893 ± 0.0033 | −0.1936 ± 0.0077 |
| Phe173 | Leu164 | −0.0364 ± 0.0108 | −0.6217 ± 0.0187 | −0.0662 ± 0.0114 | −0.4042 ± 0.0134 |
| Leu176 | Leu167 | −0.0701 ± 0.0213 | −0.0436 ± 0.0256 | 0.1918 ± 0.0137 | −0.2503 ± 0.0171 |
| Phe177 | Phe168 | −0.1931 ± 0.0494 | −0.4662 ± 0.026 | 0.6680 ± 0.0293 | −0.5791 ± 0.0819 |
| Ile180 | Leu171 | −5.3303 ± 0.1123 | −6.2815 ± 0.1427 | −2.4365 ± 0.2223 | −7.2863 ± 0.1543 |
| Asn181 | Cys172 | −4.1478 ± 0.2633 | 0.2993 ± 0.124 | 0.8583 ± 0.2391 | −2.5912 ± 0.1582 |
| Tyr197 | Tyr188 | 1.1053 ± 0.0829 | 1.7647 ± 0.076 | −4.1025 ± 0.1387 | 0.1502 ± 0.0949 |
| Ile207 | Ile198 | −3.9048 ± 0.1647 | −0.9873 ± 0.1489 | −3.1187 ± 0.1347 | −1.9079 ± 0.1410 |
| Phe208 | Ile199 | −6.1625 ± 0.2403 | −1.1994 ± 0.2342 | −2.2755 ± 0.2184 | 0.0638 ± 0.1966 |
| Gln215 | Gln206 | −7.5319 ± 0.236 | −1.7379 ± 0.2144 | −3.3143 ± 0.2539 | −1.3530 ± 0.2173 |
| Cys323 | Thr314 | −0.5525 ± 0.0492 | −2.5552 ± 0.0515 | −0.4392 ± 0.0685 | −2.3485 ± 0.0513 |
| Ile325 | Ile316 | −1.7599 ± 0.0637 | −3.1339 ± 0.0787 | −1.6549 ± 0.0398 | −4.0400 ± 0.0843 |
| Ile335 | Tyr326 | −4.5914 ± 0.1095 | −4.8286 ± 0.25 | −4.2492 ± 0.1381 | −5.9530 ± 0.1767 |
| Thr336 | Thr327 | −0.4413 ± 0.0476 | −0.3823 ± 0.0269 | −0.0784 ± 0.0246 | −0.5360 ± 0.0223 |
| Leu337 | Leu328 | −3.0775 ± 0.0836 | −1.0562 ± 0.026 | −2.2824 ± 0.0758 | −1.1358 ± 0.0269 |
| Met350 | Met341 | −0.168 ± 0.0143 | −0.438 ± 0.0118 | −0.6131 ± 0.0173 | −0.3265 ± 0.0107 |
| Phe352 | Phe343 | −0.501 ± 0.0391 | −2.0697 ± 0.1135 | −1.2125 ± 0.0482 | −2.5305 ± 0.0643 |
| Tyr407 | Tyr398 | −5.3629 ± 0.1462 | −6.0795 ± 0.1569 | −7.7790 ± 0.1624 | −8.5574 ± 0.1847 |
| Tyr444 | Tyr435 | −3.919 ± 0.2074 | −2.1508 ± 0.1615 | −7.4496 ± 0.1530 | −1.9394 ± 0.1860 |

| Compounds | R1 | R2 | R4 | R5 | R6 | MAO-A a | MAO-B a | SI b |
|---|---|---|---|---|---|---|---|---|
| M29 | H | CH3 | H |  | CH3 | 2.29 | 100 | −1.64 |
| M31 | H | (CH2)2CH3 | H |  | H | 0.50 | 64.57 | −2.11 |
| M32 | CH3 | CH3 | Cl |  | H | 81.28 | 0.60 | 2.13 |
| M43(7a) | H | CH3 | H |  | H | 0.16 | 0.36 | −0.35 |
| FR1 | –(CH2)3– | H |  | CH3 | 0.0015 | 75% (>1 uM) | <−2.82 | |
| FR2 | –(CH2)3– | H |  | CH3 | 2.82 | 12% (>10 uM) | <−0.55 | |
| FR3 | –(CH2)4– | H |  | CH3 | 0.74 | 66% (>5 uM) | <−0.83 | |
| FR4 | –(CH2)3– | H |  | H | 23% (>10 uM) | 0.018 | >2.74 | |
| FR5 | H | CH3 | H |  | H | 28% (>10 uM) | 0.015 | >2.82 |
| SP1 | H | CH3 | H |  | CH3 | 0.019 | 64% (>5 uM) | <−2.42 |
| 5d | –(CH2)3– | H |  | CH3 | 0.096 | 2.30 | −1.38 | |
| 7b | H | (CH2)2CH3 | H |  | H | 0.024 | 0.01 | 0.38 |
| 7k | H | (CH2)2CH3 | H |  | CH3 | 0.021 | 0.12 | −0.76 |
| 7c | CH3 | CH3 | Cl |  | H | 0.81 | 0.44 | 0.27 |
| 7g | H |  | H |  | H | 2.00 | 6.20 | −0.49 |
| Compounds | logP | MW | Lipinski | Lead-like | Caco-2 | CNS |
|---|---|---|---|---|---|---|
| M29 | 1.94 | 275.3 | 0 violations | 0 violations | Highly permeable | Penetrant |
| M31 | 2.21 | 289.33 | 0 violations | 0 violations | Highly permeable | Penetrant |
| M32 | 2.98 | 309.74 | 0 violations | 0 violations | Highly permeable | Penetrant |
| M43 (7a) | 3.79 | 336.37 | 0 violations | 0 violations | Highly permeable | Penetrant |
| FR1 | 3.3 | 284.31 | 0 violations | 0 violations | Highly permeable | Penetrant |
| FR2 | 2.32 | 301.34 | 0 violations | 0 violations | Highly permeable | Penetrant |
| FR3 | 4.33 | 328.36 | 0 violations | 1 violation | Highly permeable | Penetrant |
| FR4 | 4.57 | 354.78 | 0 violations | 1 violation | Highly permeable | Penetrant |
| FR5 | 3.77 | 296.32 | 0 violations | 0 violations | Highly permeable | Penetrant |
| SP1 | 3.97 | 350.39 | 0 violations | 0 violations | Highly permeable | Penetrant |
| 5d | 4.72 | 376.43 | 0 violations | 1 violation | Highly permeable | Penetrant |
| 7b | 4.49 | 364.42 | 0 violations | 1 violation | Highly permeable | Penetrant |
| 7k | 4.79 | 378.45 | 0 violations | 1 violation | Highly permeable | Penetrant |
| 7c | 4.99 | 384.84 | 0 violations | 1 violation | Highly permeable | Weak Penetrant |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Hong, R.-Y.; Guo, M.; Liu, Y.; Chen, N.; Li, X.; Kong, D.-X. Novel C7-Substituted Coumarins as Selective Monoamine Oxidase Inhibitors: Discovery, Synthesis and Theoretical Simulation. Molecules 2019, 24, 4003. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24214003
Wang D, Hong R-Y, Guo M, Liu Y, Chen N, Li X, Kong D-X. Novel C7-Substituted Coumarins as Selective Monoamine Oxidase Inhibitors: Discovery, Synthesis and Theoretical Simulation. Molecules. 2019; 24(21):4003. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24214003
Chicago/Turabian StyleWang, Dong, Ren-Yuan Hong, Mengyao Guo, Yi Liu, Nianhang Chen, Xun Li, and De-Xin Kong. 2019. "Novel C7-Substituted Coumarins as Selective Monoamine Oxidase Inhibitors: Discovery, Synthesis and Theoretical Simulation" Molecules 24, no. 21: 4003. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24214003