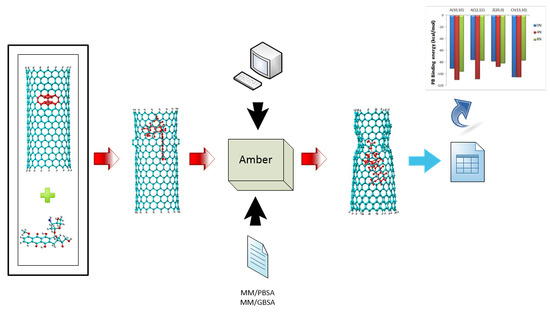
Figure 1.
(a) DOX structure representation (C27H29NO11) in frontal and lateral views; (b) 4-8-8-4 haeckelite defect representation showing bond distances in Å.
Figure 1.
(a) DOX structure representation (C27H29NO11) in frontal and lateral views; (b) 4-8-8-4 haeckelite defect representation showing bond distances in Å.
Figure 2.
DOX initial orientations when encapsulated in the regular part of an armchair Hk2 nanotube. Lateral and frontal views for: (a) v1 orientation (NH2 group pointing to the center of the nanotube), and (b) v2 orientation (inverse orientation). DOX is depicted in red for a better visualization.
Figure 2.
DOX initial orientations when encapsulated in the regular part of an armchair Hk2 nanotube. Lateral and frontal views for: (a) v1 orientation (NH2 group pointing to the center of the nanotube), and (b) v2 orientation (inverse orientation). DOX is depicted in red for a better visualization.
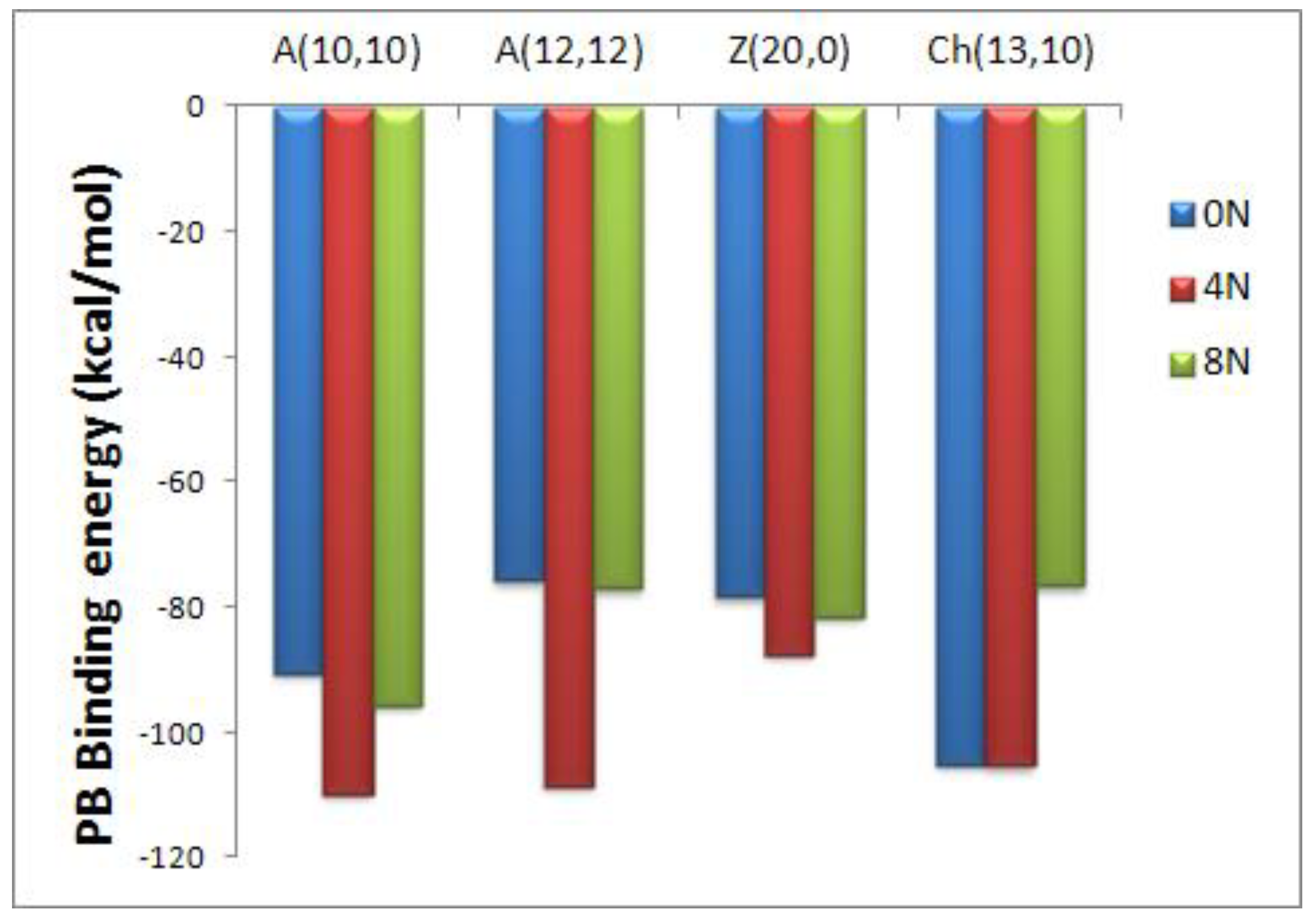
Figure 3.
Representation of the Poisson-Boltzmann (PB) binding free energies for encapsulated DOX-CNT complexes of nitrogen-doped and undoped regular armchair (10,10), armchair (12,12), zigzag (20,0) and chiral (13,10) nanotubes of diameters 14, 16, 15 and 15 Å, respectively.
Figure 3.
Representation of the Poisson-Boltzmann (PB) binding free energies for encapsulated DOX-CNT complexes of nitrogen-doped and undoped regular armchair (10,10), armchair (12,12), zigzag (20,0) and chiral (13,10) nanotubes of diameters 14, 16, 15 and 15 Å, respectively.
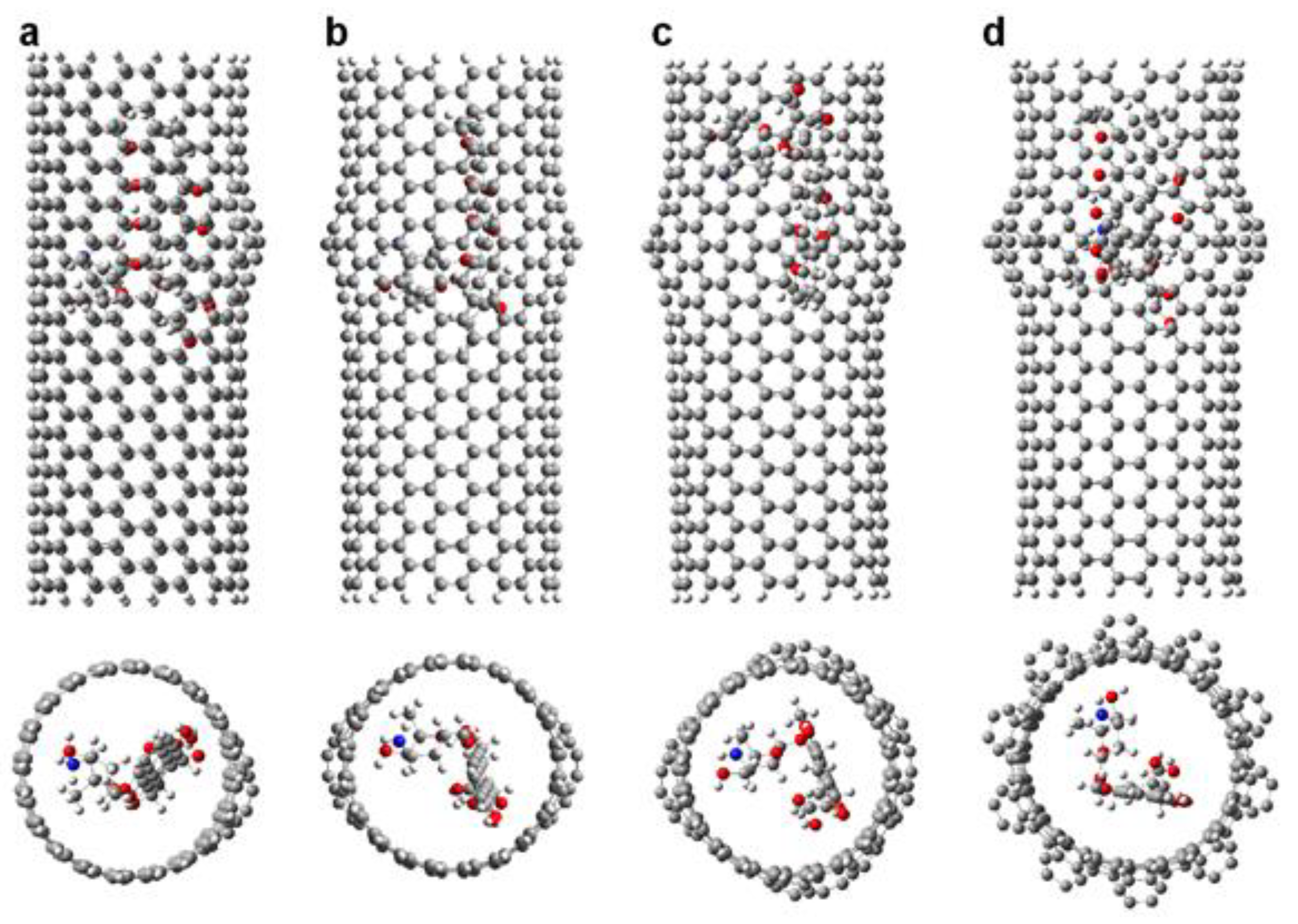
Figure 4.
Representation of the DOX-CNT complexes for armchair (10,10) nanotubes having different number of haeckelite defects. Lateral and frontal views for: (a) one defect (Hk1); (b) two defects (Hk2); (c) four defects (Hk4); (d) the maximum number of defects according to the CNT diameter (Hk), ten defects in the present case.
Figure 4.
Representation of the DOX-CNT complexes for armchair (10,10) nanotubes having different number of haeckelite defects. Lateral and frontal views for: (a) one defect (Hk1); (b) two defects (Hk2); (c) four defects (Hk4); (d) the maximum number of defects according to the CNT diameter (Hk), ten defects in the present case.
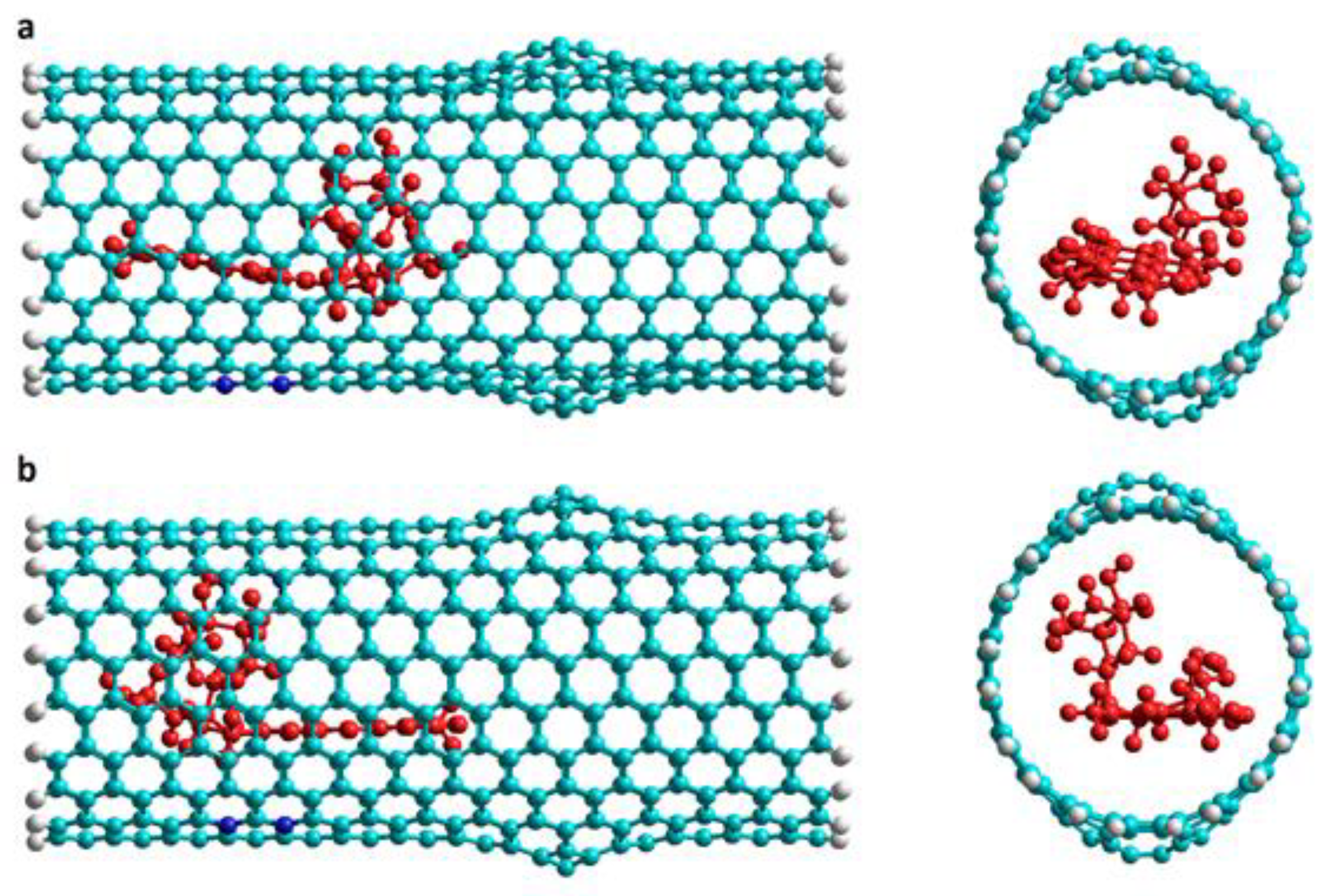
Figure 5.
Representation of the DOX-CNT complexes final structures for armchair (10,10) and zigzag (18,0) Hk1 nanotubes doped with 8N with the DOX encapsulated in the defective part of the nanotube in v1 orientation. Lateral and frontal views for (a) A(10,10)-8N-Hk1-DoxD.v1 and (b) Z(18,0)-8N-Hk1-DoxD.v1. DOX is depicted in red for a better visualization.
Figure 5.
Representation of the DOX-CNT complexes final structures for armchair (10,10) and zigzag (18,0) Hk1 nanotubes doped with 8N with the DOX encapsulated in the defective part of the nanotube in v1 orientation. Lateral and frontal views for (a) A(10,10)-8N-Hk1-DoxD.v1 and (b) Z(18,0)-8N-Hk1-DoxD.v1. DOX is depicted in red for a better visualization.
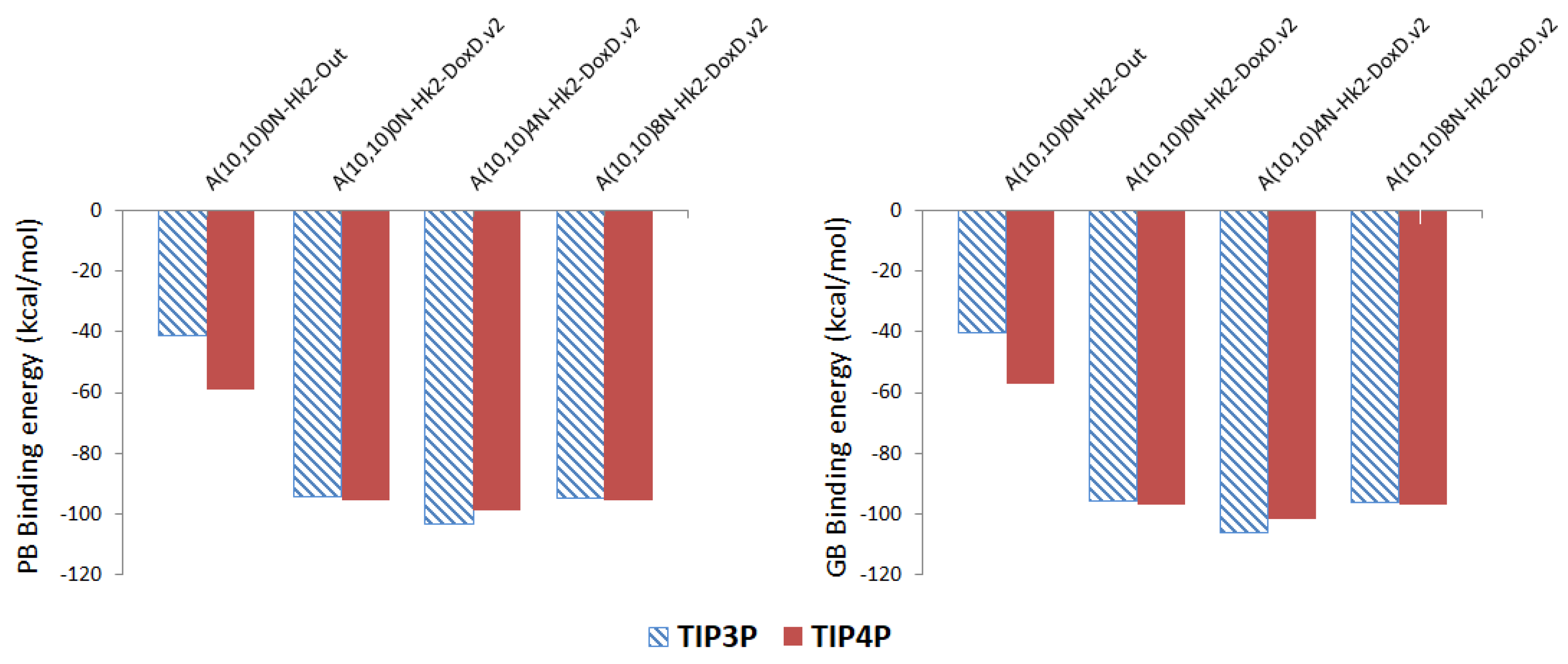
Figure 6.
Representation of the Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energies for DOX-CNT complexes of nitrogen-doped and undoped armchair (10,10) nanotubes having two haeckelite defects (Hk2) using TIP3P (hatched surface) and TIP4P (solid surface) models for water.
Figure 6.
Representation of the Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energies for DOX-CNT complexes of nitrogen-doped and undoped armchair (10,10) nanotubes having two haeckelite defects (Hk2) using TIP3P (hatched surface) and TIP4P (solid surface) models for water.
Figure 7.
Final structures of Z(18,0)0N-Hk2-DoxDIn.v1 at different simulation times. (a) Initial structure (0 ns); (b) 2 ns; (c) 5 ns; (d) 10 ns; (e) 15 ns. Left and right are side views; central are front views.
Figure 7.
Final structures of Z(18,0)0N-Hk2-DoxDIn.v1 at different simulation times. (a) Initial structure (0 ns); (b) 2 ns; (c) 5 ns; (d) 10 ns; (e) 15 ns. Left and right are side views; central are front views.
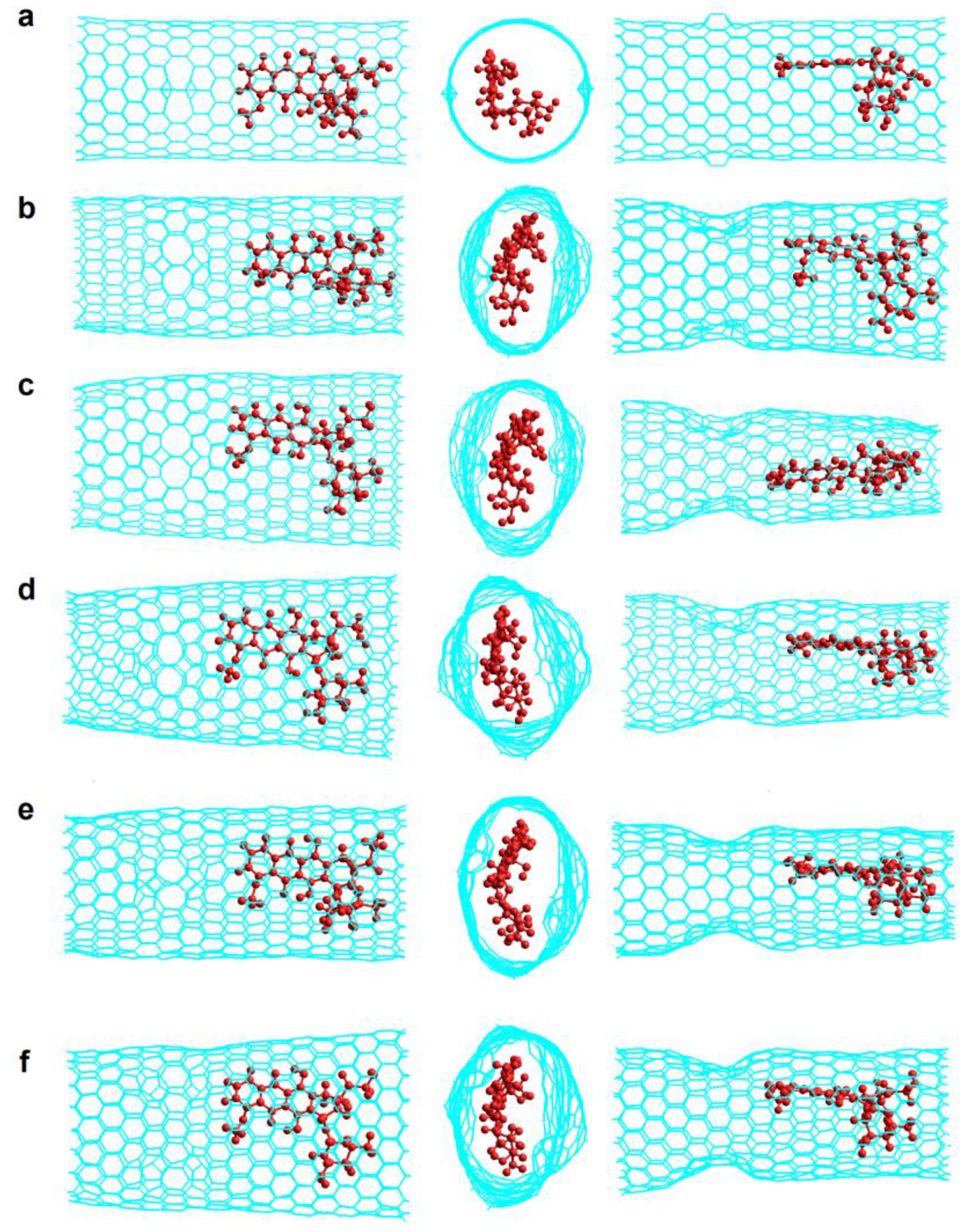
Figure 8.
Final structures of Z(18,0)0N-Hk2-DoxRIn.v2 at different simulation times. (a) Initial structure (0 ns); (b) 2 ns; (c) 5 ns; (d) 30 ns; (e) 50 ns; (f) 100 ns. Left and right, are side views; central, are front views.
Figure 8.
Final structures of Z(18,0)0N-Hk2-DoxRIn.v2 at different simulation times. (a) Initial structure (0 ns); (b) 2 ns; (c) 5 ns; (d) 30 ns; (e) 50 ns; (f) 100 ns. Left and right, are side views; central, are front views.
Figure 9.
Representation of non-covalent interactions, NCI, for final Z(18,0)4N-Hk2-DoxDIn.v1 complex structure. (a,c) are lateral views; (b) is frontal view. Blue surfaces show DOX-CNT strong interactions; in green, weak interactions and in red, repulsion interactions.
Figure 9.
Representation of non-covalent interactions, NCI, for final Z(18,0)4N-Hk2-DoxDIn.v1 complex structure. (a,c) are lateral views; (b) is frontal view. Blue surfaces show DOX-CNT strong interactions; in green, weak interactions and in red, repulsion interactions.
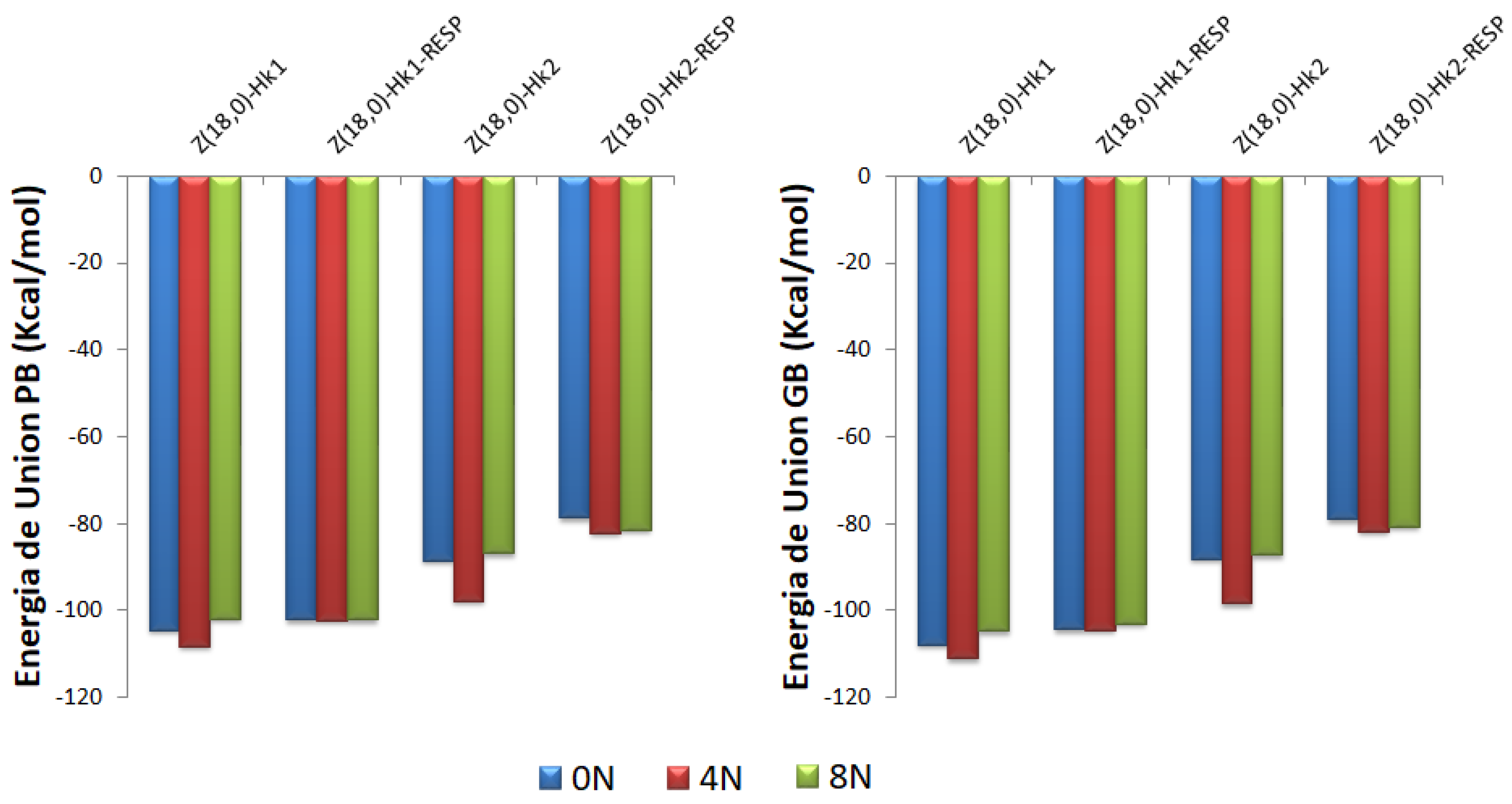
Figure 10.
Representation of the Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energies for DOX-CNT complexes of nitrogen-doped and undoped zigzag (18,0) nanotubes having one and two haeckelite defects considering Mulliken and RESP charges for DOX ligand. Each set of data is represented in the order 0N, 4N and 8N, in blue, red and green colors, respectively.
Figure 10.
Representation of the Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energies for DOX-CNT complexes of nitrogen-doped and undoped zigzag (18,0) nanotubes having one and two haeckelite defects considering Mulliken and RESP charges for DOX ligand. Each set of data is represented in the order 0N, 4N and 8N, in blue, red and green colors, respectively.
Table 1.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for DOX-CNT complexes of armchair (10,10), (12,12) and (15,15) nanotubes having 10, 12 and 15 haeckelite defects, respectively, and 28 carbon-layers, expressed in kcal/mol. D1 and D2 are the initial CNT diameter at the regular region and at the defected one, respectively, dp-NT is the equilibrium distance between the DOX planar fragment and the CNT sidewall surface, d’p-NT is the equilibrium distance between the same point of the DOX planar fragment and the opposite CNT sidewall surface, and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. All distances are expressed in Å.
Table 1.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for DOX-CNT complexes of armchair (10,10), (12,12) and (15,15) nanotubes having 10, 12 and 15 haeckelite defects, respectively, and 28 carbon-layers, expressed in kcal/mol. D1 and D2 are the initial CNT diameter at the regular region and at the defected one, respectively, dp-NT is the equilibrium distance between the DOX planar fragment and the CNT sidewall surface, d’p-NT is the equilibrium distance between the same point of the DOX planar fragment and the opposite CNT sidewall surface, and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. All distances are expressed in Å.
| Run | Type | PB | GB | D1 | D2 | Length | dp-NT | dN-NT |
|---|
| 1 | A(10,10)0N-Hk-DoxDIn.v1 | −81.3 | −80.7 | 13.3 | 14.7 | 32.5 | 4.26 | 3.04 |
| 2 | A(10,10)0N-Hk-DoxDIn.v2 | −79.5 | −79.6 | 13.3 | 14.7 | 32.5 | 4.24 | 3.15 |
| 3 | A(10,10)4N-Hk-DoxDIn.v1 | −78.7 | −79.4 | 13.3 | 14.7 | 32.5 | 3.60 | 3.58 |
| 4 | A(10,10)4N-Hk-DoxDIn.v2 | −79.4 | −79.8 | 13.3 | 14.7 | 32.5 | 3.30 | 3.71 |
| 5 | A(10,10)8N-Hk-DoxDIn.v1 | −76.4 | −76.4 | 13.3 | 14.7 | 32.5 | 3.48 | 3.03 |
| 6 | A(10,10)8N-Hk-DoxDIn.v2 | −76.2 | −77.4 | 13.3 | 14.7 | 32.5 | 3.44 | 3.45 |
| 7 | A(12,12)0N-Hk-DoxOut | −42.1 | −41.8 | 16.7 | 21.8 | 20.5 | 3.38 | 3.77 |
| 8 | A(12,12)0N-Hk-DoxIn | −70.3 | −69.9 | 16.7 | 21.8 | 20.5 | 3.59 | 4.37 |
| 9 | A(12,12)4N-Hk-DoxIn | −78.9 | −79.5 | 16.7 | 21.8 | 20.5 | 3.90 | 3.28 |
| 10 | A(12,12)8N-Hk-DoxIn | −79.4 | −79.7 | 16.7 | 21.8 | 20.5 | 3.57 | 3.64 |
| 11 | A(12,12)0N-Hk-DoxDIn.v2 | −75.4 | −74.4 | 16.8 | 21.8 | 33.1 | 3.67 | 3.64 |
| 12 | A(12,12)0N-Hk-DoxRIn.v1 | −72.2 | −71.4 | 16.9 | 21.8 | 33.0 | 3.79 | 3.15 |
| 13 | A(12,12)0N-Hk-DoxRIn.v2 | −75.1 | −74.0 | 16.9 | 21.8 | 33.0 | 3.54 | 3.22 |
| 14 | A(12,12)4N-Hk-DoxRIn.v1 | −73.7 | −72.8 | 16.7 | 21.8 | 33.1 | 3.76 | 3.50 |
| 15 | A(12,12)4N-Hk-DoxRIn.v2 | −72.3 | −72.0 | 16.7 | 21.8 | 33.1 | 3.73 | 4.47 |
| 16 | A(12,12)8N-Hk-DoxRIn.v1 | −78.1 | −77.6 | 16.7 | 21.8 | 33.1 | 3.55 | 3.04 |
| 17 | A(12,12)8N-Hk-DoxRIn.v2 | −71.6 | −70.9 | 16.7 | 21.8 | 33.1 | 4.04 | 3.33 |
| 18 | A(15,15)0N-Hk-DoxOut | −50.2 | −49.3 | 21.1 | 25.0 | 33.6 | 4.18 | 3.49 |
| 19 | A(15,15)0N-Hk-DoxDIn.v1 | −76.6 | −78.3 | 21.1 | 25.0 | 33.6 | 3.76 | 3.17 |
| 20 | A(15,15)0N-Hk-DoxDIn.v2 | −68.3 | −69.3 | 21.1 | 25.0 | 33.6 | 3.76 | 4.17 |
| 21 | A(15,15)4N-Hk-DoxDIn.v1 | −69.9 | −68.7 | 21.1 | 25.0 | 33.6 | 3.48 | 3.34 |
| 22 | A(15,15)4N-Hk-DoxDIn.v2 | −73.9 | −73.6 | 21.1 | 25.0 | 33.6 | 4.38 | 3.69 |
| 23 | A(15,15)8N-Hk-DoxDIn.v2 | −53.8 | −51.5 | 21.1 | 25.0 | 33.6 | 5.87 | 3.45 |
Table 2.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for DOX-CNT complexes of armchair (10,10) nanotubes having one, two and four haeckelite defects (Hk1, Hk2, Hk4, respectively, with ~33 Å length) expressed in kcal/mol. D1 and D2 are the initial CNT diameter at the regular region and at the defected one, respectively, dp-NT is the equilibrium distance between the DOX planar fragment and the CNT sidewall surface, d′p-NT is the equilibrium distance between the same point of the DOX planar fragment and the opposite CNT sidewall surface, and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. All distances are expressed in Å.
Table 2.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for DOX-CNT complexes of armchair (10,10) nanotubes having one, two and four haeckelite defects (Hk1, Hk2, Hk4, respectively, with ~33 Å length) expressed in kcal/mol. D1 and D2 are the initial CNT diameter at the regular region and at the defected one, respectively, dp-NT is the equilibrium distance between the DOX planar fragment and the CNT sidewall surface, d′p-NT is the equilibrium distance between the same point of the DOX planar fragment and the opposite CNT sidewall surface, and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. All distances are expressed in Å.
| Run | Type | PB | GB | D1 | D2 | dp-NT | dN-NT |
|---|
| 1 | A(10,10)0N-Hk1-DoxDIn.v1 | −91.3 | −91.6 | 13.3 | 14.6 | 3.89 | 3.33 |
| 2 | A(10,10)0N-Hk1-DoxDIn.v2 | −92.6 | −93.5 | 13.3 | 14.6 | 3.72 | 3.73 |
| 3 | A(10,10)0N-Hk1-DoxRIn.v1 | −90.4 | −91.5 | 13.3 | 14.6 | 3.59 | 3.34 |
| 4 | A(10,10)4N-Hk1-DoxDIn.v1 | −91.7 | −92.7 | 13.3 | 14.6 | 3.49 | 3.13 |
| 5 | A(10,10)4N-Hk1-DoxDIn.v2 | −91.6 | −94.0 | 13.3 | 14.6 | 3.79 | 3.13 |
| 6 | A(10,10)4N-Hk1-DoxRIn.v1 | −91.7 | −93.3 | 13.3 | 14.6 | 3.62 | 3.90 |
| 7 | A(10,10)8N-Hk1-DoxDIn.v1 | −107.1 | −111.6 | 13.3 | 14.6 | 3.69 | 3.38 |
| 8 | A(10,10)8N-Hk1-DoxDIn.v2 | −93.3 | −94.2 | 13.3 | 14.6 | 3.76 | 3.77 |
| 9 | A(10,10)8N-Hk1-DoxRIn.v1 | −92.0 | −93.1 | 13.3 | 14.6 | 3.76 | 3.57 |
| 10 | A(10,10)0N-Hk2-DoxOut | −41.1 | −40.3 | 13.1 | 15.9 | 3.39 | 5.28 |
| 11 | A(10,10)4N-Hk2-DoxOut | −43.2 | −42.5 | 13.1 | 15.9 | 3.43 | 3.22 |
| 12 | A(10,10)0N-Hk2-DoxDIn.v1 | −91.7 | −95.1 | 13.3 | 15.5 | 3.50 | 4.19 |
| 13 | A(10,10)0N-Hk2-DoxDIn.v2 | −94.6 | −95.9 | 13.3 | 15.6 | 4.02 | 3.81 |
| 14 | A(10,10)0N-Hk2-DoxRIn.v1 | −95.9 | −97.8 | 13.3 | 16.0 | 3.53 | 3.61 |
| 15 | A(10,10)0N-Hk2-DoxRIn.v2 | −102.0 | −103.4 | 13.3 | 16.0 | 3.59 | 2.93 |
| 16 | A(10,10)4N-Hk2-DoxDIn.v1 | −110.2 | −115.4 | 13.3 | 15.9 | 3.54 | 3.20 |
| 17 | A(10,10)4N-Hk2-DoxDIn.v2 | −103.5 | −106.3 | 13.3 | 15.9 | 3.68 | 3.68 |
| 18 | A(10,10)4N-Hk2-DoxRIn.v1 | −92.4 | −94.8 | 13.3 | 15.9 | 3.88 | 3.71 |
| 19 | A(10,10)4N-Hk2-DoxRIn.v2 | −102.8 | −104.4 | 13.3 | 15.9 | 3.63 | 3.43 |
| 20 | A(10,10)8N-Hk2-DoxDIn.v1 | −92.5 | −94.2 | 13.3 | 15.6 | 4.08 | 3.53 |
| 21 | A(10,10)8N-Hk2-DoxDIn.v2 | −94.6 | −96.1 | 13.3 | 15.6 | 3.85 | 3.13 |
| 22 | A(10,10)8N-Hk2-DoxRIn.v1 | −93.6 | −96.9 | 13.3 | 15.5 | 3.62 | 4.05 |
| 23 | A(10,10)8N-Hk2-DoxRIn.v2 | −93.3 | −95.1 | 13.3 | 15.9 | 3.64 | 3.55 |
| 24 | A(10,10)0N-Hk4-DoxDIn.v2 | −82.3 | −83.3 | 14.0 | 15.5 | 3.61 | 3.23 |
| 25 | A(10,10)4N-Hk4-DoxDIn.v2 | −87.5 | −90.9 | 14.1 | 15.9 | 4.10 | 3.03 |
| 26 | A(10,10)8N-Hk4-DoxDIn.v2 | −82.8 | −84.5 | 14.1 | 15.8 | 4.48 | 3.61 |
| 27 | A(10,10)0N-Hk4s-DoxDIn.v1 | −88.1 | −88.8 | 14.0 | 15.5 | 3.97 | 3.71 |
| 28 | A(10,10)0N-Hk4s-DoxDIn.v2 | −87.7 | −87.8 | 14.0 | 15.5 | 3.45 | 2.93 |
| 29 | A(10,10)4N-Hk4s-DoxDIn.v1 | −88.6 | −88.8 | 14.1 | 15.9 | 3.90 | 3.49 |
| 30 | A(10,10)4N-Hk4s-DoxDIn.v2 | −85.9 | −87.1 | 14.1 | 15.9 | 4.85 | 3.44 |
Table 3.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for DOX-CNT complexes of doped armchair (10,10) nanotubes having one and two haeckelite defects (Hk1 and Hk2, respectively) encapsulating two or three DOX molecules. PB/m and GB/m are the corresponding binding free energies per DOX molecule, all of them expressed in kcal/mol. dp-NT and d′p-NT are the equilibrium distances between the planar fragment of the first and second DOX molecules and the CNT sidewall surface respectively, and dN-NT and d’N-NT are the equilibrium distances between the DOX-nitrogen atom of the corresponding first and second DOX molecules and the CNT sidewall surface. All distances are expressed in Å.
Table 3.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for DOX-CNT complexes of doped armchair (10,10) nanotubes having one and two haeckelite defects (Hk1 and Hk2, respectively) encapsulating two or three DOX molecules. PB/m and GB/m are the corresponding binding free energies per DOX molecule, all of them expressed in kcal/mol. dp-NT and d′p-NT are the equilibrium distances between the planar fragment of the first and second DOX molecules and the CNT sidewall surface respectively, and dN-NT and d’N-NT are the equilibrium distances between the DOX-nitrogen atom of the corresponding first and second DOX molecules and the CNT sidewall surface. All distances are expressed in Å.
| Run | Type | PB | GB | PB/m | GB/m | dp-NT | dN-NT | d′p-NT | d′N-NT |
|---|
| 1 | A(10,10)4N-Hk2-2DoxOp | −197.1 | −207.2 | −98.6 | −103.6 | 4.04 | 3.16 | 4.26 | 3.71 |
| 2 | A(10,10)4N-Hk2-2DoxOpN | −201.2 | −210.5 | −100.6 | −105.3 | 3.66 | 3.63 | 3.56 | 3.56 |
| 3 | A(10,10)4N-Hk2-2DoxSec | −216.3 | −229.0 | -108.2 | −114.5 | 3.12 | 3.43 | 4.14 | 3.87 |
| 4 | A(10,10)4N-Hk2-3DoxIn | −255.3 | −270.3 | −85.1 | −90.1 | 3.20 | 3.77 | 3.09 | 3.53 |
| 5 | A(10,10)8N-Hk1-2DoxSec | −185.6 | −190.9 | −92.8 | −95.5 | 3.54 | 3.46 | 3.25 | 3.65 |
| 6 | A(10,10)8N-Hk2-2DoxSec | −201.2 | −211.2 | −100.6 | −105.6 | 3.52 | 4.73 | 3.17 | 3.68 |
Table 4.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for DOX-CNT complexes of saturated armchair (10,10) nanotubes having one and two haeckelite defects and ~33 Å length, expressed in kcal/mol. D1 and D2 are the initial CNT diameter at the regular region and at the defected one, respectively, dp-NT is the equilibrium distance between the encapsulated DOX planar fragment and the CNT sidewall surface and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. The DOX was located in the defect zone in v2 orientation. All distances are expressed in Å.
Table 4.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for DOX-CNT complexes of saturated armchair (10,10) nanotubes having one and two haeckelite defects and ~33 Å length, expressed in kcal/mol. D1 and D2 are the initial CNT diameter at the regular region and at the defected one, respectively, dp-NT is the equilibrium distance between the encapsulated DOX planar fragment and the CNT sidewall surface and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. The DOX was located in the defect zone in v2 orientation. All distances are expressed in Å.
| Run | Type | PB | GB | D1 | D2 | dp-NT | d N-NT |
|---|
| 1 | A(10,10)0N-H-Hk1 | −65.1 | −64.8 | 15.3 | 16.1 | 3.71 | 3.73 |
| 2 | A(10,10)4N-H-Hk1 | −69.8 | −70.0 | 15.3 | 16.1 | 3.95 | 6.55 |
| 3 | A(10,10)8N-H-Hk1 | −68.4 | −69.1 | 15.3 | 16.1 | 3.72 | 3.27 |
| 4 | A(10,10)0N-H-Hk2 | −64.3 | −63.9 | 15.6 | 17.7 | 3.24 | 3.65 |
| 5 | A(10,10)4N-H-Hk2 | −66.7 | −66.1 | 15.6 | 17.7 | 3.92 | 3.62 |
| 6 | A(10,10)8N-H-Hk2 | −58.0 | −60.9 | 15.6 | 17.7 | 4.14 | 4.53 |
Table 5.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for DOX-CNT complexes of saturated and unsaturated armchair (10,10) nanotubes having one and two haeckelite defects (Hk1, Hk2, respectively) expressed in kcal/mol. DOX charges were RESP charges. D is the initial CNT diameter at the defected region, dp-NT is the equilibrium distance between the DOX planar fragment and the CNT sidewall surface and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. All distances are expressed in Å.
Table 5.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for DOX-CNT complexes of saturated and unsaturated armchair (10,10) nanotubes having one and two haeckelite defects (Hk1, Hk2, respectively) expressed in kcal/mol. DOX charges were RESP charges. D is the initial CNT diameter at the defected region, dp-NT is the equilibrium distance between the DOX planar fragment and the CNT sidewall surface and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. All distances are expressed in Å.
| Run | Type | PB | GB | D | dp-NT | d N-NT |
|---|
| 1 | A(10,10)0N-Hk1-DoxDin.v1RESP | −83.5 | −83.4 | 14.6 | 3.63 | 3.27 |
| 2 | A(10,10)4N-Hk1-DoxDin.v1RESP | −83.7 | −84.8 | 14.6 | 3.77 | 5.11 |
| 3 | A(10,10)8N-Hk1-DoxDin.v1RESP | −82.5 | −83.1 | 14.6 | 3.83 | 3.43 |
| 4 | A(10,10)0N-Hk1-DoxDin.v2RESP | −81.7 | −82.9 | 14.6 | 4.09 | 4.29 |
| 5 | A(10,10)4N-Hk1-DoxDin.v2RESP | −82.6 | −84.3 | 14.6 | 3.50 | 3.28 |
| 6 | A(10,10)8N-Hk1-DoxDin.v2RESP | −83.3 | −85.1 | 14.6 | 3.76 | 4.68 |
| 7 | A(10,10)0N-Hk2-DoxOutRESP | −40.5 | −38.7 | 15.6 | 3.45 | 3.55 |
| 8 | A(10,10)4N-Hk2-DoxOutRESP | −40.4 | −38.6 | 15.9 | 3.40 | 4.22 |
| 9 | A(10,10)0N-Hk2-DoxDin.v1RESP | −83.4 | −84.4 | 15.6 | 3.35 | 3.39 |
| 10 | A(10,10)4N-Hk2-DoxDin.v1RESP | −98.6 | −101.8 | 15.9 | 3.40 | 3.74 |
| 11 | A(10,10)8N-Hk2-DoxDin.v1RESP | −83.9 | −85.8 | 15.6 | 3.61 | 3.18 |
| 12 | A(10,10)0N-Hk2-DoxDin.v2RESP | −84.6 | −86.2 | 15.6 | 3.81 | 3.18 |
| 13 | A(10,10)4N-Hk2-DoxDin.v2RESP | −84.6 | −87.1 | 15.9 | 3.82 | 3.46 |
| 14 | A(10,10)8N-Hk2-DoxDin.v2RESP | −84.5 | −86.1 | 15.6 | 3.87 | 3.19 |
| 15 | A(10,10)0N-H-DoxIn.v2RESP | −62.9 | −61.1 | 32.7 | 3.64 | 3.59 |
| 16 | A(10,10)0N-H-Hk1-DoxDin.v2RESP | −61.8 | −59.3 | 32.7 | 3.63 | 3.90 |
| 17 | A(10,10)4N-H-Hk1-DoxDin.v2RESP | −59.5 | −55.3 | 32.7 | 3.92 | 6.49 |
| 18 | A(10,10)8N-H-Hk1-DoxDin.v2RESP | −60.9 | −55.4 | 32.7 | 3.21 | 3.42 |
Table 6.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for adsorption and encapsulation DOX-CNT complexes of saturated and unsaturated zigzag (18,0) nanotubes with one and two haeckelite defects, considering RESP and Mulliken charges for DOX in v1 and v2 orientations, expressed in kcal/mol. Nanotube diameter and length are ~14 and 33 Å, respectively. Here, D is the initial CNT defect zone diameter, dp-NT is the equilibrium distance between the DOX planar fragment and the CNT sidewall surface, d′p-NT is the equilibrium distance between the same point of the DOX planar fragment and the opposite CNT sidewall surface, and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. All distances and diameter are expressed in Å.
Table 6.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for adsorption and encapsulation DOX-CNT complexes of saturated and unsaturated zigzag (18,0) nanotubes with one and two haeckelite defects, considering RESP and Mulliken charges for DOX in v1 and v2 orientations, expressed in kcal/mol. Nanotube diameter and length are ~14 and 33 Å, respectively. Here, D is the initial CNT defect zone diameter, dp-NT is the equilibrium distance between the DOX planar fragment and the CNT sidewall surface, d′p-NT is the equilibrium distance between the same point of the DOX planar fragment and the opposite CNT sidewall surface, and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. All distances and diameter are expressed in Å.
| Run | Type | PB | GB | D | dp-NT | d′p-NT | d N-NT |
|---|
| 1 | Z(18,0)0N-DoxIn.v1 | −108.6 | −112.3 | 14.82 | 3.49 | | 3.35 |
| 2 | Z(18,0)0N-Hk1-DoxOut | −39.8 | −38.9 | 14.78 | 3.38 | | 4.56 |
| 3 | Z(18,0)0N-Hk1-DoxDIn.v1 | −104.6 | −108.0 | 14.78 | 3.40 | 3.71 | 3.07 |
| 4 | Z(18,0)0N-Hk1-DoxDIn.v2 | −93.1 | −94.5 | 14.78 | 3.77 | | 3.39 |
| 5 | Z(18,0)0N-Hk1-DoxRIn.v1 | −93.6 | −95.6 | 14.78 | 3.82 | | 3.20 |
| 6 | Z(18,0)0N-Hk1-DoxRIn.v2 | −85.5 | −86.2 | 14.78 | 3.59 | | 3.66 |
| 7 | Z(18,0)4N-Hk1-DoxDIn.v1 | −108.4 | −111.1 | 14.78 | 3.79 | 3.77 | 3.08 |
| 8 | Z(18,0)8N-Hk1-DoxDIn.v1 | −106.8 | −110.5 | 14.78 | 3.64 | | 3.38 |
| 9 | Z(18,0)0N-DoxIn.v1RESP | −78.8 | −79.0 | 14.82 | 3.71 | | 4.00 |
| 10 | Z(18,0)0N-Hk1-DoxOutRESP | −39.7 | −37.7 | 14.78 | 3.54 | | 3.92 |
| 11 | Z(18,0)0N-Hk1-DoxDIn.v1RESP | −102.3 | −104.5 | 14.78 | 3.89 | 4.95 | 3.33 |
| 12 | Z(18,0)4N-Hk1-DoxDIn.v1RESP | −102.3 | −104.7 | 14.78 | 3.69 | 3.89 | 3.08 |
| 13 | Z(18,0)8N-Hk1-DoxDIn.v1RESP | −102.1 | −103.1 | 14.78 | 3.70 | 3.62 | 4.20 |
| 14 | Z(18,0)0N-Hk2-DoxDIn.v1 | −85.9 | −85.9 | 15.41 | 3.59 | | 4.77 |
| 15 | Z(18,0)0N-Hk2-DoxDIn.v2 | −99.7 | −103.6 | 15.41 | 3.22 | 4.33 | 3.25 |
| 16 | Z(18,0)0N-Hk2-DoxRIn.v1 | −92.3 | −92.4 | 15.41 | 3.96 | | 3.35 |
| 17 | Z(18,0)0N-Hk2-DoxRIn.v2 | −94.4 | −95.6 | 15.41 | 3.79 | | 3.49 |
| 18 | Z(18,0)4N-Hk2-DoxDIn.v1 | −98.0 | −98.4 | 15.41 | 3.39 | | 3.43 |
| 19 | Z(18,0)8N-Hk2-DoxDIn.v1 | −86.9 | −87.3 | 15.41 | 3.48 | | 2.97 |
| 20 | Z(18,0)0N-Hk2-DoxDIn.v1RESP | −78.7 | −79.0 | 15.41 | 3.83 | | 2.99 |
| 21 | Z(18,0)4N-Hk2-DoxDIn.v1RESP | −82.3 | −81.9 | 15.41 | 3.61 | | 3.83 |
| 22 | Z(18,0)8N-Hk2-DoxDIn.v1RESP | −81.6 | −80.9 | 15.41 | 3.57 | | 3.14 |
| 23 | Z(18,0)0N-H-Hk2-DoxDIn.v1RESP | −63.0 | −58.1 | 15.41 | 3.58 | | 3.30 |
| 24 | Z(18,0)0N-H-Hk2-DoxDIn.v2RESP | −58.8 | −54.3 | 15.41 | 3.65 | | 2.98 |
| 25 | Z(18,0)4N-H-Hk2-DoxDIn.v1RESP | −55.9 | −51.4 | 15.41 | 4.11 | | 3.36 |
| 26 | Z(18,0)4N-H-Hk2-DoxDIn.v2RESP | −61.1 | −55.7 | 15.41 | 3.59 | | 4.32 |
| 27 | Z(18,0)8N-H-Hk2-DoxDIn.v1RESP | −58.2 | −53.5 | 15.41 | 3.62 | | 4.41 |
| 28 | Z(18,0)8N-H-Hk2-DoxDIn.v2RESP | −58.3 | −47.8 | 15.41 | 3.83 | | 4.27 |
Table 7.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for encapsulation DOX-CNT complexes of zigzag (18,0) nanotubes with two haeckelite defects expressed in kcal/mol, at different simulation times expressed in ns. Nanotube diameter and length are ~14 and 33 Å, respectively. Here, dp-NT is the equilibrium distance between the DOX planar fragment and the CNT sidewall surface, and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. All distances are expressed in Å.
Table 7.
Poisson-Boltzmann (PB) and Generalized Bond (GB) binding free energy values for encapsulation DOX-CNT complexes of zigzag (18,0) nanotubes with two haeckelite defects expressed in kcal/mol, at different simulation times expressed in ns. Nanotube diameter and length are ~14 and 33 Å, respectively. Here, dp-NT is the equilibrium distance between the DOX planar fragment and the CNT sidewall surface, and dN-NT is the equilibrium distance between the DOX-nitrogen atom and the CNT sidewall surface. All distances are expressed in Å.
| Run | Simulation Time (ns) | PB | GB | dp-NT | dN-NT |
|---|
| Z(18,0)0N-Hk2-DoxRIn.v2 |
| 1 | 2 | −94.4 | −95.6 | 3.79 | 3.49 |
| 2 | 5 | −97.6 | −98.9 | 3.89 | 3.94 |
| 3 | 30 | −99.0 | −100.6 | 3.74 | 3.47 |
| 4 | 50 | −99.0 | −100.6 | 3.58 | 3.83 |
| 5 | 100 | −99.0 | −100.6 | 3.56 | 3.38 |
| Z(18,0)0N-Hk2-DoxDIn.v1 |
| 6 | 2 | −85.9 | −85.9 | 3.59 | 4.77 |
| 7 | 5 | −87.0 | −87.0 | 3.86 | 3.20 |
| 8 | 10 | −87.0 | −87.0 | 3.38 | 3.90 |
| 9 | 15 | −87.0 | −87.0 | 3.99 | 3.20 |















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}