Selective Inhibition of Human Monoamine Oxidase B by Acacetin 7-Methyl Ether Isolated from Turnera diffusa (Damiana)


 , ,
, , 
Abstract
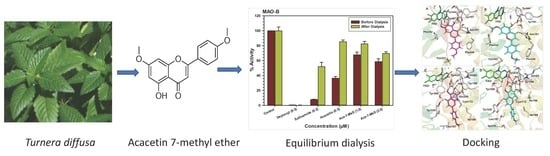
:
1. Introduction
2. Results
2.1. Determination of MAO-A and -B Inhibition Activity
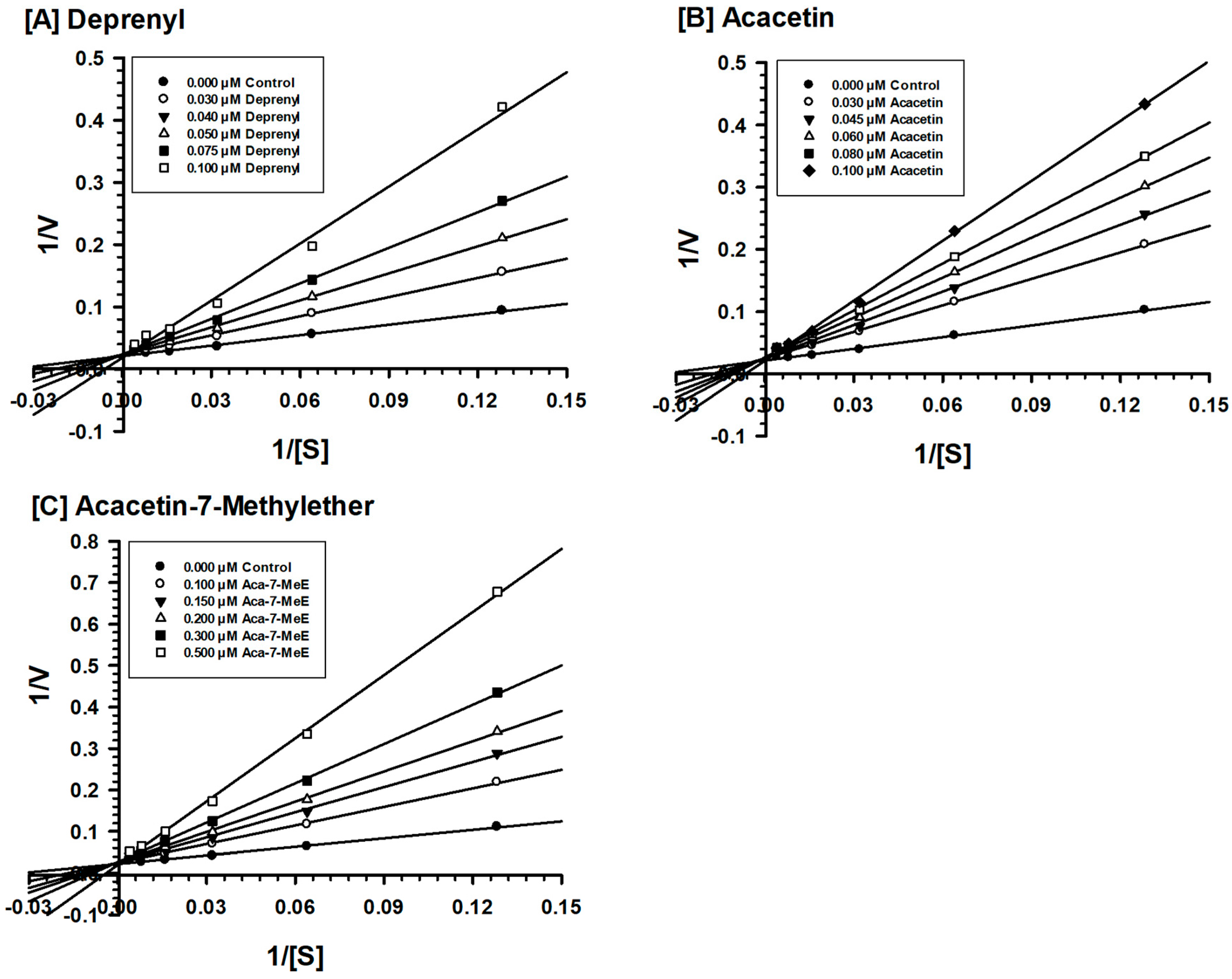
2.2. Enzyme Kinetics and Mechanism Studies
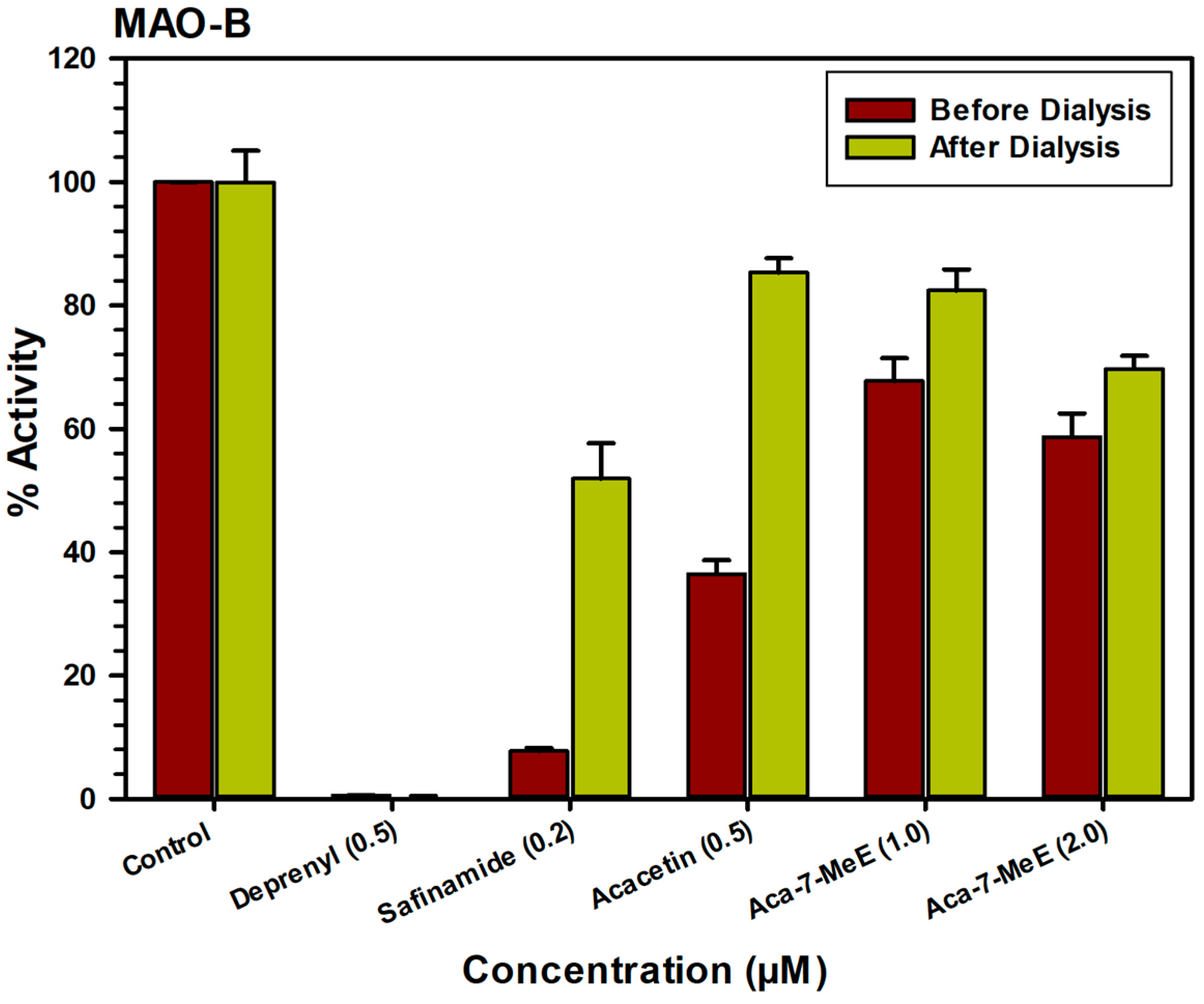
2.3. Analysis of Reversibility of Binding of Inhibitor
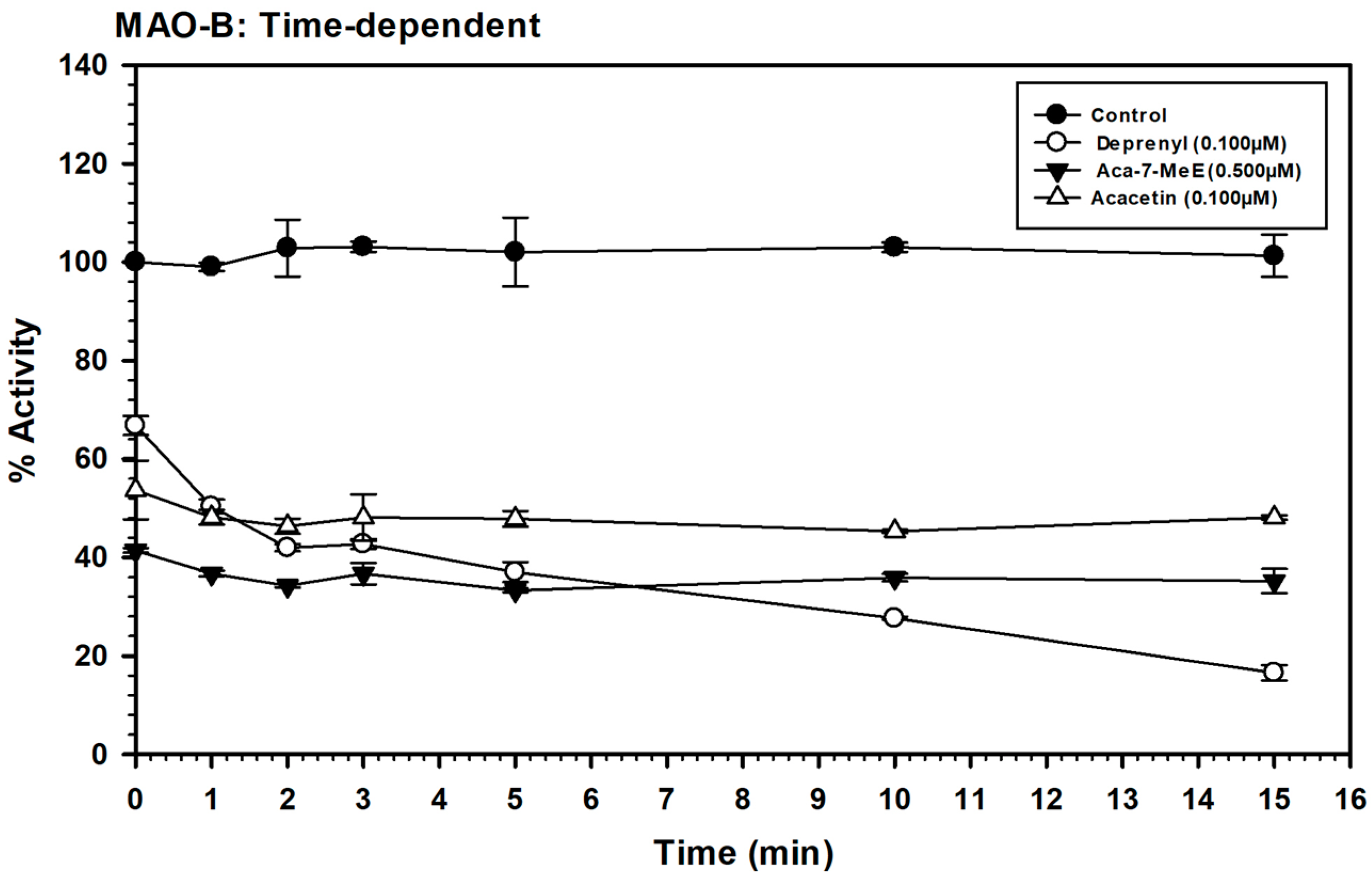
2.4. Time-Dependent Assay for Enzyme Inhibition
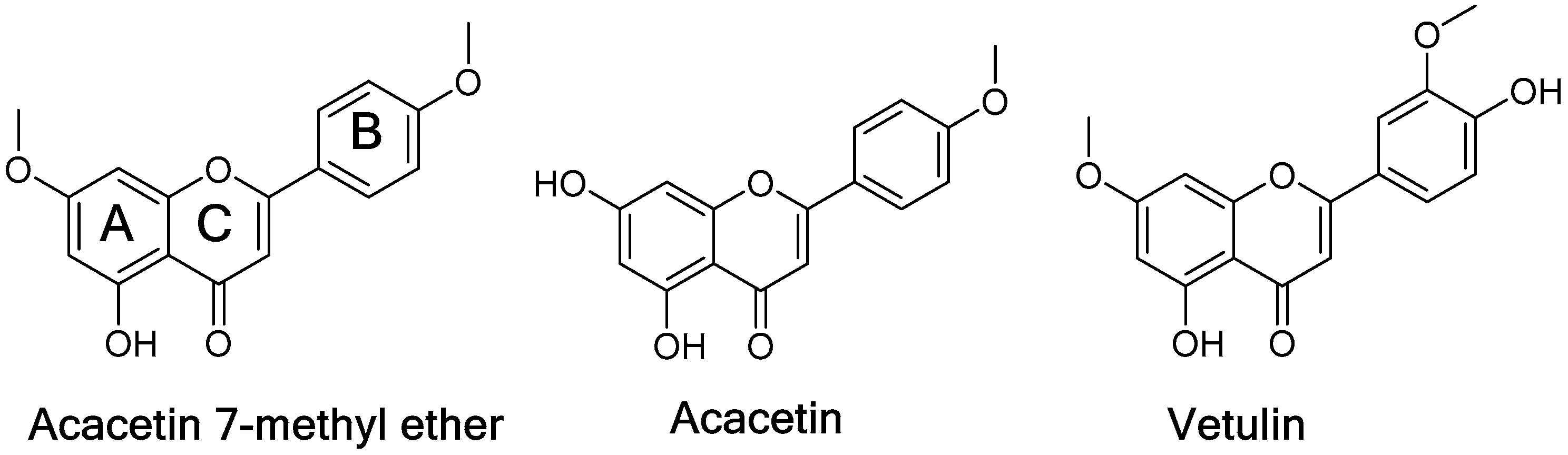
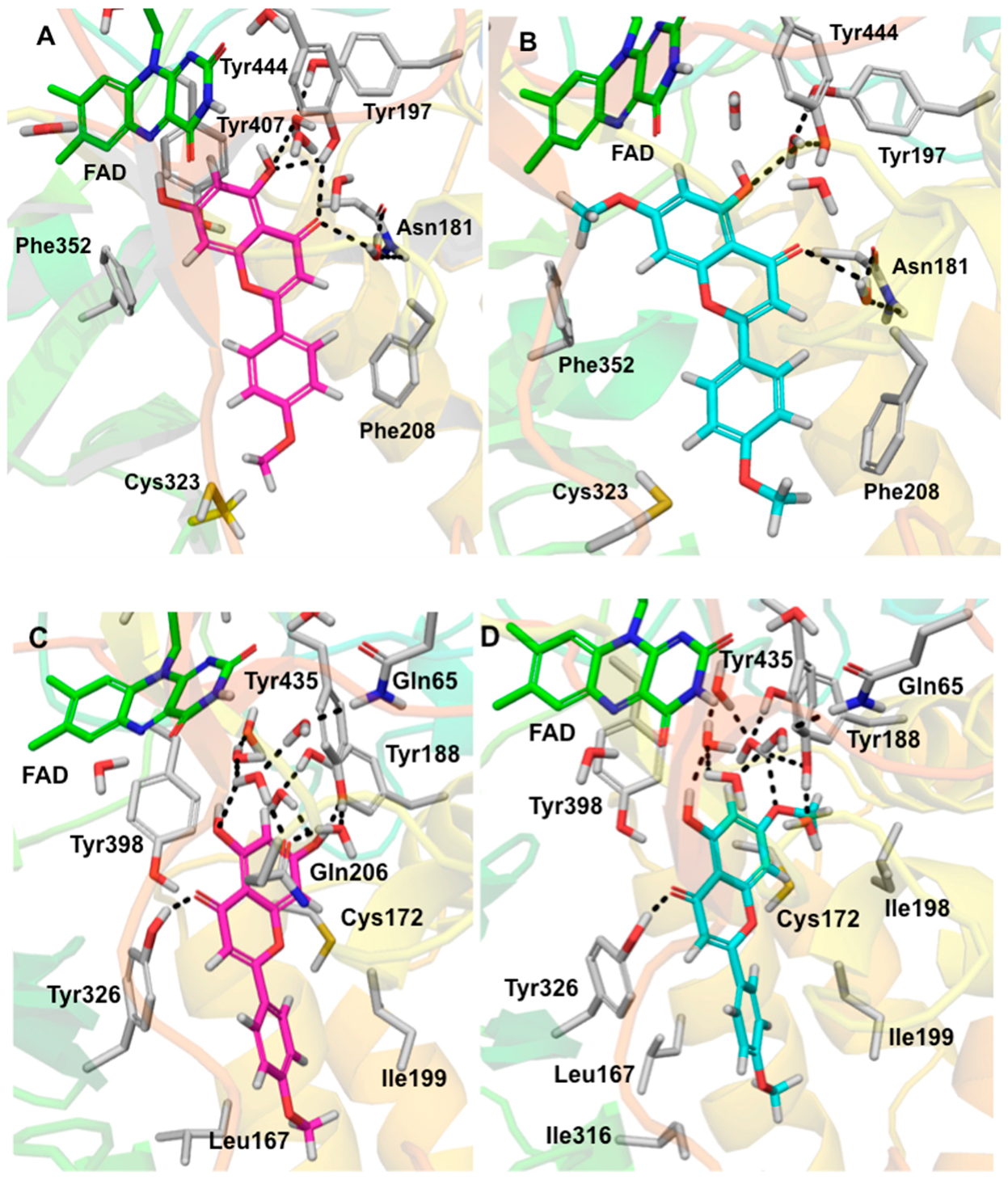
2.5. Computational Docking Study of Acacetin 7-Methyl Ether
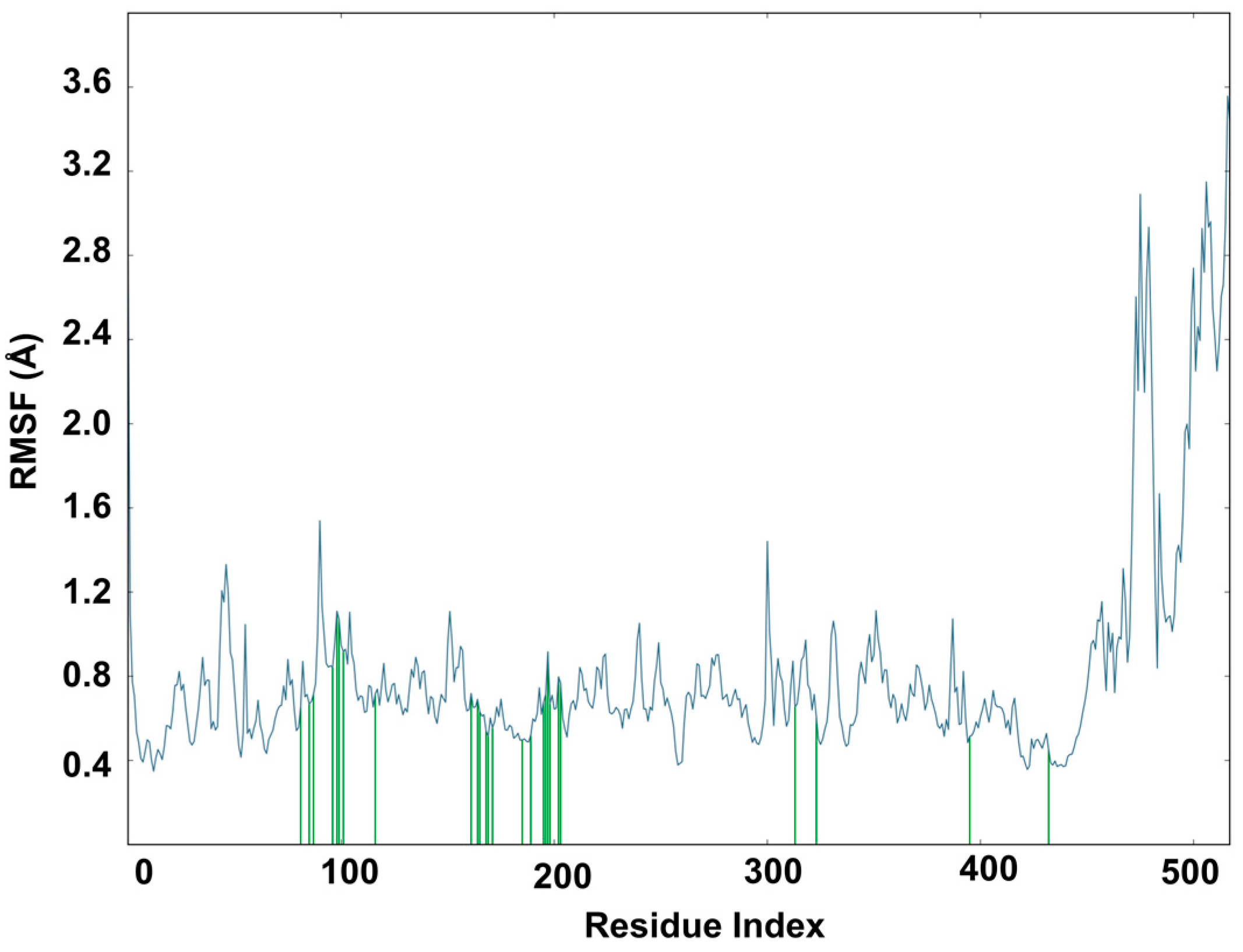
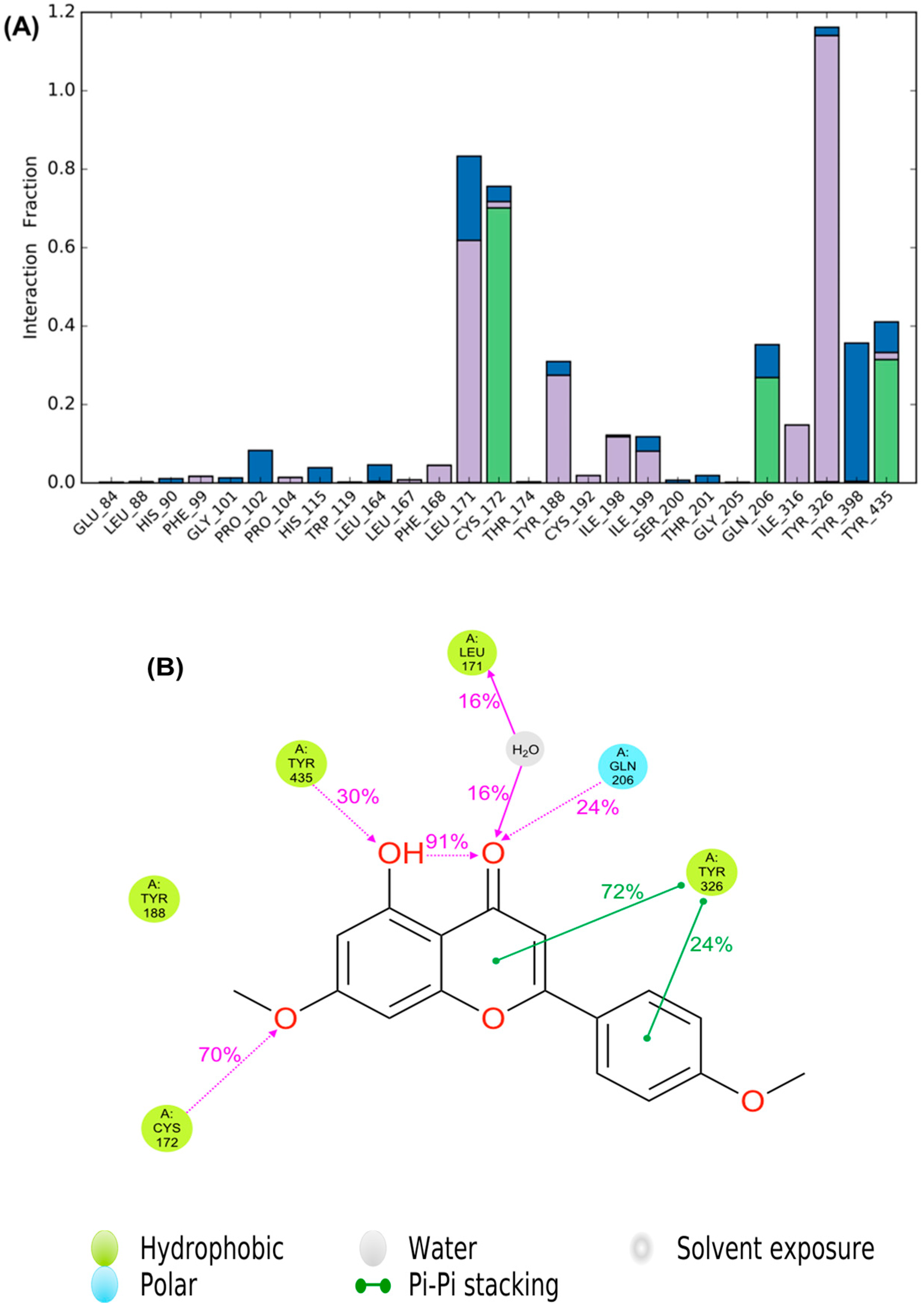
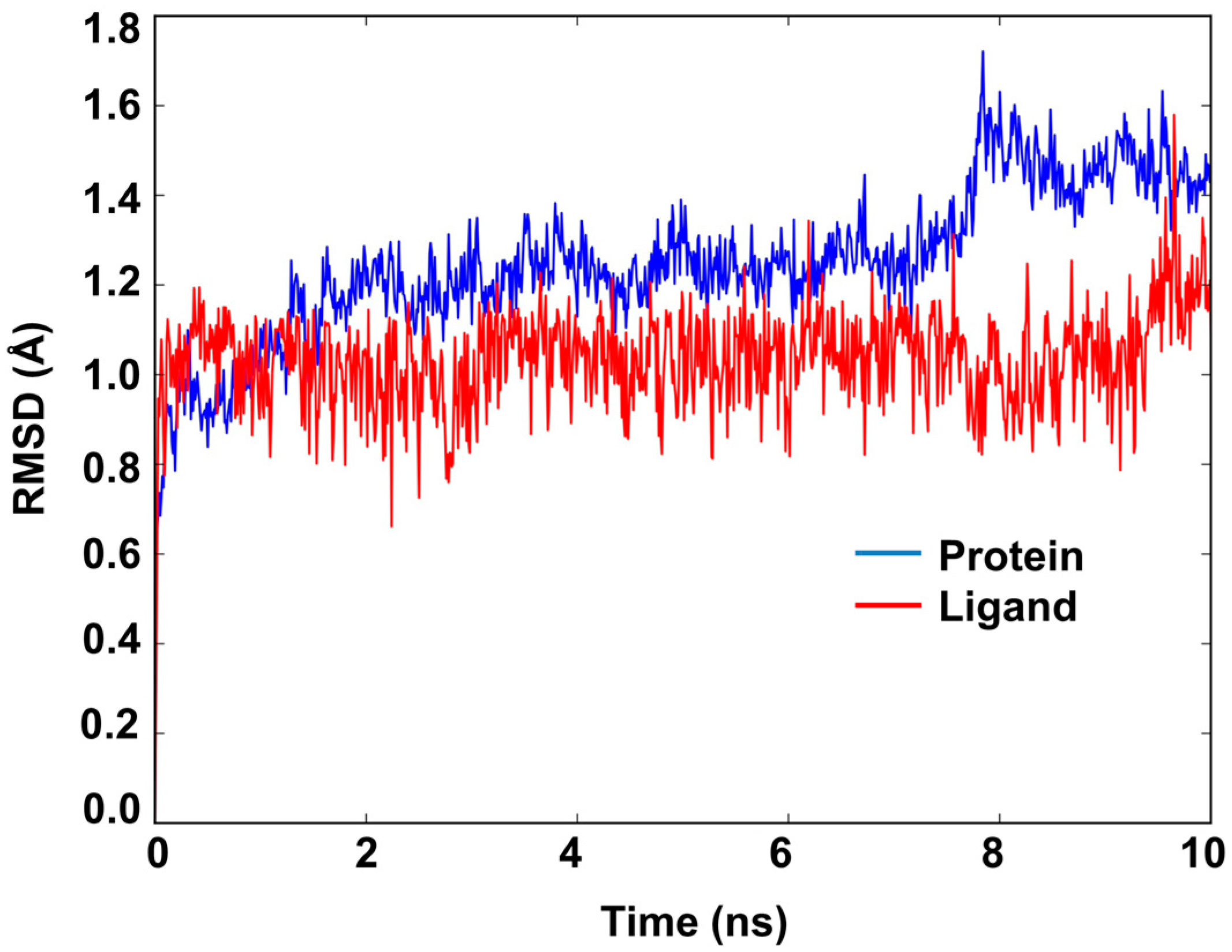
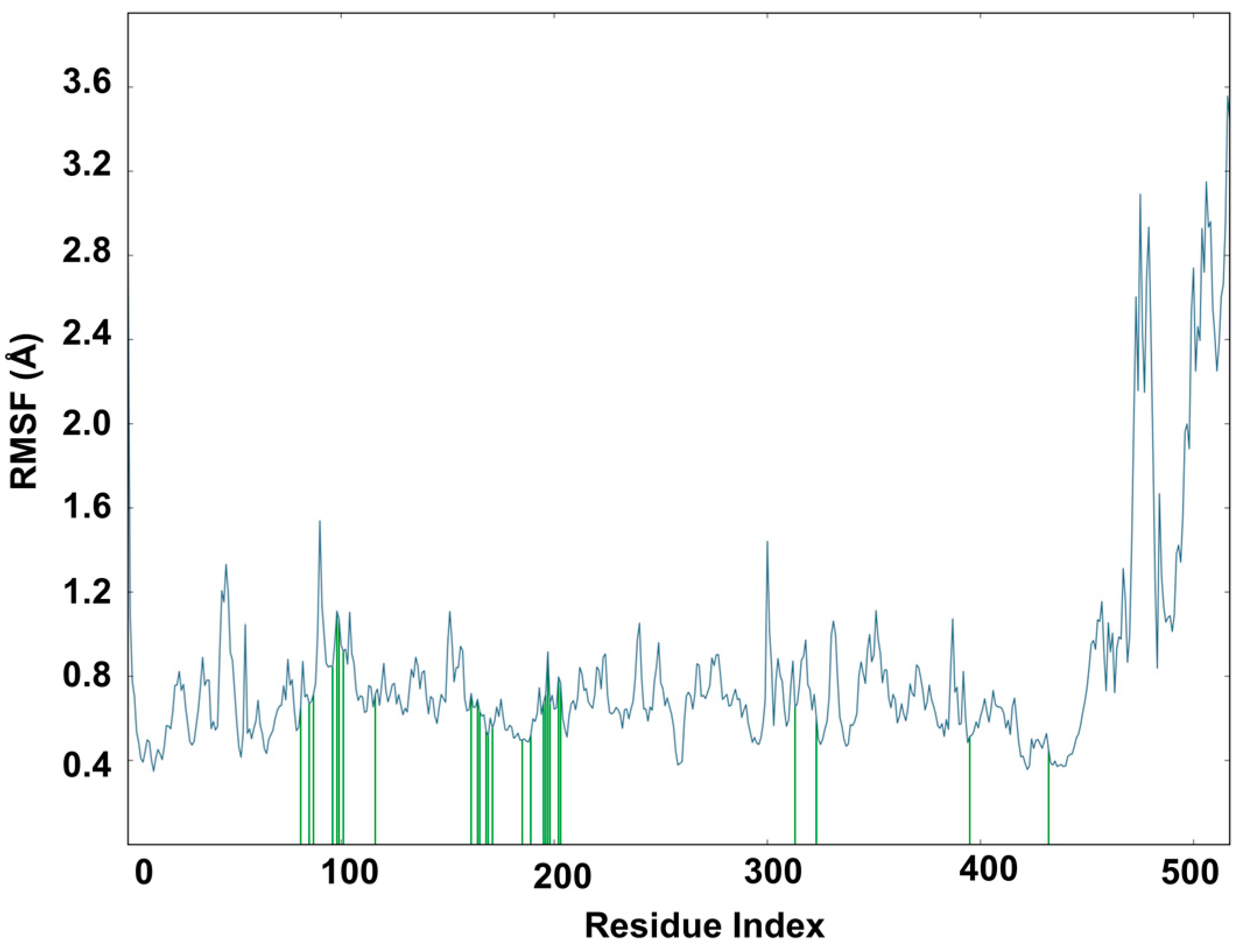
2.6. Molecular Dynamics Study of Acacetin 7-Methyl Ether and MAO-B
3. Discussion
4. Materials and Methods
4.1. Enzymes and Chemicals
4.2. MAOs Inhibition Assay
4.3. Determination of IC50 Values
4.4. Enzyme Kinetics and Mechanism Studies
4.5. Analysis of Reversibility of Binding of Inhibitor
4.6. Time-Dependent Assay for Enzyme Inhibition
4.7. Computational Methods
4.8. Molecular Dynamics (MD) Simulations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Abell, C.W.; Kwan, S.W. Molecular characterization of monoamine oxidase A and B. Prog. Nucleic Acids Res. Mol. Biol. 2001, 65, 129–156. [Google Scholar]
- Cesura, A.M.; Pletscher, A. The New Generation of Monoamine Oxidase Inhibitors. Prog. Drug Res. 1992, 38, 171–297. [Google Scholar] [PubMed]
- Yamada, M.; Yasuhara, H. Clinical pharmacology of MAO inhibitors: Safety and future. Neurotoxicology 2004, 25, 215–221. [Google Scholar] [CrossRef]
- Yudim, M.B.; Fridkin, M.; Zheng, H. Novel bifunctional drugs targeting monoamine oxidase inhibition and iron chelation as an approach to neuroprotection in Parkinson’s disease and other neurodegenerative diseases. J. Neural Transm. 2004, 111, 1455–1471. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Bakhle, Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br. J. Pharmacol. 2006, 147, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, T.; Chaparro, C. Human monoamine oxidase enzyme inhibition by coffee and beta-carbolines norharman and harman isolated from coffee. Life Sci. 2006, 78, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Borroni, E.; Bohrmann, B.; Grueninger, F.; Prinssen, E.; Nave, S.; Loetscher, H.; Chinta, S.J.; Rajagopalan, S.; Rane, A.; Siddiqui, A.; et al. Sembragiline: A Novel, Selective Monoamine Oxidase Type B Inhibitor for the Treatment of Alzheimer’s Disease. J. Pharmacol. Exp. Ther. 2017, 362, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, N.D.; Gogineni, V.; Elokely, K.M.; Leon, F.; Nunez, M.J.; Klein, M.L. Isolation of Acacetin from Calea urticifolia with Inhibitory Properties against Human Monoamine Oxidase-A and -B. J. Nat. Prod. 2016, 79, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Ryu, H.W.; Baek, S.C.; Kang, M.G.; Park, O.D.; Han, H.Y.; An, J.H.; Oh, S.R.; Kim, H. Potent inhibitions of monoamine oxidase A and B by acacetin and its 7-O-(6-O-malonylglucoside) derivative from Agastache rugosa. Int. Biol. Macromol. 2017, 104, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, K.; Zidorn, C. Ethnobotany, phytochemistry, and bioactivity of the genus Turnera (Passifloraceae) with a focus on damiana—Turnera diffusa. J. Ethnopharm. 2014, 152, 424–443. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, R.D.; Ghilardi Lago, J.H.; Rossi, L.; Fernandez Galduróz, J.C.; Rodrigues, E. Psychoactive plants described in a Brazilian literary work and their chemical compounds. Cent. Nerv. Syst. Agents Med. Chem. 2010, 10, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Pawar, R.S.; Ali, Z.; Khan, I.A. Phytochemical investigation of Turnera diffusa. J. Nat. Prod. 2007, 70, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Dasmahapatra, A.K.; Khan, S.I.; Khan, I.A. Anti-aromatase activity of the constituents from damiana (Turnera diffusa). J. Ethnopharm. 2008, 120, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Jäger, A.K.; Saaby, L. Flavonoids and the CNS. Molecules 2011, 16, 1471–1485. [Google Scholar] [Green Version]
- Moore, J.J.; Saadabadi, A. Selegiline; Pearls Publishing LLC: Treasure Island, FL, USA, 2018. [Google Scholar]
- Pandey, P.; Chaurasiya, N.D.; Tekwani, B.L.; Doerksen, R.J. Interactions of endocannabinoid virodhamine and related analogs with human monoamine oxidase-A and-B. Biochem. Pharmacol. 2018, 155, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Hubálek, F.; Binda, C.; Khalil, A.; Li, M.; Mattevi, A.; Castagnoli, N.; Edmondson, D.E. Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. J. Biol. Chem. 2005, 280, 15761–15766. [Google Scholar] [CrossRef] [PubMed]
- Desmond Molecular Dynamics System. D.E. Shaw Research; Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]
- Krishna, M.S.; Joy, B.; Sundaresan, A. Effect on oxidative stress, glucose uptake level and lipid droplet content by Apigenin 7, 4′-dimethyl ether isolated from Piper longum L. J. Food. Sci. Technol. 2015, 52, 3561–3570. [Google Scholar] [CrossRef] [PubMed]
- Sghaier, M.B.; ISkandrania, I.; Nasr, N.; Franca, M.C.D.; Chekir-Ghedira, L.; Ghedira, K. Flavonoids and sesquiterpenes from Tecurium ramosissimum promote antiproliferation of human cancer cells and enhance antioxidant activity: A structure–activity relationship study. Environ. Toxicol. Pharmacol. 2011, 32, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Thao, N.P.; Luyen, B.T.T.; Lee, S.H.; Jang, H.D.; Kim, Y.H. Anti-osteoporotic and Antioxidant Activities by Rhizomes of Kaempferia parviflora Wall. ex Baker. Nat. Prod. Sci. 2016, 22, 13–19. [Google Scholar] [CrossRef]
- Dezsi, L.; Vecsei, L. Monoamine Oxidase B Inhibitors in Parkinson’s Disease. CNS Neurol. Disord. Targets 2017, 16, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Müller, T. Monoamine oxidase-B inhibitors in the treatment of Parkinson’s disease: Clinical-pharmacological aspects. J Neural. Transm. 2018, 125, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- Hwang, O. Role of oxidative stress in Parkinson’s disease. Exp. Neurobiol. 2013, 22, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Finberg, J.P.M.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology. Front. Pharmacol. 2016, 7, 340. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Laux, G. MAO-inhibitors in Parkinson’s Disease. Exp. Neurobiol. 2011, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhu, L.; Gong, X.; Ruan, Y.; Yu, J.; Jiang, H.; Wang, Y.; Qi, X.X.; Lu, L.; Liu, Z. Sulfonation Disposition of Acacetin: In Vitro and in Vivo. J. Agric. Food Chem. 2017, 65, 4921–4931. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, N.D.; Leon, F.; Ding, Y.; Gomez-Betancur, I.; Benjumea, D.; Walker, L.A.; Cutler, S.J.; Tekwani, B.L. Interactions of Desmethoxyyangonin, a Secondary Metabolite from Renealmia alpinia, with Human Monoamine Oxidase-A and Oxidase-B. Evid. Based Complement. Altern. Med. 2017, 2017, 4018724. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Hanscom, S.; Gagne, P.; Crespi, C.; Patten, C. A Fluorescent-Based, High-Throughput Assay for Detecting Inhibitors of Human Monoamine Oxidase A and B; BD Biosciences Discovery Labware, Inc.: Woburn, MA, USA, 2002. [Google Scholar]
- Son, S.Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Aldeco, M.; Geldenhuys, W.J.; Tortorici, M.; Mattevi, A.; Edmondson, D.E. Molecular insights into human monoamine oxidase B inhibition by the glitazone antidiabetes drugs. ACS Med. Chem. Lett. 2011, 3, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Maestro, version 10.6; Schrödinger, LLC: New York, NY, USA, 2016.
- LigPrep, version 3.8; Schrödinger, LLC: New York, NY, USA, 2016.
- Schrödinger, version 2016-2; Schrödinger, LLC: New York, NY, USA, 2016.
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/ receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Hubaĺek, F.; Li, M.; Edmondson, D.E.; Mattevi, A. Crystal Structure of Human Monoamine Oxidase B, a Drug Target Enzyme Monotopically Inserted into the Mitochondrial Outer Membrane. FEBS Lett. 2004, 564, 225–228. [Google Scholar] [CrossRef]
- Maestro, version 11.5.011; Schrodinger, LLC: New York, NY, USA, 2018.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.H. Exploring the protein folding free energy landscape: Coupling replica exchange method with P3ME/RESPA algorithm. J. Mol. Graph. Model. 2004, 22, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Li, G.S.; Martins-Costa, M.T.C.; Millot, C.; Ruiz-Lopez, M.F. AM1/TIP3P molecular dynamics simulation of imidazole proton-relay processes in aqueous solution. Chem. Phys. Lett. 1998, 297, 38–44. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nose-Hoover Thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant- Pressure Molecular-Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors after execution of Intellectual Property and Material Transfer Agreements with the University of Mississippi. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MAO-A IC50 (µM) * | MAO-B IC50 (µM) * | SI MAO-A/-B |
|---|---|---|---|
| Acacetin 7-methyl ether | >100.00 | 0.198 ± 0.001 | >505.051 |
| Vetulin | 18.799 ± 0.291 | 0.447 ± 0.010 | 42.056 |
| Apigenin-7-O-β-d-(6-O-p-coumaroyl) glucoside | 22.508 ± 4.440 | 22.001 ± 1.759 | 1.023 |
| Echinaticin | 21.830 ± 4.367 | 13.404 ± 0.148 | 1.703 |
| Tetraphyllin B | - | - | - |
| Tricin-7-glucoside | 50.901 ± 0.506 | 27.444 ± 0.819 | 1.854 |
| Diffusavone | 19.091 ± 1.450 | 14.518 ± 0.214 | 1.314 |
| Turneradiffusin | 61.427 ± 3.568 | 37.250 ± 2.933 | 1.649 |
| Rhamnosylorientin | 34.909 ± 3.887 | 25.541 ± 2.020 | 1.366 |
| Rhamnosylvitexin | 40.940 ± 6.810 | 26.286 ± 0.277 | 1.557 |
| Turneradin | 19.169 ± 0.802 | 13.027 ± 2.142 | 1.471 |
| Acacetin | 0.115 ± 0.004 | 0.050 ± 0.0025 | 2.30 |
| Clorgyline | 0.0052 ± 0.0001 | 2.30 ± 0.0570 | - |
| Deprenyl | 23.00 ± 1.00 | 0.051 ± 0.002 | 450.980 |
| Safinamide # (ref) | 90.00 ± 2.470 | 0.060 ± 0.005 | 1500.00 |
| Compounds | Monoamine Oxidase-B | |
|---|---|---|
| Ki (nM) * | Type of Inhibition | |
| Acacetin 7-methyl ether | 45.0 ± 3.0 | Competitive/Partially Reversible |
| Acacetin | 36 ± 4.0 | Competitive/Reversible |
| Deprenyl | 29 ± 6.3 | Mixed/Irreversible |
| Compound | IC50 (µM) * | Glide Docking Score (kcal/mol) | Binding Free-Energies (kcal/mol) | |||
|---|---|---|---|---|---|---|
| MAO-A | MAO-B | MAO-A | MAO-B | MAO-A | MAO-B | |
| Acacetin | 0.115 | 0.049 | −10.685 | −11.890 | −56.303 | −67.205 |
| Acacetin 7-methyl ether | >100 | 0.198 | −9.085 | −10.708 | −31.791 | −67.494 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaurasiya, N.D.; Zhao, J.; Pandey, P.; Doerksen, R.J.; Muhammad, I.; Tekwani, B.L. Selective Inhibition of Human Monoamine Oxidase B by Acacetin 7-Methyl Ether Isolated from Turnera diffusa (Damiana). Molecules 2019, 24, 810. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24040810
Chaurasiya ND, Zhao J, Pandey P, Doerksen RJ, Muhammad I, Tekwani BL. Selective Inhibition of Human Monoamine Oxidase B by Acacetin 7-Methyl Ether Isolated from Turnera diffusa (Damiana). Molecules. 2019; 24(4):810. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24040810
Chicago/Turabian StyleChaurasiya, Narayan D., Jianping Zhao, Pankaj Pandey, Robert J. Doerksen, Ilias Muhammad, and Babu L. Tekwani. 2019. "Selective Inhibition of Human Monoamine Oxidase B by Acacetin 7-Methyl Ether Isolated from Turnera diffusa (Damiana)" Molecules 24, no. 4: 810. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24040810