
Cross-Species Analysis of Glycosaminoglycan Binding Proteins Reveals Some Animal Models Are “More Equal” than Others



Abstract
:
1. Introduction
2. Sequence and Structure Conservation in Heparin Binding Proteins
2.1. Antithrombin III (AT)
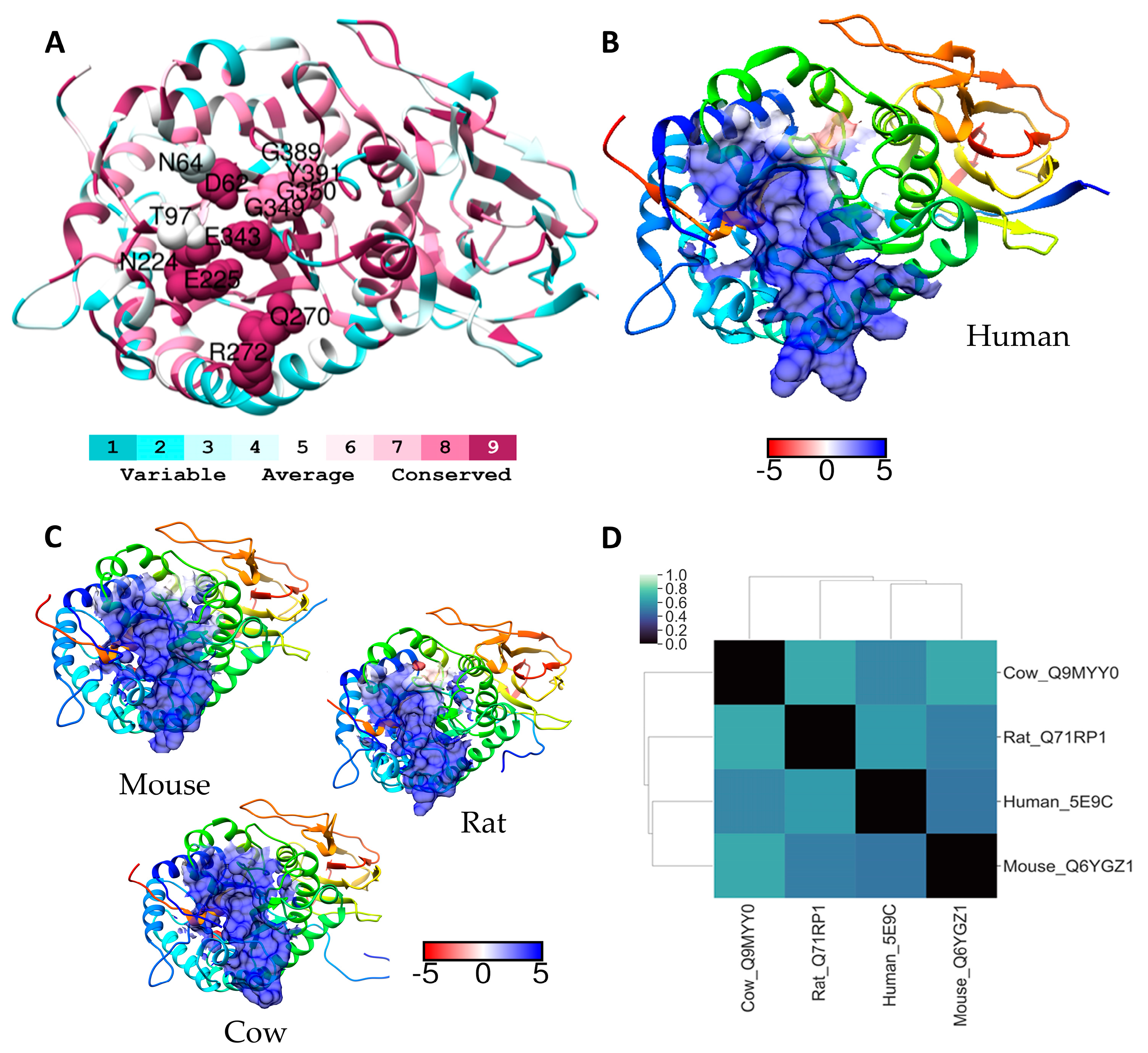
2.2. Heparanase
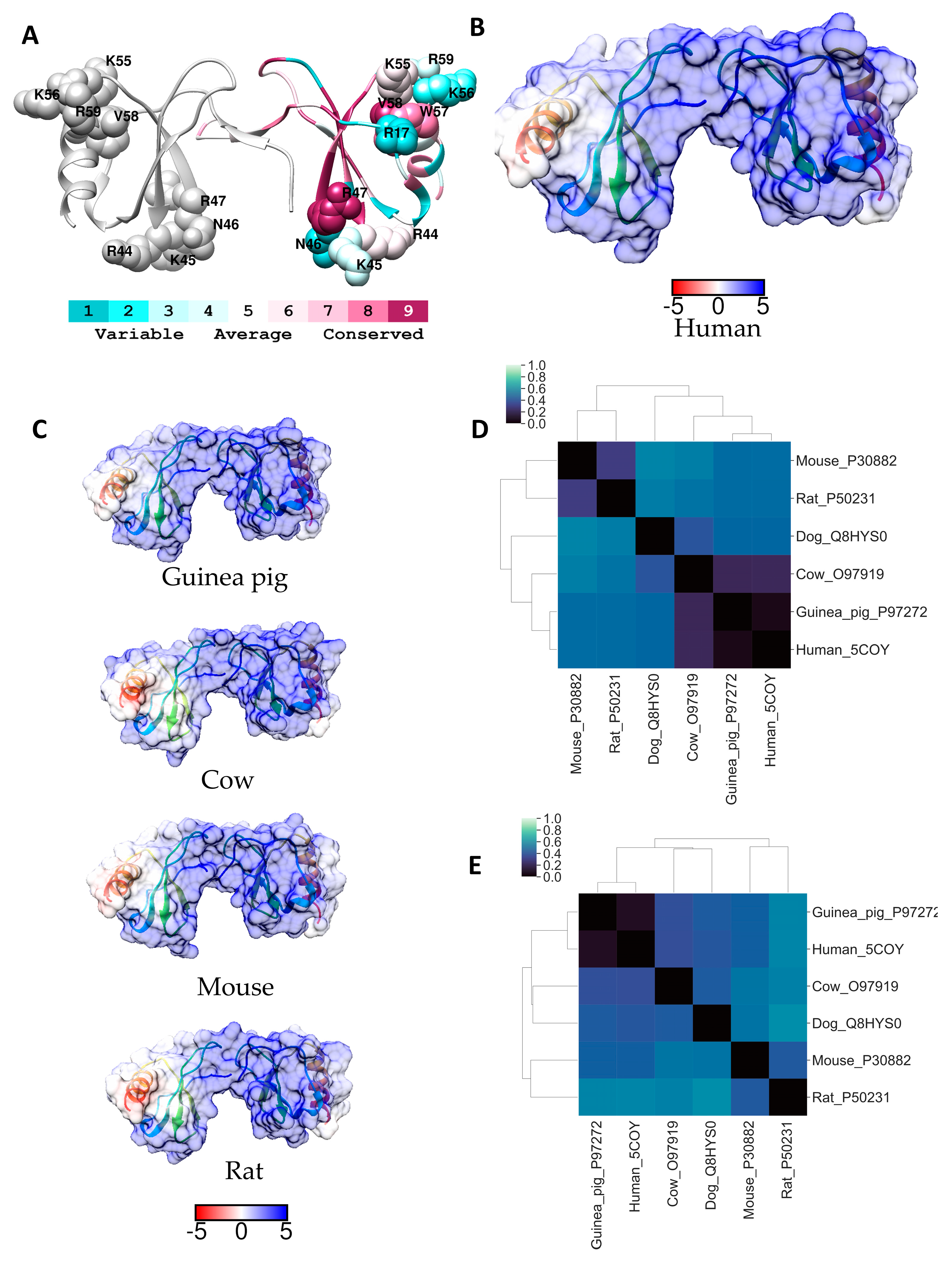
2.3. RANTES (CCL5)
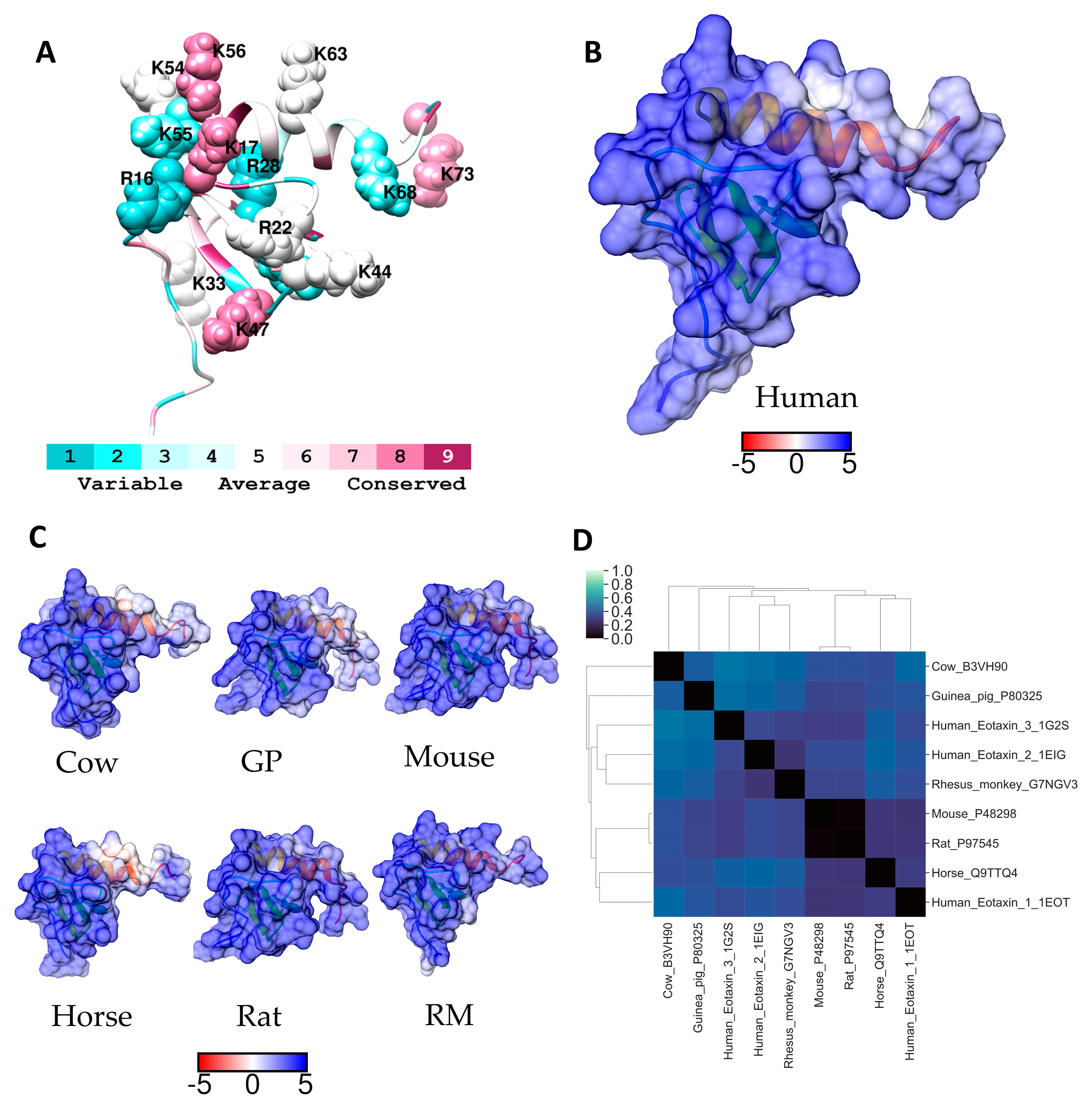
2.4. Eotaxin-1 (CCL11)
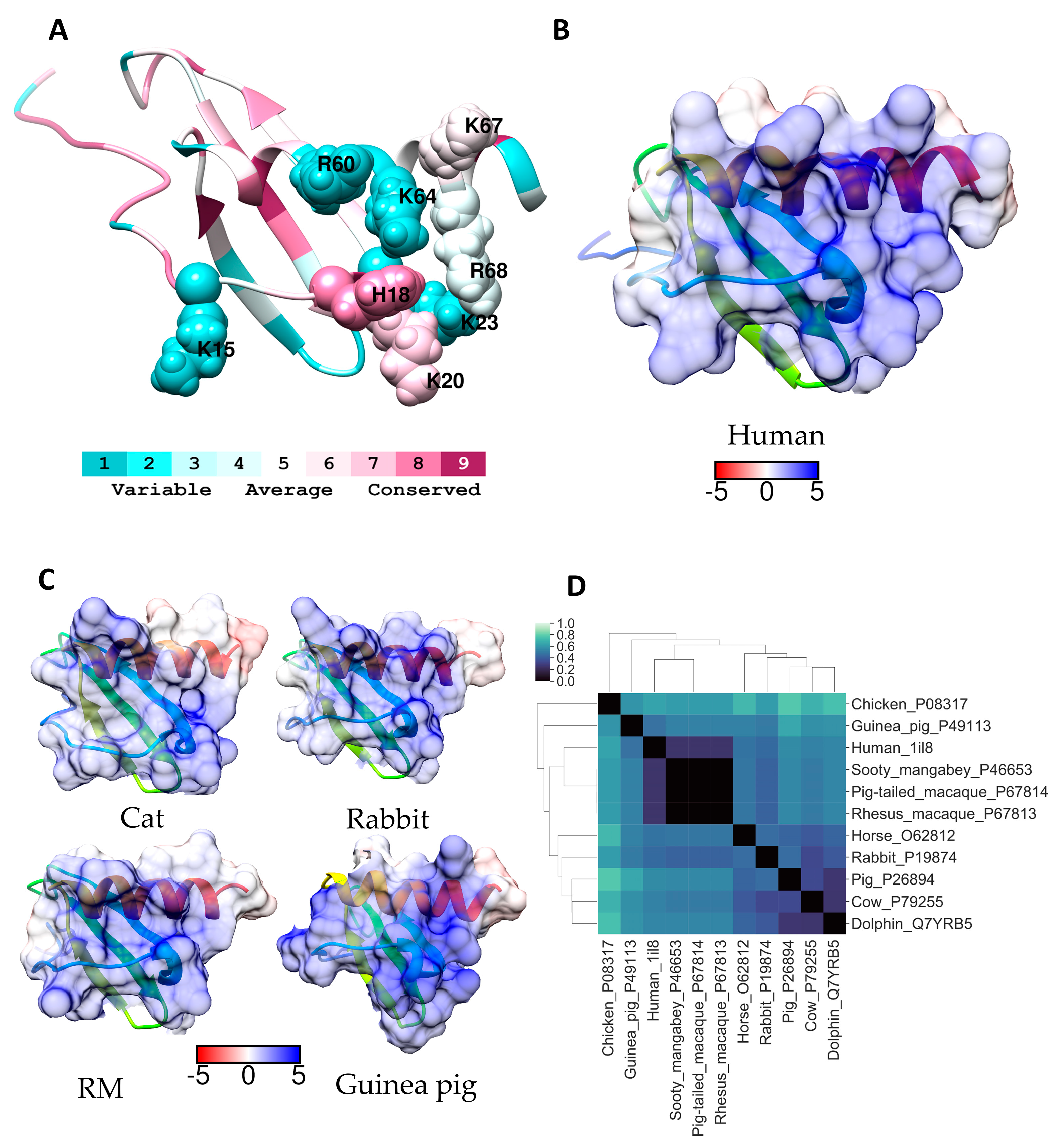
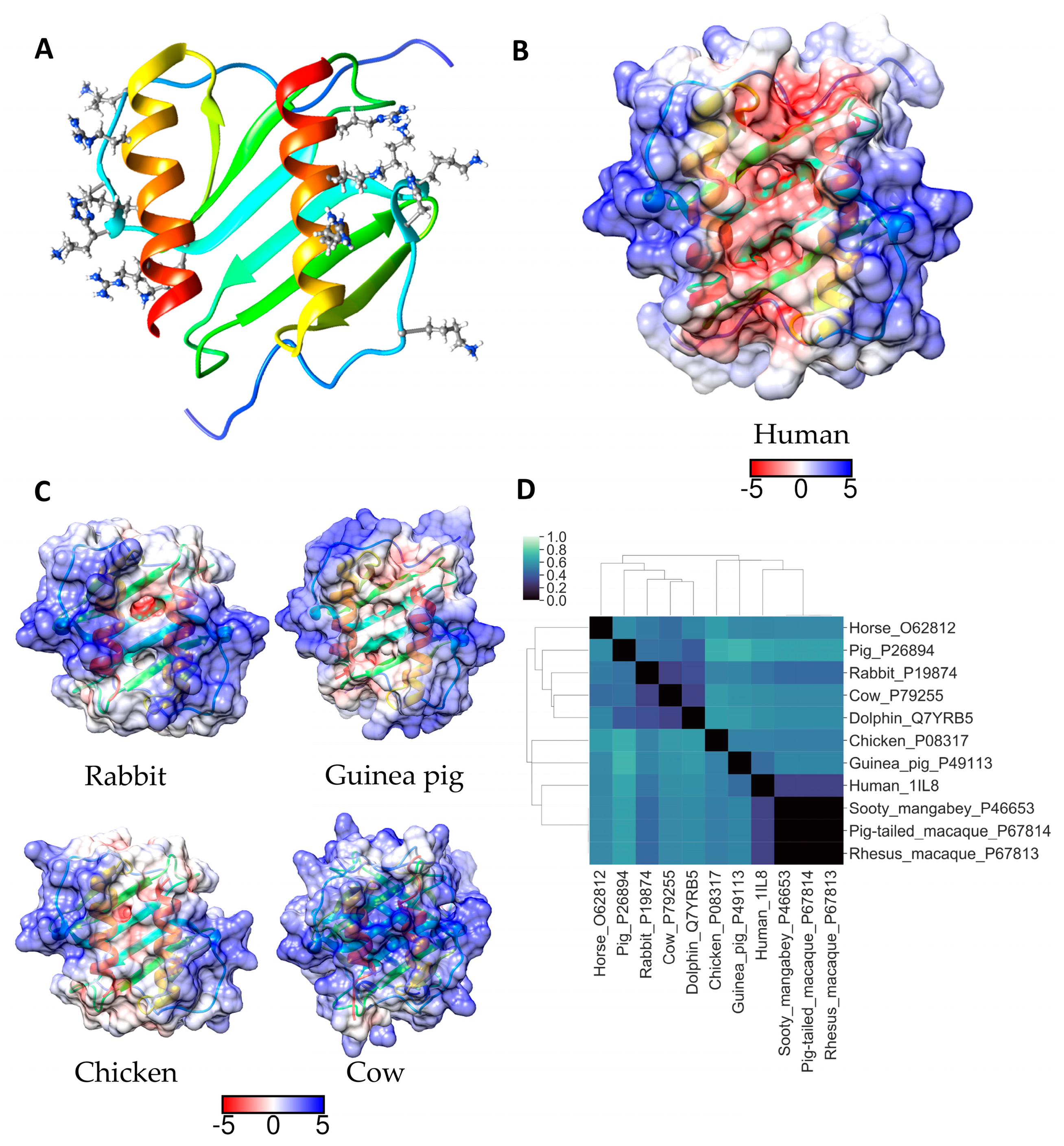
2.5. IL-8 (CXCL8)
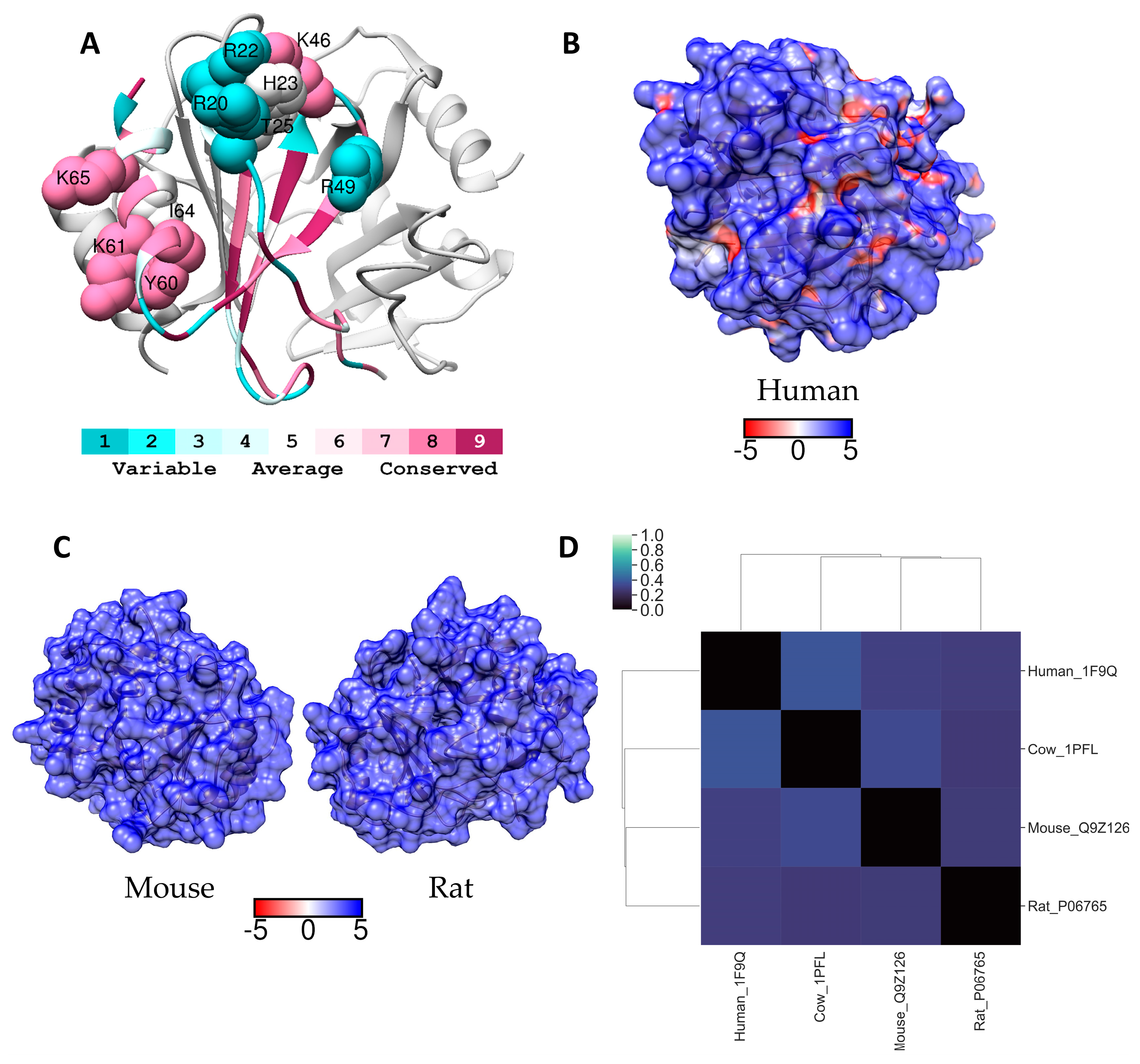
2.6. PF4 (CXCL4)
3. Methods
3.1. Sequence Selection
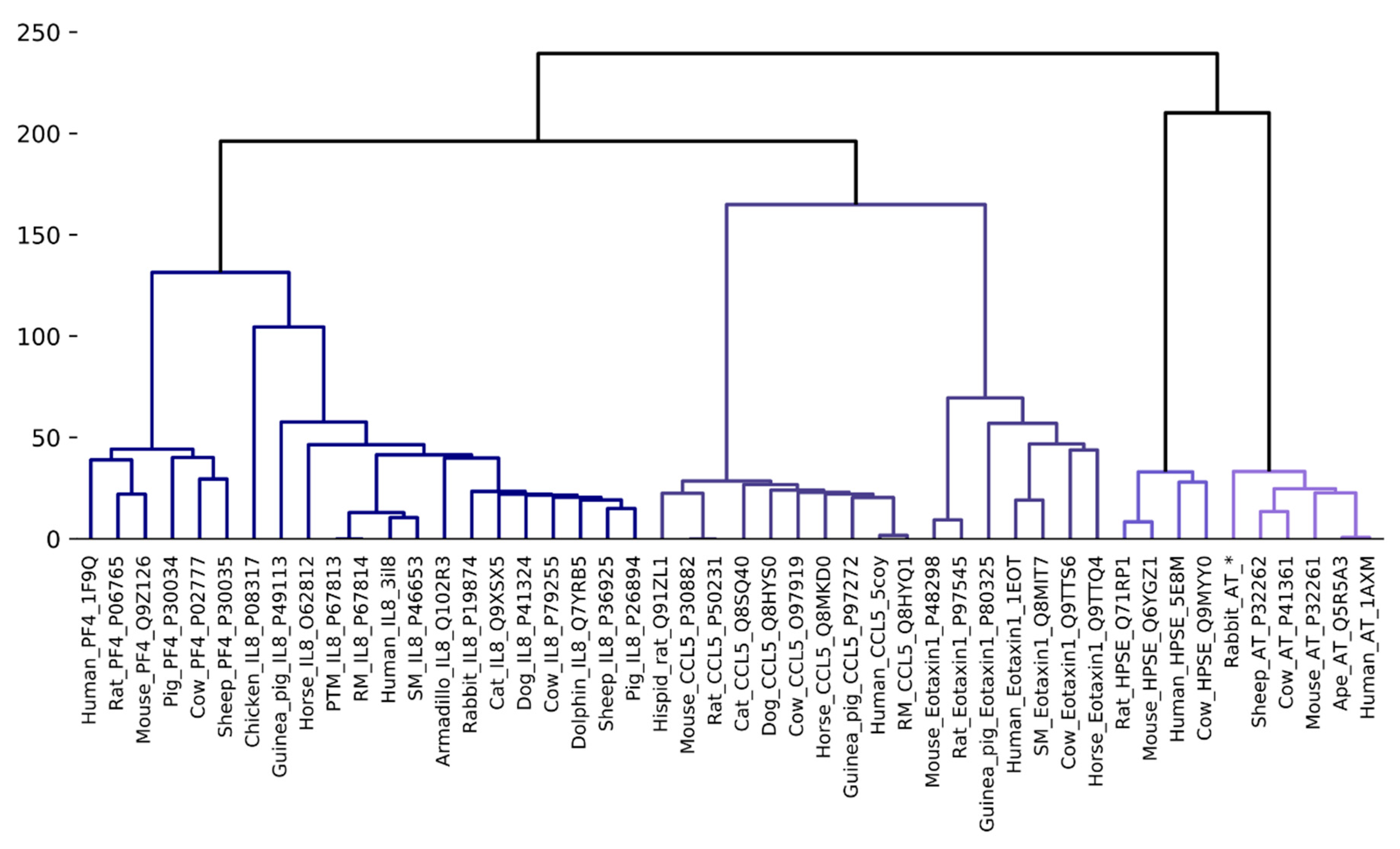
3.2. Phylogenetic Analysis and Clustering
3.3. Homology Modelling
3.4. Electrostatic Potential Surface Difference Calculations of GAG Binding Sites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gandhi Neha, S.; Mancera Ricardo, L. The Structure of Glycosaminoglycans and their Interactions with Proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coombe, D.R.; Kett, W.C. Heparan sulfate-protein interactions: Therapeutic potential through structure-function insights. Cell Mol. Life Sci. 2005, 62, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.; Coombe, D.R. Heparin Mimetics: Their Therapeutic Potential. Pharmaceuticals 2017, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Nahain, A.A.; Ignjatovic, V.; Monagle, P.; Tsanaktsidis, J.; Ferro, V. Heparin mimetics with anticoagulant activity. Med. Res. Rev. 2018, 38, 1582–1613. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.A.; Hawkins, D.W.; Peters, P.C.; Petitou, M.; Herbert, J.-M.; van Boeckel, C.A.A.; Meuleman, D.G. Fondaparinux, a Synthetic Pentasaccharide: The First in a New Class of Antithrombotic Agents—The Selective Factor Xa Inhibitors. Cardiovasc. Drug Rev. 2002, 20, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Ersdal-Badju, E.; Lu, A.; Zuo, Y.; Picard, V.; Bock, S.C. Identification of the Antithrombin III Heparin Binding Site. J. Biol. Chem. 1997, 272, 19393–19400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitou, M.; van Boeckel, C.A. A synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew. Chem. Int. Ed. Engl. 2004, 43, 3118–3133. [Google Scholar] [CrossRef] [PubMed]
- Herault, J.P.; Donat, F.; Barzu, T.; Crepon, B.; Bernat, A.; Lormeau, J.C.; Herbert, J.M. Pharmacokinetic study of three synthetic AT-binding pentasaccharides in various animal species-extrapolation to humans. Blood Coagul. Fibrinolysis 1997, 8, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Heyman, B.; Yang, Y. Mechanisms of heparanase inhibitors in cancer therapy. Exp. Hematol. 2016, 44, 1002–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khachigian, L.M.; Parish, C.R. Phosphomannopentaose Sulfate (PI-88): Heparan Sulfate Mimetic with Clinical Potential in Multiple Vascular Pathologies. Cardiovasc. Drug Rev. 2004, 22, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Prechel, M.M.; Walenga, J.M. Emphasis on the Role of PF4 in the Incidence, Pathophysiology and Treatment of Heparin Induced Thrombocytopenia. Thrombosis J. 2013, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proudfoot, A.E.I.; Johnson, Z.; Bonvin, P.; Handel, T.M. Glycosaminoglycan Interactions with Chemokines Add Complexity to a Complex System. Pharmaceuticals 2017, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Severin, I.C.; Soares, A.; Hantson, J.; Teixeira, M.; Sachs, D.; Valognes, D.; Scheer, A.; Schwarz, M.K.; Wells, T.N.C.; Proudfoot, A.E.I.; et al. Glycosaminoglycan analogs as a novel anti-inflammatory strategy. Front. Immunol. 2012, 3, 293. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.R.B.; Mosier, P.D.; Desai, U.R.; Rajarathnam, K. Solution NMR characterization of chemokine CXCL8/IL-8 monomer and dimer binding to glycosaminoglycans: Structural plasticity mediates differential binding interactions. Biochem. J. 2015, 472, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Schlorke, D.; Franz, S.; Arnhold, J. Functional aspects of the interaction between interleukin-8 and sulfated glycosaminoglycans AU—Pichert, Annelie. Biomatter 2012, 2, 142–148. [Google Scholar]
- Ellyard, J.I.; Simson, L.; Bezos, A.; Johnston, K.; Freeman, C.; Parish, C.R. Eotaxin Selectively Binds Heparin: An Interaction that Protects Eotaxin from Proteolysis and Potentiates Chemotactic Activity in Vivo. J. Biol. Chem. 2007, 282, 15238–15247. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kett, W.C.; Severin, I.C.; Agyekum, I.; Duan, J.; Amster, I.J.; Proudfoot, A.E.I.; Coombe, D.R.; Woods, R.J. The Interaction of Heparin Tetrasaccharides with Chemokine CCL5 Is Modulated by Sulfation Pattern and pH. J. Biol. Chem. 2015, 290, 15421–15436. [Google Scholar] [CrossRef] [PubMed]
- Amerio, P.; Frezzolini, A.; Feliciani, C.; Verdolini, R.; Teofoli, P.; De Pità, O.; Puddu, P. Eotaxins and CCR3 Receptor in Inflammatory and Allergic Skin Diseases: Therapeutical Implications. Curr. Drug Targets Inflamm. Allergy 2003, 2, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Aun, M.V.; Bonamichi-Santos, R.; Arantes-Costa, F.M.; Kalil, J.; Giavina-Bianchi, P. Animal models of asthma: Utility and limitations. J. Asthma Allergy 2017, 10, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Veliça, P.; Davies, N.J.; Rocha, P.P.; Schrewe, H.; Ride, J.P.; Bunce, C.M. Lack of functional and expression homology between human and mouse aldo-keto reductase 1C enzymes: Implications for modelling human cancers. Mol. Cancer 2009, 8, 121. [Google Scholar] [CrossRef] [PubMed]
- Coombe, D.R.; Stevenson, S.M.; Kinnear, B.F.; Gandhi, N.S.; Mancera, R.L.; Osmond, R.I.; Kett, W.C. Platelet endothelial cell adhesion molecule 1 (PECAM-1) and its interactions with glycosaminoglycans: 2. Biochemical analyses. Biochemistry 2008, 47, 4863–4875. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.; Gabdoulline, R.R.; Wade, R.C. Cross-species analysis of the glycolytic pathway by comparison of molecular interaction fields. Mol. Biosyst. 2010, 6, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Lortat-Jacob, H.; Grosdidier, A.; Imberty, A. Structural diversity of heparan sulfate binding domains in chemokines. Proc. Natl. Acad. Sci. USA 2002, 99, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Viola, C.M.; Brzozowski, A.M.; Davies, G.J. Structural characterization of human heparanase reveals insights into substrate recognition. Nat. Struct. Mol. Biol. 2015, 22, 1016. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.R.B.; Sawant, K.V.; Rajarathnam, K. Heparin-bound chemokine CXCL8 monomer and dimer are impaired for CXCR1 and CXCR2 activation: Implications for gradients and neutrophil trafficking. Open Biol. 2017, 7, 170168. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yarovoi, S.V.; Zhu, Z.; Rauova, L.; Hayes, V.; Lebedeva, T.; Liu, Q.; Poncz, M.; Arepally, G.; Cines, D.B.; et al. Atomic description of the immune complex involved in heparin-induced thrombocytopenia. Nat. Commun. 2015, 6, 8277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayo, K.H.; Ilyina, E.; Roongta, V.; Dundas, M.; Joseph, J.; Lai, C.K.; Maione, T.; Daly, T.J. Heparin binding to platelet factor-4. An NMR and site-directed mutagenesis study: Arginine residues are crucial for binding. Biochem. J. 1995, 312 Pt 2, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Gettins, P.G.W. Serpin Structure, Mechanism, and Function. Chem. Rev. 2002, 102, 4751–4804. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Adams, T.E.; Nangalia, J.; Esmon, C.T.; Huntington, J.A. Molecular basis of thrombin recognition by protein C inhibitor revealed by the 1.6-A structure of the heparin-bridged complex. Proc. Natl. Acad. Sci. USA 2008, 105, 4661–4666. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, W.; Brothers, A.; Wells, M.; Hatton, M.; Clarke, B.; Blajchman, M. Molecular cloning and expression of rabbit antithrombin III. Blood 1992, 79, 2330–2339. [Google Scholar] [PubMed]
- Fu, L.; Li, K.; Mori, D.; Hirakane, M.; Lin, L.; Grover, N.; Datta, P.; Yu, Y.; Zhao, J.; Zhang, F.; et al. Enzymatic Generation of Highly Anticoagulant Bovine Intestinal Heparin. J. Med. Chem. 2017, 60, 8673–8679. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Higashi, N.; Taka, T.; Nakajima, M.; Irimura, T. Cell Surface Localization of Heparanase on Macrophages Regulates Degradation of Extracellular Matrix Heparan Sulfate. J. Immunol. 2004, 172, 3830–3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masola, V.; Bellin, G.; Gambaro, G.; Onisto, M. Heparanase: A Multitasking Protein Involved in Extracellular Matrix (ECM) Remodeling and Intracellular Events. Cells 2018, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.; Lewis, K.D. PI-88: A novel inhibitor of angiogenesis. Expert Opin. Investig. Drugs 2008, 17, 1769–1776. [Google Scholar]
- Joyce, J.A.; Freeman, C.; Meyer-Morse, N.; Parish, C.R.; Hanahan, D. A functional heparan sulfate mimetic implicates both heparanase and heparan sulfate in tumor angiogenesis and invasion in a mouse model of multistage cancer. Oncogene 2005, 24, 4037. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I.; Fritchley, S.; Borlat, F.; Shaw, J.P.; Vilbois, F.; Zwahlen, C.; Trkola, A.; Marchant, D.; Clapham, P.R.; Wells, T.N.C. The BBXB Motif of RANTES Is the Principal Site for Heparin Binding and Controls Receptor Selectivity. J. Biol. Chem. 2001, 276, 10620–10626. [Google Scholar] [CrossRef] [PubMed]
- Deshauer, C.; Morgan, A.M.; Ryan, E.O.; Handel, T.M.; Prestegard, J.H.; Wang, X. Interactions of the Chemokine CCL5/RANTES with Medium-Sized Chondroitin Sulfate Ligands. Structure 2015, 23, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Abayev, M.; Rodrigues, J.; Srivastava, G.; Arshava, B.; Jaremko, L.; Jaremko, M.; Naider, F.; Levitt, M.; Anglister, J. The solution structure of monomeric CCL5 in complex with a doubly sulfated N-terminal segment of CCR5. FEBS J. 2018, 285, 1988–2003. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Watson, C.; Sharp, J.S.; Handel, T.M.; Prestegard, J.H. Oligomeric structure of the chemokine CCL5/RANTES from NMR, MS, and SAXS data. Structure 2011, 19, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.G.; Triandafillou, C.G.; Huang, T.-Y.; Zulueta, M.M.L.; Banerjee, S.; Dinner, A.R.; Hung, S.-C.; Tang, W.-J. Structural basis for oligomerization and glycosaminoglycan binding of CCL5 and CCL3. Proc. Natl. Acad. Sci. USA 2016, 113, 5000–5005. [Google Scholar] [CrossRef] [PubMed]
- Segerer, S.; Johnson, Z.; Rek, A.; Baltus, T.; von Hundelshausen, P.; Kungl, A.J.; Proudfoot, A.E.I.; Weber, C.; Nelson, P.J. The basic residue cluster 55KKWVR59 in CCL5 is required for in vivo biologic function. Mol. Immunol. 2009, 46, 2533–2538. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, P.; Bardi, G.; Clark-Lewis, I.; Baggiolini, M.; Uguccioni, M. Eotaxin is a natural antagonist for CCR2 and an agonist for CCR5. Blood 2001, 97, 1920–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millard, C.J.; Ludeman, J.P.; Canals, M.; Bridgford, J.L.; Hinds, M.G.; Clayton, D.J.; Christopoulos, A.; Payne, R.J.; Stone, M.J. Structural basis of receptor sulfotyrosine recognition by a CC chemokine: The N-terminal region of CCR3 bound to CCL11/eotaxin-1. Structure 2014, 22, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Sweeney, M.D.; Saad, O.M.; Crown, S.E.; Hsu, A.R.; Handel, T.M.; Leary, J.A. Chemokine-glycosaminoglycan binding: Specificity for CCR2 ligand binding to highly sulfated oligosaccharides using FTICR mass spectrometry. J. Biol. Chem. 2005, 280, 32200–32208. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.P.; Rajarathnam, K.; Kim, K.-S.; Clark-Lewis, I.; Sykes, B.D. Solution Structure of Eotaxin, a Chemokine That Selectively Recruits Eosinophils in Allergic Inflammation. J. Biol. Chem. 1998, 273, 22471–22479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dykstra, A.B.; Sweeney, M.D.; Leary, J.A. Structural Evidence for the Tetrameric Assembly of Chemokine CCL11 and the Glycosaminoglycan Arixtra. Biomolecules 2013, 3, 905–922. [Google Scholar] [CrossRef] [PubMed]
- Stillie, R.; Farooq, S.M.; Gordon, J.R.; Stadnyk, A.W. The functional significance behind expressing two IL-8 receptor types on PMN. J. Leukoc. Biol. 2009, 86, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, L.; Rajarathnam, K. Structural basis of chemokine receptor function--a model for binding affinity and ligand selectivity. Biosci. Rep. 2006, 26, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.R.B.; Rajarathnam, K. Solution NMR characterization of WT CXCL8 monomer and dimer binding to CXCR1 N-terminal domain. Prot. Sci. 2014, 24, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.W.; Raghuwanshi, S.K.; Grant, D.J.; Jala, V.R.; Rajarathnam, K.; Richardson, R.M. Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J. Immunol. 2009, 183, 3425–3432. [Google Scholar] [CrossRef] [PubMed]
- Frevert, C.W.; Kinsella, M.G.; Vathanaprida, C.; Goodman, R.B.; Baskin, D.G.; Proudfoot, A.; Wells, T.N.; Wight, T.N.; Martin, T.R. Binding of interleukin-8 to heparan sulfate and chondroitin sulfate in lung tissue. Am. J. Respir. Cell Mol. Biol. 2003, 28, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Skelton, N.J.; Quan, C.; Reilly, D.; Lowman, H. Structure of a CXC chemokine-receptor fragment in complex with interleukin-8. Structure 1999, 7, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Modi, W.S.; Yoshimura, T. Isolation of novel GRO genes and a phylogenetic analysis of the CXC chemokine subfamily in mammals. Mol. Biol. Evol. 1999, 16, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanino, Y.; Coombe, D.R.; Gill, S.E.; Kett, W.C.; Kajikawa, O.; Proudfoot, A.E.I.; Wells, T.N.C.; Parks, W.C.; Wight, T.N.; Martin, T.R.; et al. Kinetics of Chemokine-Glycosaminoglycan Interactions Control Neutrophil Migration into the Airspaces of the Lungs. J. Immunol. 2010, 184, 2677–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayo, K.H.; Chen, M.J. Human platelet factor 4 monomer-dimer-tetramer equilibria investigated by proton NMR spectroscopy. Biochemistry 1989, 28, 9469–9478. [Google Scholar] [CrossRef] [PubMed]
- Krauel, K.; Weber, C.; Brandt, S.; Zahringer, U.; Mamat, U.; Greinacher, A.; Hammerschmidt, S. Platelet factor 4 binding to lipid A of Gram-negative bacteria exposes PF4/heparin-like epitopes. Blood 2012, 120, 3345–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhailov, D.; Young, H.C.; Linhardt, R.J.; Mayo, K.H. Heparin dodecasaccharide binding to platelet factor-4 and growth-related protein-alpha. Induction of a partially folded state and implications for heparin-induced thrombocytopenia. J. Biol. Chem. 1999, 274, 25317–25329. [Google Scholar] [CrossRef] [PubMed]
- Stringer, S.E.; Gallagher, J.T. Specific Binding of the Chemokine Platelet Factor 4 to Heparan Sulfate. J. Biol. Chem. 1997, 272, 20508–20514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.-H.; Greinacher, A.; Delcea, M. Quantitative description of thermodynamic and kinetic properties of the platelet factor 4/heparin bonds. Nanoscale 2015, 7, 10130–10139. [Google Scholar] [CrossRef] [PubMed]
- Kreimann, M.; Brandt, S.; Krauel, K.; Block, S.; Helm, C.A.; Weitschies, W.; Greinacher, A.; Delcea, M. Binding of anti-platelet factor 4/heparin antibodies depends on the thermodynamics of conformational changes in platelet factor 4. Blood 2014, 124, 2442–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvarna, S.; Qi, R.; Arepally, G.M. Optimization of a murine immunization model for study of PF4/heparin antibodies. J. Thromb. Haemost. 2009, 7, 857–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, S.; Krauel, K.; Jaax, M.; Renne, T.; Helm, C.A.; Hammerschmidt, S.; Delcea, M.; Greinacher, A. Polyphosphates form antigenic complexes with platelet factor 4 (PF4) and enhance PF4-binding to bacteria. Thromb. Haemost. 2015, 114, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Jaax, M.E.; Krauel, K.; Marschall, T.; Brandt, S.; Gansler, J.; Furll, B.; Appel, B.; Fischer, S.; Block, S.; Helm, C.A.; et al. Complex formation with nucleic acids and aptamers alters the antigenic properties of platelet factor 4. Blood 2013, 122, 272–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33 (Suppl. S2), W299–W302. [Google Scholar] [CrossRef] [PubMed]
- Berezin, C.; Glaser, F.; Rosenberg, J.; Paz, I.; Pupko, T.; Fariselli, P.; Casadio, R.; Ben-Tal, N. ConSeq: The identification of functionally and structurally important residues in protein sequences. Bioinformatics 2004, 20, 1322–1324. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Perkins, J.A.; Showell, H.J.; Matsushima, K.; Williams, T.J.; Collins, P.D. A comparative study of the neutrophil stimulatory activity in vitro and pro-inflammatory properties in vivo of 72 amino acid and 77 amino acid IL-8. J. Immunol. 1992, 148, 106–111. [Google Scholar] [PubMed]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.E.S.; Mohan, R.R.; Gorham, R.D., Jr.; Kieslich, C.A.; Morikis, D. AESOP: A Python Library for Investigating Electrostatics in Protein Interactions. Biophys. J. 2017, 112, 1761–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.-Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics 2006, 15, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | GAG Binding Residues | Other Interactions | Ref. |
|---|---|---|---|
| Antithrombin (AT) | 46RR47 K136 235RK236K275 121FF122 K125Rl29 132RK133 228K | Thrombin | [6] |
| Heparanase | 389G 64N 391Y 97T 62N 224N E225 E343 Q270 R272 349GG350 | PI-88 and PG545 (GAG mimetics) | [24] |
| RANTES (CCL5) | 17R 44RKNR47 55KKWVR59 | CCR1, CCR3, CCR5, oligomerization | [17] |
| Eotaxin-1 (CCL11) | 44KLAK47 54KKK56 | CCR3 | [a] |
| IL-8 (CXCL8) | 15K18H20K23K60R64K68R | CXCR1, oligomerization | [25] |
| PF4 (CXCL4) | 20R22PR23 25T 46K49R 60YK61 64IK65 | CXCR3B, oligomerization | [26,27] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boittier, E.D.; Gandhi, N.S.; Ferro, V.; Coombe, D.R. Cross-Species Analysis of Glycosaminoglycan Binding Proteins Reveals Some Animal Models Are “More Equal” than Others. Molecules 2019, 24, 924. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050924
Boittier ED, Gandhi NS, Ferro V, Coombe DR. Cross-Species Analysis of Glycosaminoglycan Binding Proteins Reveals Some Animal Models Are “More Equal” than Others. Molecules. 2019; 24(5):924. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050924
Chicago/Turabian StyleBoittier, Eric D., Neha S. Gandhi, Vito Ferro, and Deirdre R. Coombe. 2019. "Cross-Species Analysis of Glycosaminoglycan Binding Proteins Reveals Some Animal Models Are “More Equal” than Others" Molecules 24, no. 5: 924. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050924