Investigation of 8-Aza-7-Deaza Purine Nucleoside Derivatives


1
College of Chemistry and Molecular Engineering, Zhengzhou University, 100 Science Avenue, Zhengzhou 450001, China
2
Zhengzhou Granlen PharmaTech, Ltd., 1300 Eastern Hanghai Road, Zhengzhou 450016, China
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(5), 983; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050983
Submission received: 22 February 2019
/
Accepted: 5 March 2019
/
Published: 11 March 2019
(This article belongs to the Section Organic Chemistry)
Abstract
:Glycosylation of 6-amino-4-methoxy-1H-pyrazolo[3,4-d]pyrimidine and its iodo- and bromo- analogues with the protected ribofuranose and 2′-deoxyribofuranose under different conditions resulted in the synthesis of N9- and N8-glycosylated purine nucleosides. Five key intermediate nucleosides, having 6-methoxy, 7-iodo, and 2-bromo groups, were further derivatized to 23 final 8-aza-7-deazapurine nucleoside derivatives. The structures of N9- and N8-glycosylated products were assigned based on UV and NMR spectra. HMBC analysis of 2D NMR spectra and X-ray crystallographic studies of the representative compounds unambiguously verified the connection of ribose ring to N9- or N8-position of the purine ring. The anticancer activity of these new compounds was evaluated.
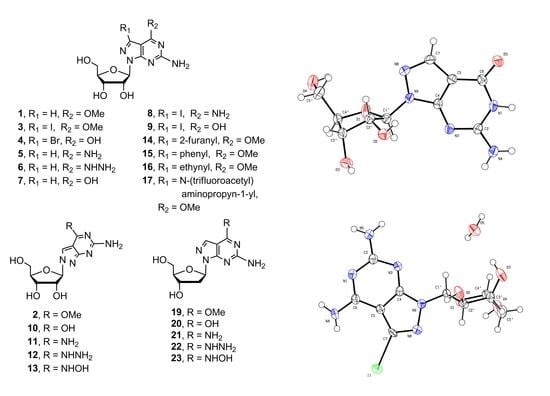
1. Introduction
8-Aza-7-deazapurines as well as their nucleosides exhibit a wide range of antiparasitic, antitumor, and antiviral activity [1,2]. A classical example of these biologically active analogues is allopurinol, which is an approved drug for the treatment of gout and it is also active against several Leishmania and trypanosome species [3]. 8-Aza-7-deazapurine-2,6-diamine and its riboside 5 are powerful purine nucleoside antagonists [4] (Figure 1), and 2′-deoxy-2′-β-fluoro-8-aza-7-deazapurine nucleosides display significant activity against HBV [5]. 8-Aza-7-deazapurines closely resemble the structure of purine with only C8 and N7 exchanged, which make their nucleosides excellent substrates of bio-enzymes [6,7] and ideal mimics of the canonical purine constituents of DNA or RNA. It has been demonstrated that the thermal stability of DNA duplexes increases significantly when 7-halogeno/alkynyl 8-aza-7-deazapurines replace purine [8]. Some N8-2′-deoxy-adenine analogues show ambiguous base pairing when incorporated into oligonucleotide duplexes opposite to the four canonical DNA-constituents [9]. Compounds 5 (pK = 5.8) and 11 (pK = 6.4) (Figure 1) are possibly protonated in neutral conditions when incorporated into RNA, thereby forming a stable mismatched base pair [10]. The RNA containing 7-deaza-8-azainosine was found to be a weaker toll-like receptor 8 activator than guanosine-containing RNA [11]. 7-Alkyne-8-aza-7-deaza nucleosides was used for azide-alkyne “Click” conjugation of DNA [12,13]. Furthermore, some other properties of such oligonucleotides have also been studied in drug discovery related fields [14,15].
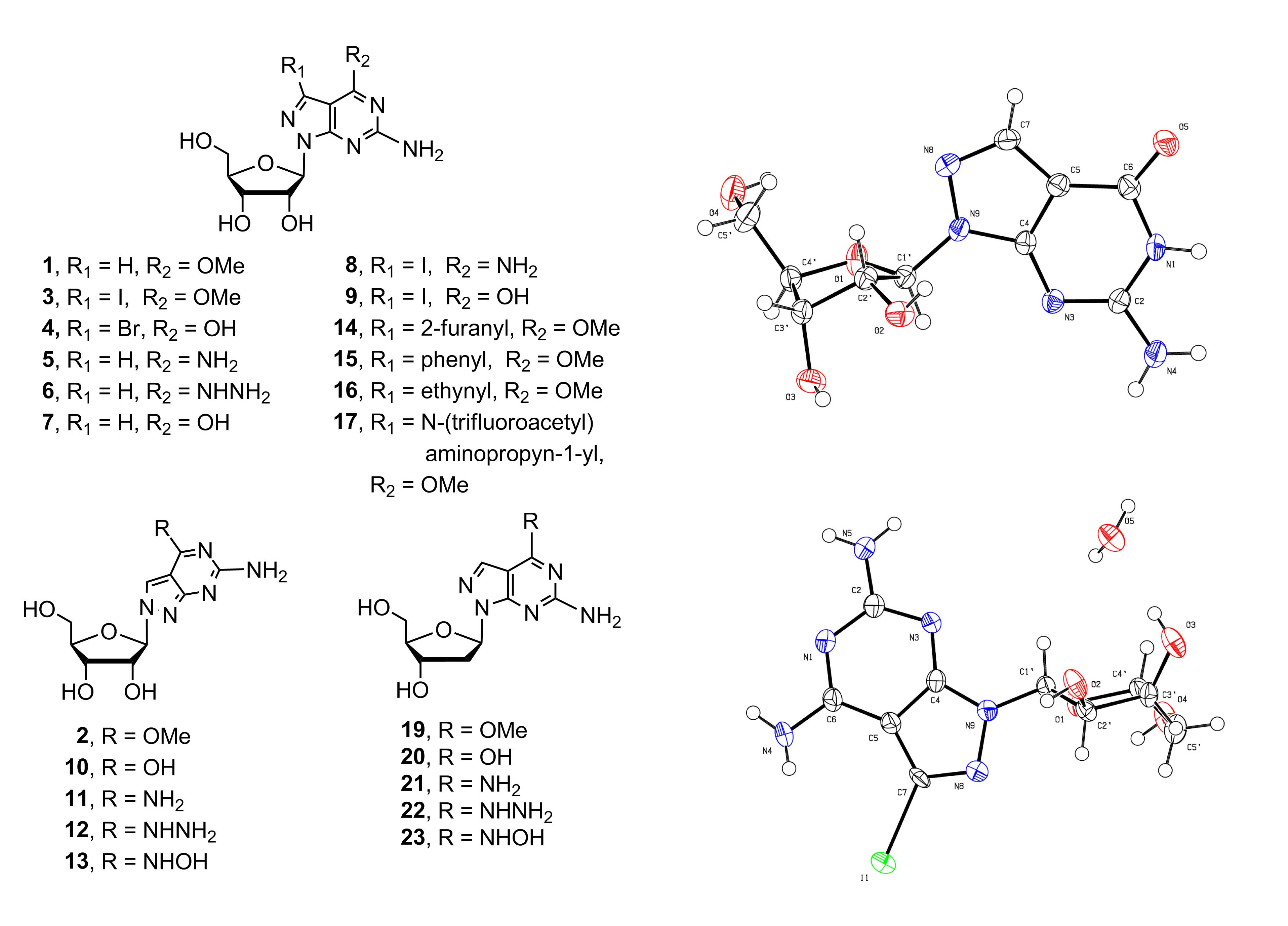
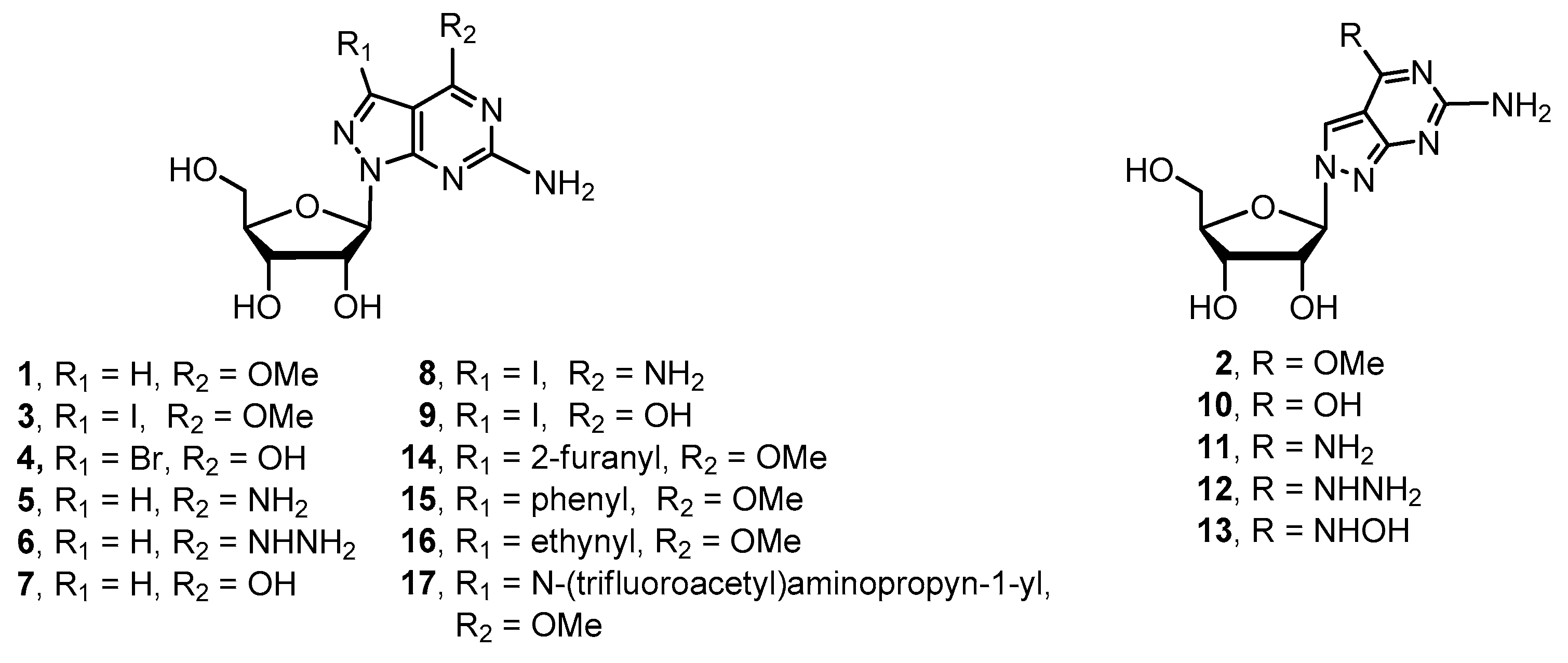
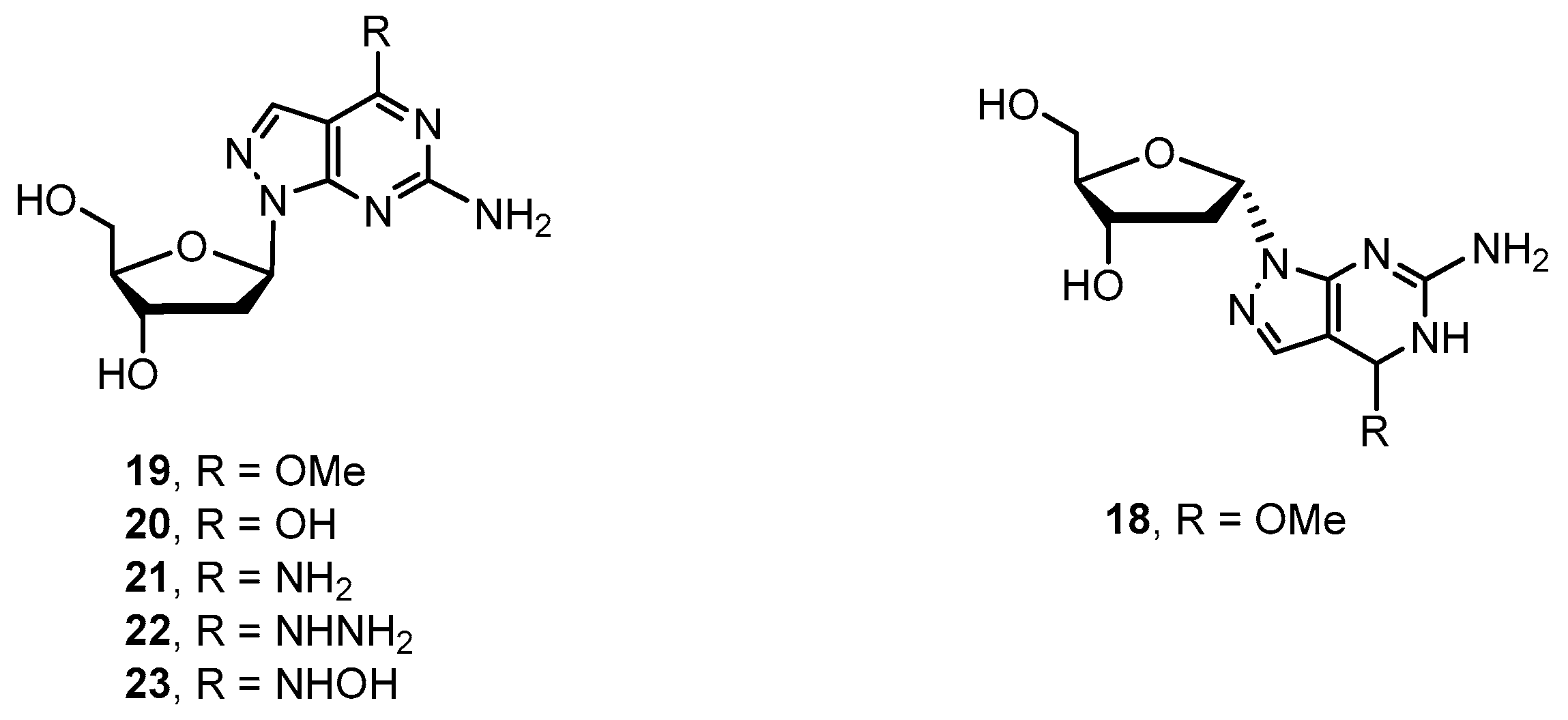
Various 8-aza-7-deaza purine nucleosides have been synthesized in the past fewer years. However, there still remains a space for the design and development of new nucleoside-based therapeutics. We decided to investigate 8-aza-7-deaza purine nucleoside derivatives modified at the positions 6 and 7 of the purine ring. Herein, we report the synthesis of 23 modified 8-aza-7-deaza purine nucleoside derivatives, of which 11 are novel compounds, including 2-amino-6-substituted-ribonucleosides 1–3 and 5–13 (Figure 1), 2-amino-6-methoxy/oxo-7-substituted purine ribonucleosides 4 and 14–17 (Figure 1) and 2-amino-6-substituted 2′-deoxy nucleosides 18–23 (Figure 2). Different glycosylation protocols were studied for N9- and N8-glycosylation. In addition, the glycosylation sites and anomeric configuration of the newly synthesized compounds were investigated and assigned on the basis of 1H-NMR, 13C-NMR, 2D NMR, UV spectra, and X-ray crystallographic analysis of the representative compounds (see Supplementary Materials). The anticancer activity of these compounds was also evaluated.
2. Results and Discussion
2.1. Synthesis of 8-Aza-7-deazapurine Ribo-Nucleosides
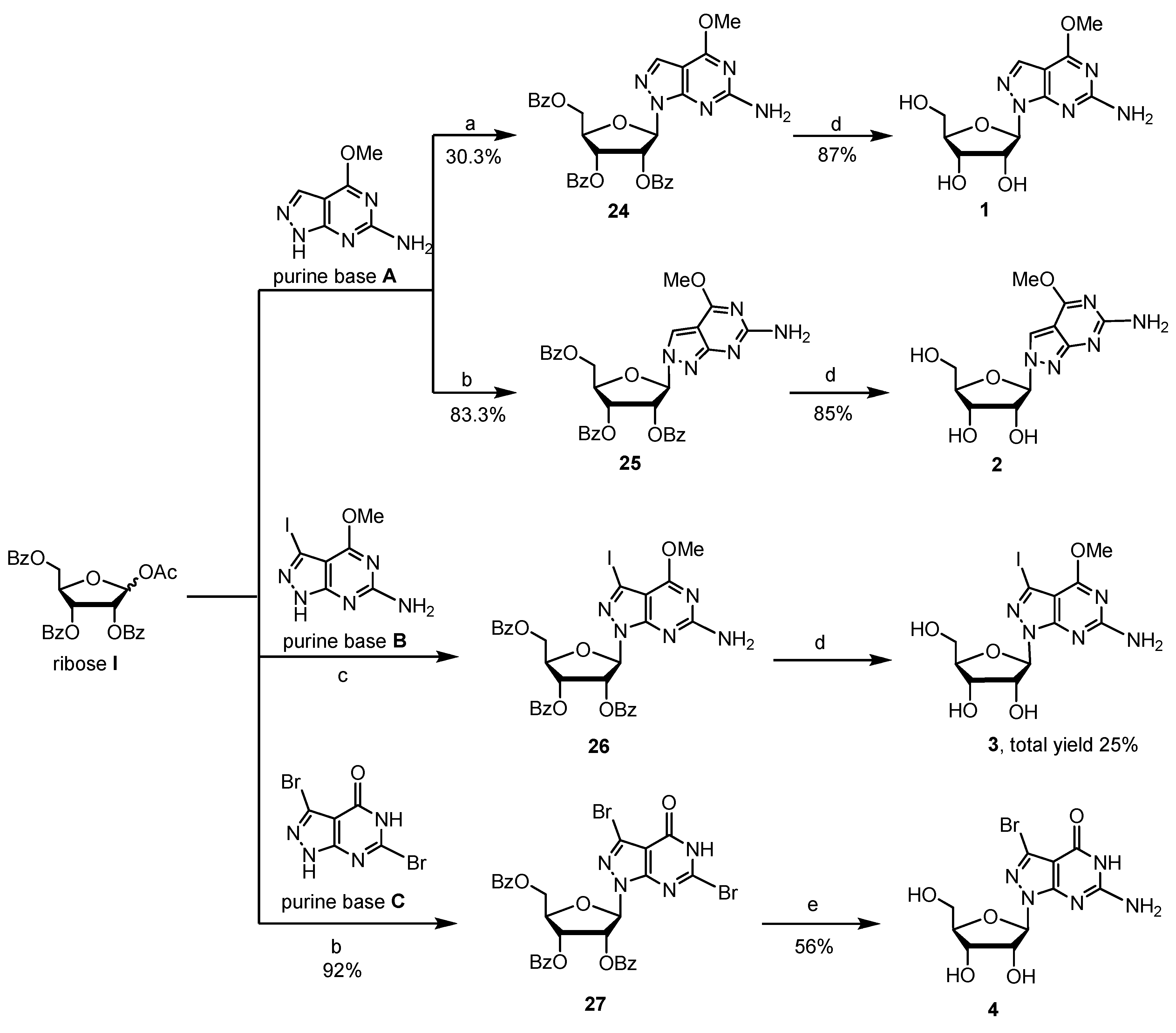
6-Amino-4-methoxy-1H-pyrazolo[3,4-d]pyrimidine (purine base A) [16], 6-amino-3-iodo-4-methoxy-1H-pyrazolo[3,4-d]pyrimidine (purine base B) [17], and 3,6-dibromo-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (purine base C) [18] (Scheme 1) were made utilizing reported protocols, and selected as the heterocyclic bases of 8-aza-7-deazapurine nucleosides to be studied. These purines were chosen for a number of advantages including the switch in the nitrogen atom from position 7 to 8, which changes the binding mode and strength of purine nucleosides in the duplex nucleic acids and other biological systems, substantially altering the biological properties and application. The 2-amino-group on both 8-aza-7-deazapurine bases A and B does not need to be protected during the glycosylation process. The 6-methoxy group on purine bases A and B is selected to serve as a protecting group during glycosylation process. It is stable enough not to be cleaved in subsequent processes while active enough to be substituted by strong nucleophiles, therefore, converting to the desired corresponding 2-amino-8-aza-7-deazaguanosine derivatives when needed. The 6-oxo on purine base C does not need to be protected for glycosylation. The iodination of purine bases A to B adds high versatility for further derivatization on position 7 via Heck, Stille, Suzuki, Sonogashira, and related reactions. This will expand research in the related fields. To the best of our knowledge, the direct glycosylation of 2-amino-8-aza-7-deazapurine bases A and B with routinely used l-O-acetyl-2,3,5-tri-O-benzoyl-d-ribofuranose (ribose I) has not been reported.
A number of glycosylation protocols have been routinely used for the synthesis of different classes of nucleoside derivatives [10,19,20]. The silyl-Hilbert–Johnson glycosylation processes of silylated nucleobases with protected ribosides under Lewis acid conditions are very efficient for most cases. We glycosylated purine base A with ribose I (Scheme 1) under historic stannic chloride condition with no success. We then switched to the stronger promoter trimethylsilyl triflate (TMSOTf) and also explored silylating agents hexamethyldisilazane (HMDS) and N,O-bis(trimethylsilyl)acetamide (BSA) in acetonitrile, nitromethane and 1,2-dichloroethane. It was found that the HMDS-silylated purine base A was glycosylated with ribose I using TMSOTf in freshly distilled 1,2-dichloroethane overnight at room temperature giving the best result. The N9-glycosylated compound 24 (Scheme 1) was isolated in 30.3% yield as the major product, and only very trace amount of N8-glycosylated compound 25 was observed on TLC but could not be isolated.
We would like to develop a high-yield protocol that would lead to the large-scale synthesis of the key intermediates and diversified final 8-aza-7-deazaguanosine derivatives. We performed the reported protocol [10] using boron trifluoride ether solution as the Lewis acid promotor at an elevated temperature. The glycosylation of ribose I with purine base A in acetonitrile worked excellently. However, the N8-glycosylated product 25 was isolated in 83.3% high yield, and almost no N9-glycosylated compound 24 was observed under this condition (Scheme 1). The N9-glycosylated product 24 was the major product under room temperature glycosylation condition while the N8-glycosylated product 25 was the thermodynamically dominant product formed at an elevated temperature. Therefore, two different compounds were made under different conditions from the same materials.
Next, we set out to study the glycosylation of purine base B, which introduces a 7-iodo moiety that enables further derivatization. The glycosylation of iodo-purine base B with ribose I under both SnCl4 and TMSOTf conditions resulted in the complicated mixtures, and only trace amount of N9-glycosylated product 26 could be detected. We then utilized boron trifluoride-ether as the catalyst for this glycosylation in acetonitrile at room temperature. The desired N9-glycosylated product 26 was isolated in about 28% yield (Scheme 1). The purine base C, without 6-oxo-protection, was also glycosylated with ribose I, resulting in the protected nucleoside 27. The two bromo-atoms on this molecule are expected to have different reactivity allowing for chemo-selective functionalization towards the synthesis of novel nucleoside derivatives.
The glycosylated products 24, 25, and 26 thus obtained were deprotected giving the corresponding 8-aza-7-deazapurine nucleosides 1–3. These 6-methoxy derivatives were heated with ammonium hydroxide solution overnight providing the corresponding 2-amino-8-aza-7-deazaadenosine derivatives 5, 8, and 11 in excellent yields (Scheme 2). The 6-methoxy compounds 1, 2, and 3 were also treated with aqueous potassium hydroxide solution to undergo deprotection, yielding the corresponding 8-aza-7-deazaguanosine derivatives 7, 9, and 10. Similarly, key intermediates 1 and 2 were heated with hydrazine solution to give the corresponding 6-hydrozino-purine nucleosides 6 and 12. The 6-hydroxyamino purine nucleoside 13 was obtained by treatment of compound 2 with hydroxylamine at 60 °C. Therefore, the 6-methoxy group was effectively used as the protecting group during the glycosylation process, and it can be further substituted with various nucleophiles to form 6-modified purine nucleoside derivatives. When compound 27 was deprotected with NH3/MeOH at 120 °C, the bromo-atom at position 2 was substituted with NH2 group during the process, resulting in the corresponding 7-bromo-8-aza-7-deazaguanosine analogue 4. Therefore, the bromo-atom at position 2 is more reactive against nucleophile than the bromo-atom at position 7.
The site of glycosylation and anomeric configuration of 24, 25, and 26 was assigned on the basis of 1H-NMR, 13C-NMR, 2D NMR, UV spectra, and X-ray crystallographic analysis of the representative compounds.
The nucleoside products 1 and 3 have very similar UV absorption compared to the corresponding free purine bases A and B (Table 1). This indicates that the ribofuranosyl group was glycosylated at the N9 position of purine bases A and B because the structure still maintains the same aromatic conjugate system [1]. However, the N8-glycosylated nucleosides showed very different UV spectra because the ribose-base bond connection dramatically altered the aromatic conjugate system. Careful UV spectral comparison of the N9- and N8-glycosylated nucleoside pairs shows the difference clearly. The N8-glycosylated products 2, 10, 11, and 12 have UV maximum absorbances λmax at longer wavelength than corresponding N9-glycosylated products 1, 7, 5, and 6 (Figure 3).
We studied the 2D NMR (HMBC heteronuclear multiple bond correlation) of N9-glycosylated 8-aza-7-deazaguanosine 7 and N8-glycosyalted 8-aza-7-deazaguanosine derivatives 10 (Figure 4). 1′-H proton (5.88 ppm) on the ribose ring has a long-range coupling correlation with C4 (157.7 ppm) of the purine base for compound 7 while compound 10 does not have this coupling correlation. 1′-H (5.66 ppm) on the ribose ring has a long-range coupling correlation with C7 (128.2 ppm) of the purine base for compound 10 but compound 7 does not have the same coupling correlation. In addition, the 7-H on the purine ring of compound 10 is closer to the 1′-C of the ribose ring comparing to the distance of compound 7. Therefore, the 7-H (8.525 ppm) on the purine ring of compound 10 has the long-range coupling correlation with 1′-C (94.057 ppm) of the ribose ring. However, compound 7 does not have the same coupling. Therefore, the 1′-C of the ribose ring is connected to N9 of the purine base on compound 7 (1′-H vs C4 coupling) while 1′-C of the ribose ring is connected to N8 of the purine base on the compound 10 (1′-H vs. C7 coupling and 7H vs 1′-C coupling). The data clearly further verified that compound 7 is the N9-glycosylated 8-aza-7-deazaguanosine, and compound 10 is the N8-glycosylated 8-aza-7-deazaguanosine derivative.
X-ray Crystallographic Study
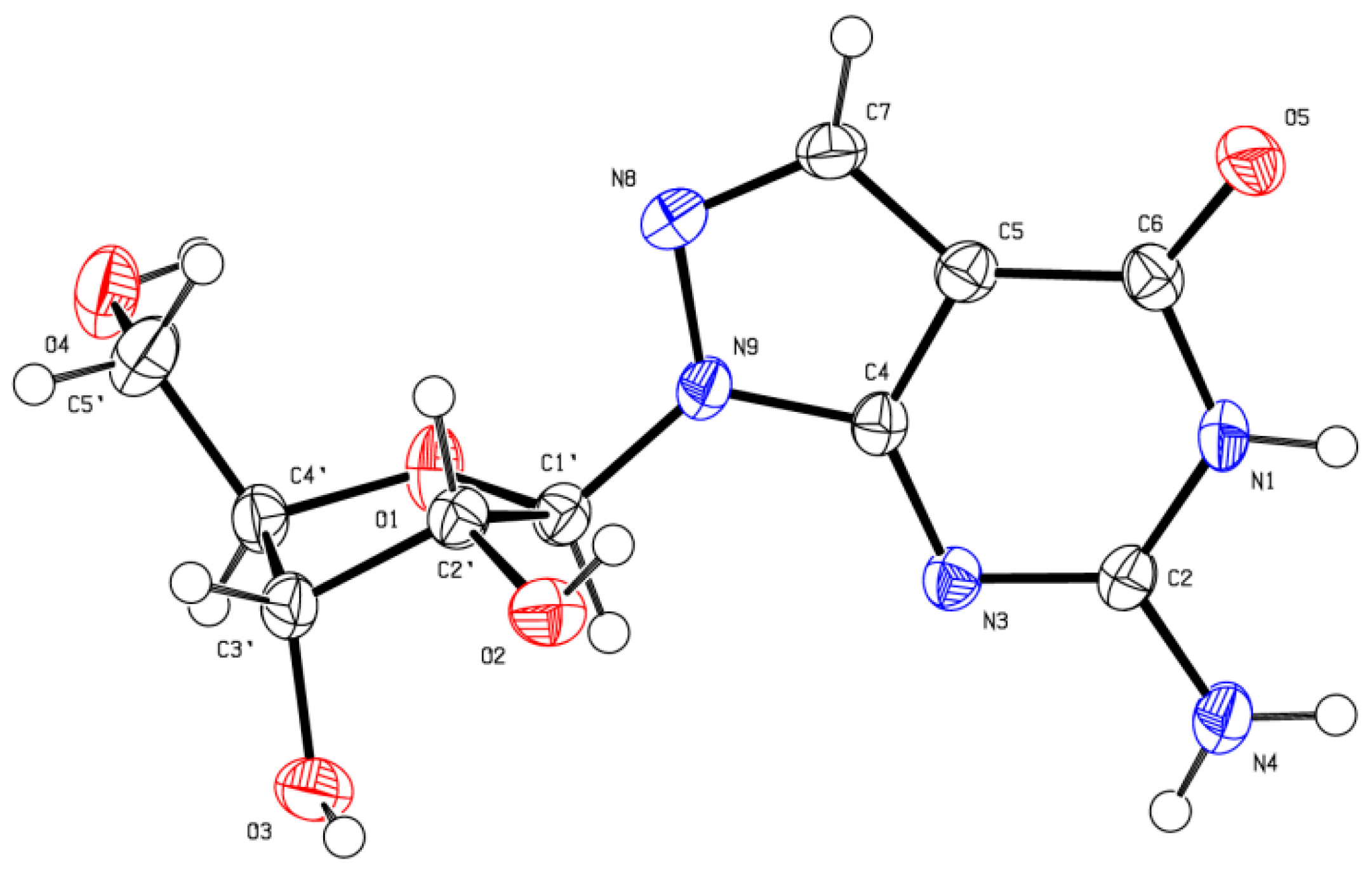
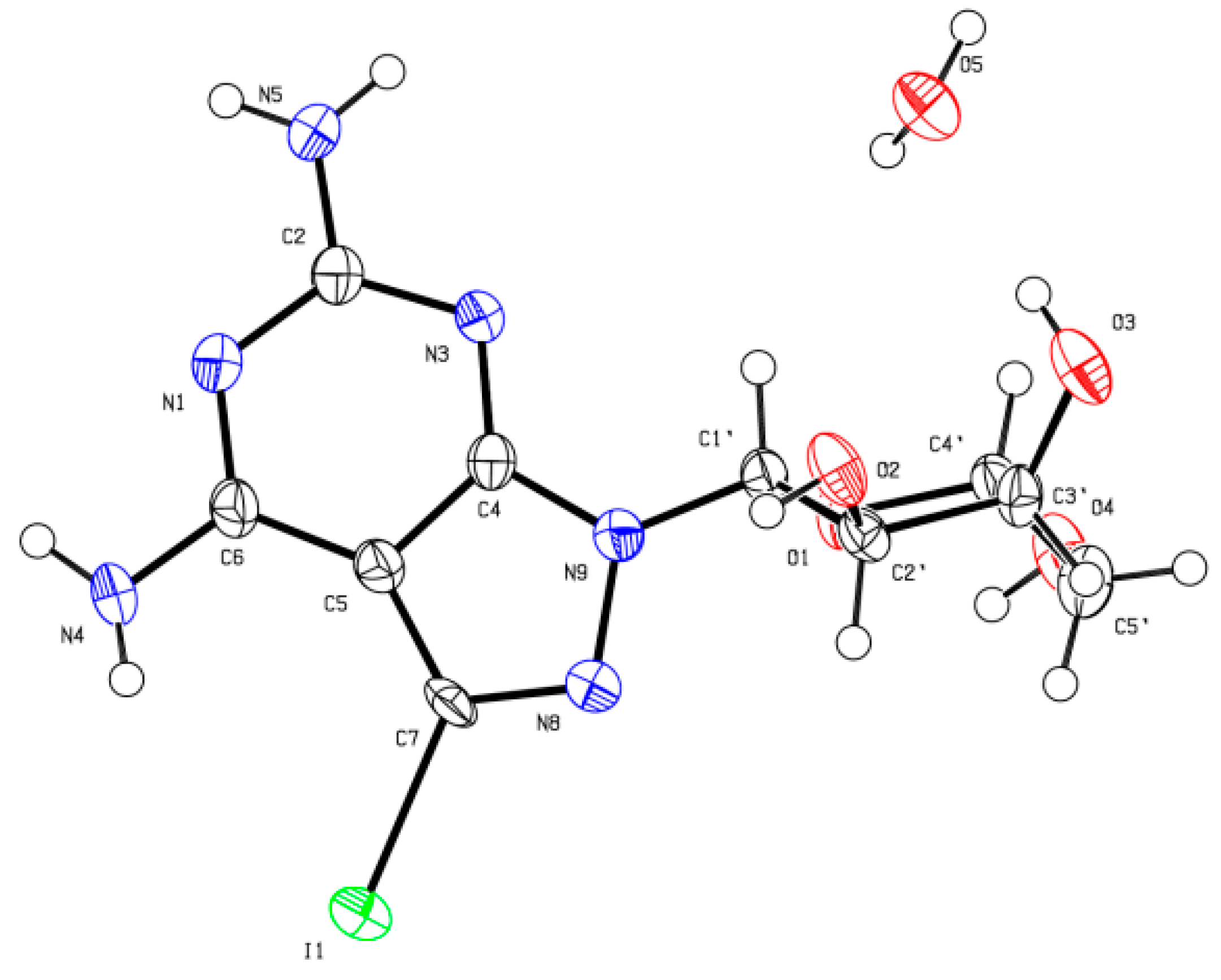
Slow crystallization of compounds 7 and 8 from water gave X-ray quality crystals. A suitable crystal was selected and mounted on a Xcalibur, Eos, Gemini diffractometer. The crystal was kept at 293(2) K during data collection. Using Olex2, the structure was solved with the ShelXS structure solution program using Direct Methods and refined with the ShelXL refinement package using Least Squares minimization. The resulted crystal structures are shown in Figure 5 and Figure 6. The results confirmed the β-D-anomeric configuration and the site of glycosylation as N9. Consequently, the structures of the corresponding products 7 and 8 were further verified.
Furthermore, the iodinated intermediate 3 is a valuable starting point for the introduction of aromatic or alkynyl substituents by the Pd catalyzed cross-coupling reaction. The 7-iodine intermediate 3 was coupled with 2-(tributylstannyl)furan catalyzed by bis(triphenylphosphine) palladium(II) chloride at 90 °C in DMF providing derivative 14 in 90% yield. Compound 3 was reacted with phenylboronic acid catalyzed by tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) in DME–water (2:1) mixture resulting in the desired 7-substituted product 15 in 80% yield. The 7-alkynylated products 16 and 17 were obtained in high yields by Sonogashira coupling reaction of 3 with alkynyl reagents (Scheme 3). The 8-aza-7-deazaguanine derivatives having 7-iodo (compound 9) and 7-bromo (compound 4) gave extremely low yields under Pd-catalyzed cross-coupling reaction conditions. Therefore, protection of 6-oxo group by methoxy group is required to activate the 7-halo atom for C-C coupling reactions.
2.2. Synthesis of 8-Aza-7-deaza-2′-deoxy Purine Nucleosides
2′-Deoxy-ribofuranosyl halide was utilized to glycosylate with various 7-dezaz-, 8-aza-7-deaza- and other purine related heterocyclic bases [21,22,23,24]. We did the glycosylation of purine base A with 2-deoxy-3,5-di-O-(p-toluoy1)-α-d-erythro-pentafuranosyl chloride (ribose II) [24,25] under the typical potassium conditions in the presence of cryptand TDA-1 catalyst. The desired N9-β-glycosylated compound 29 was obtained in 52% yield, and the α-anomer 28 was also isolated in 11% yield (Scheme 4) [16]. Surprisingly, N8-glycosylated compound was not observed. Deprotection of compounds 28 and 29 was accomplished with sodium methoxide at ambient temperature to give product 18 and 19 in 89% and 91% yields, respectively (Scheme 4). The 6-methoxy group of compound 19 was reacted with sodium hydroxide, ammonium hydroxide, hydrazine, and hydroxylamine as described above giving the corresponding 8-aza-7-deaza-2′-deoxy-purine nucleoside derivatives 20–23 in good yield.
3. Biological Evaluation
Newly synthesized compounds were tested for inhibitory activity against human lung carcinoma cell line A549 and the human breast cancer cell line MDA-MB-231 (Table 2). We found that the 7-iodine substituted derivative 8 showed the best inhibitory activity against A549 with an IC50 value of 7.68 μM. Compounds 14 and 16 showed slightly better activity than other modified derivatives, which did not show detectable activity against the tested tumor cell lines. By comparing the activity of tested analogs with different substituents, it suggested that electron-donating group at 6-position of the base may improve the activity. Further biological evaluation of these modified nucleosides is in progress and will be reported in due course.
4. Materials and Methods
4.1. Materials and Reagents
Starting materials and reagents were obtained from commercial suppliers and were used without further purification unless otherwise stated. 1H NMR spectra were obtained with a Bruker Avance 400 spectrometer using DMSO-d6 or CDCl3 (ppm) downfield with respect to an internal δ as solvents. Chemical shifts are reported as standard of tetramethylsilane (TMS). Product purity was tested by an Agilent 1260 analytical HPLC system. LC-MS spectra were measured on an Agilent 6120 LC–MS spectrometer. High-resolution mass spectra were obtained on a Micro-Q-TOF mass spectrometer. Single-crystal structure was determined by a Xcalibur, Eos, Gemini X-ray single crystal diffractometer. TLC was performed on silica gel GF254. Flash chromatography was performed on silica gel 200–300 mesh (Yantai Silica Gel Co. LTD). 6-Amino-4-methoxy-1H-pyrazolo[3–d]pyrimidine (purine base A), 6-amino-3-iodo-4-methoxy-1H-pyrazolo[3,4-d]pyrimidine (purine base B) and 3,6-dibromo-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (purine base C) were prepared following literature procedures [16,17,18]. The synthesis of N-(2-propynyl)-2,2,2-trifluoroacetamide was accomplished from propargylamine according to the reported protocol [26].
4.2. Chemical Synthesis
4.2.1. Procedure for Preparation of Glycosylated Products 24–26 and Key Intermediate 3
Synthesis of 6-amino-4-methoxy-1-(2,3,5-tri-O-benzoyl-β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (24): A suspension of 6-amino-4-methoxy-1H-pyrazolo[3.4-d]pyrimidine (purine base A, 9.0 g, 54.5 mmol, 1.0 eq) and a catalytic amount of ammonium sulfate in hexamethyldisilazane (HMDS, 150 mL) was refluxed for 6 h. The excess hexamethyldisilazane was removed by evaporation under reduced pressure, and the residue was dissolved in 1,2-dichloroethane (200 mL). l-O-Acetyl-2,3,5-tri-O-benzoyl-d-ribofuranose (ribose I) (35.7 g, 70.8 mmol, 1.3 eq) was added at room temperature. The reaction mixture was cooled to 0 °C, and trimethylsilyl trifluoromethanesulfonate (TMSOTf, 29.6 mL, 163.5 mmol, 3.0 eq) was added dropwise for 30 min with stirring. The reaction mixture was stirred at room temperature overnight. Upon completion of the reaction as monitored by TLC, the mixture was diluted with dichloromethane (200 mL) and washed with saturated sodium bicarbonate solution. The aqueous layer was extracted with dichloromethane. The combined organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography to afford 10.0 g main product N9 β-isomer 24 as a white solid in 30.3% yield with an HPLC purity of 96%; Rf = 0.3 (petroleum ether–ethyl acetate = 1:1).
Synthesis of 6-amino-4-methoxy-2-(2,3,5-tri-O-benzoyl-β-d-ribofuranosyl)-2H-pyrazolo[3,4-d]pyrimidine (25): 6-Amino-4-methoxy-1H-pyrazolo[3,4-d]pyrimidine (purine base A; 8.0 g, 48.4 mmol, 1.0 eq) was suspended in 200 mL dry acetonitrile and 1-O-acetyl-2,3,5-tri-O-benzoyl-d-ribofuranose (ribose I) (36.6 g, 72.6 mmol, 1.5 eq) was added. The reaction mixture was heated to reflux at 95 °C, and the freshly distilled BF3·OEt2 (12.2 mL, 96.8 mmol, 2.0 eq) was then added with stirring. The reaction mixture became clear immediately and then slowly became dark. The mixture was stirred at this temperature for 15 min. Upon the completion of the reaction as monitored by TLC, the reaction mixture was cooled to room temperature and concentrated under reduced pressure. The resulting residue was purified by column chromatography to afford 25.0 g N8-product 25 as a white solid in 83.3% yield with an HPLC purity of 96.8%. Rf = 0.6 (dichloromethane–methanol = 30:1). UV–vis (MeOH) λmax: 225 nm; ESI-MS m/z: 610.6 [M + H]+.
Synthesis of 6-amino-3-iodo-4-methoxy-1-(2,3,5-tri-O-benzoyl-β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]-pyrimidine (26) and 6-amino-3-iodo-4-methoxy-1-(β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (3): 6-Amino-3-iodo-4-methoxy-1H-pyrazolo[3,4-d]pyrimidine (purine base B, 5.0 g, 17.2 mmol, 1.0 eq) and 1-O-acetyl-2,3,5-tri-O-benzoyl-d-ribofuranose (ribose I) (13.0 g, 25.8 mmol, 1.5 eq) were suspended in 200 mL of anhydrous acetonitrile. The freshly distilled BF3·OEt2 solution (3.2 mL, 34.4 mmol, 2.0 eq) was added in 30 min with stirring at room temperature, and the reaction mixture was stirred at the same temperature for 5 h. Upon the completion of the reaction as monitored by TLC, the reaction mixture was poured into 500 mL of saturated sodium bicarbonate solution and extracted with ethyl acetate. The organic phase was separated, and the aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate. The drying agent was filtered off, and the filtrate was concentrated under reduced pressure to afford crude product 26, which was used directly without further purification. Rf = 0.2 (dichloromethane–methanol = 30:1). The crude compound 26 was dissolved in 100 mL of MeOH and 10 mL of THF solution. Then sodium methoxide (2.8 g, 51.6 mmol, 3.0 eq) was added, and the mixture was stirred at room temperature for 5 h. Upon completion of the reaction as monitored by TLC, the mixture was neutralized with 2 N HCl solution and evaporated under reduced pressure. The resulting residue was purified by column chromatography to afford 1.8 g desired product 3 as a white solid in 25% overall yield with an HPLC purity of 98.5%. Rf = 0.2 (dichloromethane–methanol = 10:1). UV–vis (MeOH) λmax: 232, 278 nm; 1H NMR (DMSO-d6, 400 MHz) δ 7.02 (s, 2H, NH2), 5.90 (d, J = 4.8 Hz, 1H, 1′-H), 5.33 (d, 1H, J = 6.0 Hz, 2′-OH), 5.08 (d, J = 5.2 Hz, 1H, 3′-OH), 4.74 (t, J = 5.6 Hz, 1H, 5′-OH), 4.45–4.55 (m, 1H, 2′-H), 4.08–4.15 (m, 1H, 3′-H), 3.09 (s, 3H, CH3), 3.80–3.87 (m, 1H, 4′-H), 3.38–3.58 (m, 2H, CH2); ESI-MS m/z: 424.3 [M + H]+, 446.2 [M + Na]+.
4.2.2. Procedure for Preparation of Glycosylated Product 27 and Compound 4
Synthesis of3,6-dibromo-4-hydro-1-(2,3,5-tri-O-benzoyl-β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (27): 3,6-Dibromo-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (purine base C; 14.2 g, 48.4 mmol, 1.0 eq) was suspended in 300 mL dry acetonitrile and 1-O-acetyl-2,3,5-tri-O-benzoyl-d-ribofuranose (ribose I) (36.6 g, 72.6 mmol, 1.5 eq) was added. The reaction mixture was heated to reflux at 95 °C, and the freshly distilled BF3·OEt2 (12.2 mL, 96.8 mmol, 2.0 eq) was then added with stirring. The reaction mixture became clear immediately and then slowly became dark. The mixture was kept stirred at this temperature for 20 min. Upon the completion of the reaction as monitored by TLC, the reaction mixture was cooled to room temperature and concentrated under reduced pressure. The resulting residue was purified by column chromatography to afford 33 g product 27 as a white solid in 92% yield with an HPLC purity of 98%. Rf = 0.5 (dichloromethane–methanol = 20:1). UV–vis (MeOH) λmax: 231, 275 nm; 1H NMR (DMSO-d6, 400 MHz) δ 8.06–8.16 (m, 2H, Ar-H), 7.91–8.01 (m, 4H, Ar-H), 7.51–7.61 (m, 3H, Ar-H), 7.34–7.46 (m, 6H, Ar-H), 6.62 (d, J = 2.8 Hz, 1H, 1′-H), 6.27–6.36 (m, 1H, 2′-H), 6.17–6.26 (m, 1H, 3′-H), 4.72–4.89 (m, 2H, 4′-H, CH2), 4.57–4.70 (m, 1H, CH2); ESI-MS m/z: 761.0 [M + Na]+.
Synthesis of 6-amino-3-bromo-1,5-dihydro-1-(β-d-ribofuranosyl)-4H-pyrazolo[3,4-d]pyrimidin-4-one (4): Compound 27 (9.0 g, 12.2 mmol) was dissolved in NH3/MeOH (300 mL, saturated at 0 °C) in high pressure reactor, and the reaction mixture was heated at 120 °C overnight. Upon completion of the reaction as monitored by TLC, the reaction solvent was evaporated under reduced pressure. The resulting precipitate was filtered off and recrystallized from water to afford 2.5 g compound 4 as a white solid in 56% yield with an HPLC purity of 98%. Rf = 0.3 (dichloromethane–methanol = 1:1). UV–vis (MeOH) λmax: 223, 255 nm; ESI-MS m/z: 363 [M + H]+.
4.2.3. Procedure for Preparation of Key Intermediates 1 and 2
Synthesis of 6-amino-4-methoxy-1-(β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (1): Compound 24 (14.5 g, 23.8 mmol) was dissolved in a mixture of methanol (200 mL) and THF (50 mL). Sodium methoxide (3.9 g, 71.4 mmol, 3.0 eq) was then added, and the mixture was stirred at room temperature. Upon completion of the reaction as monitored by TLC, the mixture was neutralized with 2 N HCl solution. Then the reaction mixture was evaporated under reduced pressure, and the resulting residue was purified by column chromatography to afford 6.2 g desired product 1 as a white solid in 87% yield with an HPLC purity of 98.2%. Rf = 0.2 (dichloromethane–methanol = 10:1). UV–vis (MeOH) λmax: 222, 252, 276 nm; 1H NMR (DMSO-d6, 400 MHz) δ 7.90 (s, 1H, Ar-H), 6.86 (s, 2H, NH2), 5.97 (d, J = 4.8 Hz, 1H, 1′-H), 5.32 (d, J = 5.6 Hz, 1H, 2′-OH), 5.05 (d, J = 5.6 Hz, 1H, 3′-OH), 4.74 (t, J = 5.6 Hz, 1H, 5′-OH), 4.46–4.56 (m, 1H, 2′-H), 4.13–4.21 (m, 1H, 3′-H), 3.97 (s, 3H, CH3), 3.82–3.89 (m, 1H, 4′-H), 3.49–3.60 (m, 1H, 5′-H), 3.36–3.47 (m, 1H, 5′-H); ESI-MS m/z: 298.1 [M + H]+, 320.1 [M + Na]+.
Synthesis of 6-amino-4-methoxy-2-(β-d-ribofuranosyl)-2H-pyrazolo[3,4-d]pyrimidine (2): As described for 1, 6-amino-4-methoxy-2-(2,3,5-tri-O-benzoyl-β-d-ribofuranosyl)-2H-pyrazolo[3,4-d]pyrimidine 25 (15 g, 24.6 mmol) was converted to 6.2 g of compound 2 as a white solid in 85% yield with an HPLC purity of 97.2%. Rf = 0.15 (dichloromethane–methanol = 10:1). UV–vis (MeOH) λmax: 222, 266, 296 nm; 1H NMR (DMSO-d6, 400 MHz) δ 8.53 (s, 1H, Ar-H), 6.46 (s, 2H, NH2), 5.75 (d, J = 3.2 Hz, 1H, 1′-H), 4.28–4.34 (m, 1H, 2′-H), 4.15 (t, J = 5.2 Hz, 1H, 3′-H), 3.92–4.01 (m, 4H, 4′-H, CH3), 3.66–3.72 (m, 1H, 5′-H), 3.51–3.57 (m, 1H, 5′-H); ESI-MS m/z: 297.9 [M + H]+. HRMS calcd for C11H16N5O5 298.1151 [M + H]+, found 298.1153.
4.2.4. Procedure for Preparation of Target Compounds 5–17
Synthesis of 4,6-diamino-1-(β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (5): Compound 1 (2.0 g, 6.7 mmol) was dissolved in 25% ammonium hydroxide (40 mL) in high pressure reactor, and the reaction mixture was heated at 80 °C overnight. Upon completion of the reaction as monitored by TLC, the reaction solvent was evaporated under reduced pressure. The resulting precipitate was filtered off and recrystallized from water to afford 900 mg of compound 5. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by column chromatography to afford 520 mg more desired product 5 as a white solid. Totally, 1.42 g desired product was obtained in 75% yield with an HPLC purity of 96.8%. Rf = 0.2 (dichloromethane–methanol = 4:1). UV–vis (MeOH) λmax: 223, 254, 275 nm; 1H NMR (DMSO-d6, 400 MHz) δ 7.97 (s, 1H, Ar-H), 7.38 (br, 2H, NH2), 6.34 (s, 2H, NH2), 5.91 (d, J = 4.4 Hz, 1H, 1′-H), 5.21–5.38 (br, 1H, 2′-OH), 5.02–5.20 (br, 1H, 3′-OH), 4.45–4.55 (m, 1H, 2′-H), 4.08–4.20 (m, 1H, 3′-H), 3.80–3.97 (m, 1H, 4′-H), 3.38–3.58 (m, 2H, CH2); ESI-MS m/z: 283.1 [M + H]+.
Synthesis of 6-amino-4-hydrazino-1-(β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]-pyrimidine (6): Compound 1 (2.0 g, 6.7 mmol) was dissolved in 80% hydrazine hydrate solution (30 mL), and the reaction mixture was heated at 70 °C for 3 h. Upon completion of the reaction as monitored by TLC, the reaction solvent was evaporated under reduced pressure. The resulting precipitate was filtered off and recrystallized from water to afford 850 mg compound 6. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by column chromatography to afford 550 mg desired product 6 as a white solid. Totally, 1.4 g desired product was obtained in 72% yield with an HPLC purity of 96%. Rf = 0.1 (dichloromethane–methanol = 3:1). UV–vis (MeOH) λmax: 225, 275 nm; 1H NMR (DMSO-d6, 400 MHz) δ 8.98, 8.50 (br, 1H, NH), 8.10, 7.86 (br, 1H, NH), 6.23 (br, 1H, NH), 6.01 (br, 1H, NH), 5.93 (d, J = 4.4 Hz, 1H, 1′-H), 5.25 (d, J = 5.6 Hz, 1H, 2′-OH), 5.01 (d, J = 4.0 Hz, 1H, 3′-OH), 4.78–4.96(m, 1H, 5′-OH), 4.55–4.74 (m, 1H, NH), 4.40–4.54 (m, 1H, 2′-H), 4.07–4.24 (m, 1H, 3′-H), 3.76–3.92 (m, 1H, 4′-H), 3.36–3.58 (m, 2H, 5′-H); ESI-MS m/z: 298.1 [M + H]+. HRMS calcd for C10H16N7O4 298.1264 [M + H]+, found 298.1265.
Synthesis of 6-amino-1,5-dihydro-1-(β-d-ribofuranosyl)-4H-pyrazolo[3,4-d]pyrimidin-4-one (7): Compound 1 (2.0 g, 6.7 mmol) was dissolved in 2N NaOH solution (50 mL). The reaction mixture was stirred at room temperature overnight. Upon completion of the reaction as monitored by TLC, the reaction mixture was neutralized with 6 N HCl under cooling condition and then concentrated under reduced pressure. The resulting precipitate was filtered off and recrystallized from water to yielding 1.2 g of compound 7 as a white solid in 62% yield with an HPLC purity of 96.3%. Rf = 0.2 (dichloromethane–methanol = 4:1). UV–vis (MeOH) λmax: 219, 254 nm; 1H NMR (DMSO-d6, 400 MHz) δ 10.61 (s, 1H, Ar-NH), 7.85 (s, 1H, Ar-H), 6.70 (s, 2H, NH2), 5.88 (d, J = 4.4 Hz, 1H, 1′-H), 5.31 (d, 1H, J = 5.6 Hz, 2′-OH), 5.05 (d, J = 4.2 Hz, 1H, 3′-OH), 4.73 (t, J = 5.6 Hz, 1H, 5′-OH), 4.42–4.52 (m, 1H, 2′-H), 4.10–4.20 (m, 1H, 3′-H), 3.80–3.90 (m, 1H, 4′-H), 3.38–3.62 (m, 2H, CH2); 13C NMR (100 MHz, DMSO-d6) δ: 157.7 (C(4)), 155.9 (C(6)), 154.9 (C(2)), 135.4 (C(7)), 99.6 (C(5)), 87.5 (C(1′)), 84.8 (C(4′)), 73.0 (C(2′)), 70.8 (C(3′)), 62.4 (C(5′)); ESI-MS m/z: 284.1 [M + H]+.
Synthesis of 4,6-diamino-3-iodo-1-(β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (8): As described for 5, 6-amino-3-iodo-4-methoxy-1-(β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine 3 (1.3 g, 3.0 mmol) was converted to 860 mg of compound 8 as a white solid in 69% yield with an HPLC purity of 95.3%. Rf = 0.3 (dichloromethane–methanol = 6:1). UV–vis (MeOH) λmax: 229, 263, 278 nm; 1H NMR (DMSO-d6, 400 MHz) δ 6.37 (s, 2H, NH2), 5.85 (d, J = 4.8 Hz, 1H, 1′-H), 5.29 (d, 1H, J = 5.6 Hz, 2′-OH), 5.04 (d, J = 4.2 Hz, 1H, 3′-OH), 4.78 (t, J = 5.6 Hz, 1H, 5′-OH), 4.43–4.54 (m, 1H, 2′-H), 4.06–4.16 (m, 1H, 3′-H), 3.80–3.86 (m, 1H, 4′-H), 3.37–3.60 (m, 2H, CH2); ESI-MS m/z: 409.0 [M + H]+, 431.0 [M + Na]+.
Synthesis of 6-amino-3-iodo-1,5-dihydro-1-(β-d-ribofuranosyl)-4H-pyrazolo[3,4-d]pyrimidin-4-one (9): As described for 7, 6-amino-3-iodo-4-methoxy-1-(β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine 3 (1.7 g, 4.0 mmol) was converted to 1.0 g compound 9 as a white solid in 65% yield with an HPLC purity of 97%. Rf = 0.2 (dichloromethane–methanol = 6:1). UV–vis (MeOH) λmax: 220, 232, 256 nm; ESI-MS m/z: 410.1 [M + H]+.
Synthesis of 6-amino-2,5-dihydro-2-(β-d-ribofuranosyl)-4H-pyrazolo[3,4-d]pyrimidin-4-one (10): As described for 7, 6-amino-4-methoxy-2-(β-d-ribofuranosyl)-2H-pyrazolo[3,4-d]pyrimidine 2 (1.0 g, 3.4 mmol) was converted to 650 mg of compound 10 as a white solid in 67.5% yield with an HPLC purity of 98%. Rf = 0.15 (dichloromethane–methanol = 1:1). UV–vis (MeOH) λmax: 225, 280 nm; 1H NMR (DMSO-d6, 400 MHz) δ 10.45 (s, 1H, Ar-H), 8.52 (s, 1H, NH), 6.28 (s, 2H, NH2), 5.66 (d, J = 3.2 Hz, 1H, 1′-H), 5.49 (d, 1H, J = 5.2 Hz, 2′-OH), 5.11 (d, J = 5.6 Hz, 1H, 3′-OH), 5.01 (t, J = 5.2 Hz, 1H, 5′-OH), 4.25–4.35 (m, 1H, 2′-H), 4.08–4.18 (m, 1H, 3′-H), 3.89–3.96 (m, 1H, 4′-H), 3.61–3.64 (m, 1H, CH), 3.46–3.57 (m, 1H, CH); 13C NMR (100 MHz, DMSO-d6) δ: 160.8 (C(4)), 159.3 (C(6)), 153.7 (C(2)), 128.2 (C(7)), 102.2 (C(5)), 94.0 (C(1′)), 85.1 (C(4′)), 74.7 (C(2′)), 70.0 (C(3′)), 61.3 (C(5′)); ESI-MS m/z: 284.0 [M + H]+.
Synthesis of 4,6-diamino-2-(β-d-ribofuranosyl)-2H-pyrazolo[3,4-d]pyrimidine (11): As described for 5, 6-amino-4-methoxy-2-(β-d-ribofuranosyl)-2H-pyrazolo[3,4-d]pyrimidine 2 (1.0 g, 3.4 mmol) was converted to 670 mg of compound 11 as a white solid in 70% yield with an HPLC purity of 96%. Rf = 0.15 (dichloromethane–methanol = 10:1). UV–vis (MeOH) λmax: 225, 262, 302 nm; 1H NMR (DMSO-d6, 400 MHz) δ 8.42 (s, H, Ar-H), 7.79 (br, 2H, NH2), 6.58 (br, 2H, NH2), 5.73 (s, 1H, 1′-H), 4.90–5.54 (br, 3H, 2′-OH, 3′-OH, 5′-OH), 4.34 (br, 1H, 2′-H), 4.11 (br, 1H, 3′-H), 3.97 (br, 1H, 4′-H), 3.45–3.74 (m, 2H, CH2); ESI-MS m/z: 283.2 [M + H]+.
Synthesis of 6-amino-4-hydrozino-2-(β-d-ribofuranosyl)-2H-pyrazolo[3,4-d]pyrimidine (12): As described for 6, 6-amino-4-methoxy-2-(β-d-ribofuranosyl)-2H-pyrazolo[3,4-d]pyrimidine 2 (1.0 g, 3.4 mmol) was converted to 695 mg of compound 12 as a white solid in 69% yield with an HPLC purity of 96%. Rf = 0.1 (dichloromethane–methanol = 1:2). UV–vis (MeOH) λmax: 223, 264, 298 nm; 1H NMR (DMSO-d6, 400 MHz) δ 8.80, 8.35 (br, 1H, NH), 6.89 (br, H, Ar-H), 6.64 (br, 1H, NH), 5.68–5.84 (m, 1H, 1′-H), 5.38–5.60 (m, 1H, 2′-OH), 4.92–5.24 (m, 2H, 3′-OH, 5′-OH), 4.26–4.43 (m, 1H, 2′-H), 4.04–4.25 (m, 1H, 3′-H), 3.86–4.02 (m, 1H, 4′-H), 3.16–3.70 (m, 5H, CH2, NHNH2), 3.46–3.57 (m, 1H, CH);ESI-MS m/z: 298.8 [M + H]+. HRMS calcd for C10H16N7O4 298.1264 [M + H]+, found 298.1263.
Synthesis of 6-amino-4-hydroxyamino-2-(β-d-ribofuranosyl)-2H-pyrazolo[3,4-d]pyrimidine (13): Compound 2 (1.0 g, 3.4 mmol) was dissolved in 50% hydroxylamine solution (30 mL, 506 mmol), and the reaction mixture was heated at 60 °C. Upon completion of the reaction as monitored by TLC, the reaction solvent was evaporated under reduced pressure. The resulting precipitate was filtered off and recrystallized from water to yielding 506 mg of compound 13 as a white solid in 50% yield with an HPLC purity of 96%. Rf = 0.2 (dichloromethane–methanol = 1:1). UV–vis (MeOH) λmax: 223, 285 nm; 1H NMR (DMSO-d6, 400 MHz) δ 9.97 (s, 1H, NH), 9.86 (s, 1H, OH), 8.16 (s, 1H, Ar-H), 6.81 (br, 2H, NH2), 5.59 (d, J = 3.6 Hz, 1H, 1′-H), 5.46 (d, 1H, J = 5.2 Hz, 2′-OH), 5.14 (d, J = 5.2 Hz, 1H, 3′-OH), 4.94–5.08 (m, 1H, 5′-OH), 4.26–4.33 (m, 1H, 2′-H), 4.07–4.16 (m, 1H, 3′-H), 3.87–3.94 (m, 1H, 4′-H), 3.56–3.64 (m, 1H, CH), 3.45–3.53 (m, 1H, CH); ESI-MS m/z: 298.8 [M + H]+, 320.7 [M + Na]+. HRMS calcd for C10H14N6O5Na 321.0923 [M + Na]+, found 321.0920.
Synthesis of 6-amino-3-(furan-2-yl)-4-methoxy-1-(β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (14): To a stirred mixture of 6-amino-3-iodo-4-methoxy-1-(β-d-ribofuranosyl)-1H- pyrazolo[3,4-d]pyrimidine 3 (600 mg, 1.4 mmol) and bis(triphenylphosphine)-palladium (II) chloride (49 mg, 0.07 mmol, 0.05 eq) in 30 mL of anhydrous DMF was added 2-(tributylstannyl)furan (1.75 g, 4.9 mmol, 3.5 eq) under an nitrogen atmosphere. The reaction mixture was stirred at 90 °C for 18 h and the concentrated to dryness under reduced pressure. The resulting residue was purified by column chromatography to afford 450 mg desired product 14 as a pale-yellow solid in 90% yield with an HPLC purity of 96%. Rf = 0.5 (dichloromethane–methanol = 10:1). 1H NMR (DMSO-d6, 400 MHz) δ 7.77–7.84 (m, 1H, Ar-H), 7.21 (d, J = 3.2 Hz, 1H, Ar-H), 6.96 (s, 2H, NH2), 6.61–6.69 (m, 1H, Ar-H), 6.02 (d, 1H, J = 4.4 Hz, 1′-H), 5.34 (d, 1H, J = 6.0 Hz, 2′-OH), 5.09 (d, J = 4.2 Hz, 1H, 3′-OH), 4.76 (t, J = 5.6 Hz, 1H, 5′-OH), 4.54–4.61 (m, 1H, 2′-H), 4.17–4.26 (m, 1H, 3′-H), 4.05 (s, 1H, CH3), 3.84–3.91 (m, 1H, 4′-H), 3.41–3.64 (m, 2H, CH2); ESI-MS m/z: 364.2 [M + H]+. HRMS calcd for C15H18N5O6 364.1257 [M + H]+, found 364.1258.
Synthesis of 6-amino-4-methoxy-3-phenyl-1-(β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (15): To a stirred mixture of 6-amino-3-iodo-4-methoxy-1-(β-d-ribofuranosyl)-1H-pyrazolo[3,4-d] pyrimidine 3 (800 mg, 1.9 mmol), phenylboronic acid (346 mg, 2.8 mmol, 1.5 eq) and potassium carbonate (387 mg, 2.8 mmol, 1.5 eq) in 60 mL of DME–water (2:1) was added Pd(PPh3)4 (115 mg, 0.09 mmol, 0.05 eq). The reaction mixture was stirred at 90 °C for 4 h. Upon completion of the reaction as monitored by TLC, the mixture was concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column to provided 570 mg of product 15 as a white solid in 80% yield with an HPLC purity of 96%. Rf = 0.6 (dichloromethane–methanol = 10:1). 1H NMR (DMSO-d6, 400 MHz) δ 7.90–8.00 (m, 2H, Ar-H), 7.39-7.54 (m, 3H, Ar-H), 6.92 (s, 2H, NH2), 6.07 (d, 1H, J = 4.0 Hz, 1′-H), 5.36 (d, 1H, J = 5.6 Hz, 2′-OH), 5.08 (d, J = 5.6 Hz, 1H, 3′-OH), 4.76 (t, J = 6.0 Hz, 1H, 5′-OH), 4.54–4.62 (m, 1H, 2′-H), 4.21–4.31 (m, 1H, 3′-H), 3.99 (s, 3H, CH3), 3.85–3.92 (m, 1H, 4′-H), 3.43–3.65 (m, 2H, CH2); ESI-MS m/z: 374.1 [M + H]+. HRMS calcd for C17H20N5O5 374.1464 [M + H]+, found 374.1461.
Synthesis of 6-amino-3-ethynyl-4-methoxy-1-(β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (16): To a stirred mixture of compound 3 (600 mg, 1.42 mmol), Pd (PPh3)4 (169 mg, 0.14 mmol, 0.1 eq), CuI (53 mg, 0.28 mmol, 0.2 eq) and triethylamine (0.6 mL, 4.26 mmol, 3.0 eq) in DMF (20 mL) was added trimethylsilylacetylene (1.2 mL, 8.52 mmol, 6.0 eq). The reaction mixture was stirred at room temperature overnight. Upon completion of the reaction as monitored by TLC, the mixture was concentrated under reduced pressure. The resulting residue was directly deprotected by treatment with K2CO3 (39 mg, 0.3 mmol, 0.2 eq) in MeOH (30 mL) at room temperature for 2 h. Upon completion of the reaction as monitored by TLC, the mixture was concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column to provided 300 mg of product 16 as a white solid in 65% overall yield with an HPLC purity of 96%. Rf = 0.45 (dichloromethane–methanol = 10:1). 1H NMR (DMSO-d6, 400 MHz) δ 7.03 (s, 2H, NH2), 5.95 (d, 1H, J = 4.2 Hz, 1′-H), 5.37 (d, 1H, J = 6.0 Hz, 2′-OH), 5.10 (d, J = 4.2 Hz, 1H, 3′-OH), 4.75 (t, J = 6.0 Hz, 1H, 5′-OH), 4..45–4.55 (m, 2H, 2′-H, CH), 4.10–4.17 (m, 1H, 3′-H), 3.98 (s, 3H, CH3), 3.81–3.90 (m, 1H, 4′-H), 3.37–3.58 (m, 2H, CH2). HRMS calcd for C13H16N5O5 322.1151 [M + H]+, found 322.1150.
Synthesis of 6-amino-4-methoxy-3-[N-(trifluoroacetyl)aminopropyn-1-yl]-1-(β-d-ribofuranosyl)-1H- pyrazolo[3,4-d]pyrimidine (17): The mixture of compound 3 (500 mg, 1.18 mmol), Pd (PPh3)4 (145 mg, 0.12 mmol, 0.1 eq), CuI (44.5 mg, 0.25 mmol, 0.2 eq), N-(2-propynyl)-2,2,2-trifluoroacetamide (1.07 g, 7.08 mmol, 6.0 eq) and triethylamine (0.5 mL, 3.54 mmol, 3.0 eq) in DMF (20 mL) was stirred at room temperature overnight. Upon completion of the reaction as monitored by TLC, the mixture was concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column to provided 310 mg of product 17 as a white solid in 60% yield with an HPLC purity of 95%. Rf = 0.40 (dichloromethane–methanol = 10:1). 1H NMR (DMSO-d6, 400 MHz) δ 10.16 (s, 1H, NH), 7.03 (s, 2H, NH2), 5.95 (d, 1H, J = 4.2 Hz, 1′-H), 5.36 (d, 1H, J = 6.0 Hz, 2′-OH), 5.10 (d, J = 4.2 Hz, 1H, 3′-OH), 4.74 (t, J = 5.6 Hz, 1H, 5′-OH), 4.44–4.55 (m, 1H, 2′-H), 4.07–4.17 (m, 1H, 3′-H), 3.96 (s, 3H, CH3), 3.76–3.88 (m, 1H, 4′-H), 3.37–3.56 (m, 2H, CH2). ESI-MS m/z: 447.1 [M + H]+.
4.2.5. Procedure for Preparation of Glycosylated Products 28–29
Synthesis of 6-amino-4-methoxy-1-(2-deoxy-3,5-di-(O-p-toluoyl)-α-d-ribofuranosyl)-1H-pyrazolo [3,4-d]pyrimidine (28) and 6-amino-4-methoxy-1-(2-deoxy-3,5-di-(O-p-toluoyl)-β-d-ribofuranosyl)-1H- pyrazolo[3,4-d]pyrimidine (29): To a stirred suspension of tris[2-(2-methoxyethoxy)ethyl]amine (TDA-1) (411 mg, 1.3 mmol, 0.03 eq) and finely grounded KOH (6.6 g, 118.7 mmol, 2.8 eq) in anhydrous acetonitrile (200 mL) was added 6-amino-4-methoxy-1H-pyrazolo-[3.4-d]pyrimidine (purine base A, 7.0 g, 42.4 mmol, 1.0 eq). The reaction mixture was stirred for another 15 min. 2-Deoxy-3,5-di-O-(p-toluoyl)-α-D-erythro-pentfuranosyl chloride (ribose II) (24.7 g, 63.6 mmol, 1.5 eq) was added, and the reaction mixture was stirred at room temperature for an additional 30 min. Upon completion of the reaction as monitored by TLC, the reaction mixture was diluted with ethyl acetate (100 mL) and washed with saturated ammonium chloride solution. The aqueous layer was extracted with ethyl acetate. The combined organic layer was washed with saturated brines solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified by column chromatography to afford two products. The compound eluting first was β-isomer 29 (11.4 g, 52%) as a white solid with an HPLC purity of 97.1%. Rf = 0.4 (petroleum ether–ethyl acetate = 3:1). The compound eluting last was α-isomer 28 (2.4 g, 11%) as a white foam with an HPLC purity of 96%. Rf = 0.35 (petroleum ether–ethyl acetate = 3:1).
4.2.6. Procedure for Preparation of Key Intermediates 18 and 19
Synthesis of 6-amino-4-methoxy-1-(2-deoxy-α-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (18): As described for 1, 6-amino-4-methoxy-1-(2-deoxy-3,5-di-(O-p-toluoyl)-α-d-ribofuranosyl) -1H-pyrazolo[3,4-d]pyrimidine 28 (2.4 g, 4.6 mmol) was converted to 1.2 g of compound 18 as a white solid in 89% yield with an HPLC purity of 95%. Rf = 0.35 (dichloromethane–methanol = 10:1). 1H NMR (400 MHz, CDCl3) δ 7.94 (s, 1H, Ar-H), 6.88 (s, 2H, NH2), 6.27–6.45 (m, 1H, 1′-H), 5.60 (d, 1H, J = 8.0 Hz, 2′-OH), 4.72 (t, J = 5.6 Hz, 1H, 5′-OH), 4.10–4.20 (m, 1H, 3′-H), 3.98 (s, 3H, CH3), 3.87–3.95 (m, 1H, 4′-H), 3.35–3.56 (m, 2H, 2′-H) 2.51–2.73 (m, 2H, CH2);
Synthesis of 6-amino-4-methoxy-1-(2-deoxy-β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (19): As described for 1, 6-amino-4-methoxy-1-(2-deoxy-3,5-di-(O-p-toluoyl)-β-d-ribofuranosyl)- 1H-pyrazolo[3,4-d] pyrimidine 29 (4.0 g, 7.7 mmol) was converted to 2.0 g of compound 19 as a white solid in 91% yield with an HPLC purity of 97%. Rf = 0.4 (dichloromethane–methanol = 10:1). UV–vis (MeOH) λmax: 224, 251, 285 nm; 1H NMR (DMSO-d6, 400 MHz) δ 7.88 (s, 1H, Ar-H), 6.84 (s, 2H, NH2), 6.41 (s, 1H, 1′-H), 5.23 (s, 1H, 2′-OH), 4.73 (br, 1H, 5′-OH), 4.39 (br, 1H, 3′-H), 3.97 (s, 3H, CH3), 3.78 (s, 1H, 4′-H), 3.41–3.56 (m, 1H, 2′-H) 2.62–3.18 (m, 2H, 2′-H, CH2), 2.12–2.23 (m, 1H, CH2); ESI-MS m/z: 282.8 [M + H]+.
4.2.7. Procedure for Preparation of Target Compounds 20–23
Synthesis of 6-amino-1,5-dihydro-1-(2-deoxy-β-d-ribofuranosyl)-4H-pyrazolo[3,4-d]pyrimidin-4-one (20): As described for 7, 6-amino-4-methoxy-1-(2-deoxy-β-d-ribofuranosyl)-1H-pyrazolo[3,4-d] pyrimidine 19 (1.2 g, 4.2 mmol) was converted to 990 mg of compound 20 as a white solid in 88% yield with an HPLC purity of 95%. Rf = 0.15 (dichloromethane–methanol = 5:1). UV–vis (MeOH) λmax: 257 nm; ESI-MS m/z: 267.9 [M + H]+, 289.9 [M + Na]+.
Synthesis of 4,6-diamino-1-(2-deoxy-β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (21): As described for 5, 6-amino-4-methoxy-1-(2-deoxy-β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine 19 (1.0 g, 3.5 mmol) was converted to 620 mg of compound 21 as a white solid in 65% yield with an HPLC purity of 96%. Rf = 0.2 (dichloromethane–methanol = 5:1). UV–vis (MeOH) λmax: 225, 257, 278 nm; ESI-MS m/z: 267.7 [M + H]+.
Synthesis of 6-amino-4-hydrazino-1-(2-deoxy-β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (22): As described for 6, 6-amino-4-methoxy-1-(2-deoxy-β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine 19 (1.0 g, 3.5 mmol) was converted to 690 mg of compound 22 as a white solid in 70% yield with an HPLC purity of 96%. Rf = 0.15 (dichloromethane–methanol = 5:1). UV–vis (MeOH) λmax: 226, 276 nm; 1H NMR (DMSO-d6, 400 MHz) δ 9.04, 8.49 (br, 1H, NH), 8.09, 7.80 (br, 1H, NH), 6.36 (s, 1H, Ar-H), 6.05, 6.15 (br, 2H, NH2), 5.17 (d, 1H, J = 4.4 Hz, NH), 4.83 (t, 1H, J = 5.6 Hz, 1′-H), 4.45–4.76 (br, 2H, 3′-OH, 5′-OH), 4.33–4.42 (m, 1H, 3′-H), 3.72–3.81 (m, 1H, 2′-H), 3.45–3.55 (m, 1H, CH), 3.35–3.41 (m, 1H, CH), 2.67–2.76 (m, 1H, 2′-H), 2.07–2.17 (m, 1H, 2′-H); ESI-MS m/z: 282.8 [M + H]+.
Synthesis of 6-amino-4-hydroxyamino-1-(2-deoxy-β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine (23): As described for 13, 6-amino-4-methoxy-1-(2-deoxy-β-d-ribofuranosyl)-1H-pyrazolo[3,4-d]pyrimidine 19 (1.0 g, 3.5 mmol) was converted to 560 mg of compound 23 as a white solid in 56% yield with an HPLC purity of 95%. Rf = 0.2 (dichloromethane–methanol = 4:1). UV–vis (MeOH) λmax: 223, 276 nm; 1H NMR (DMSO-d6, 400 MHz) δ 9.75, 9.53 (br, 1H, NH), 7.50 (s, 1H, Ar-H), 6.68 (br, 1H, NH), 6.25 (br, 1H, NH), 5.18 (d, 1H, J = 4.4 Hz, OH), 4.78 (t, 1H, J = 5.6 Hz, 1′-H), 4.32–4.45 (m, 1H, 3′-H), 3.72–3.85 (m, 1H, 2′-H), 3.35–3.53 (m, 2H, CH2), 2.61–2.75 (m, 1H, 2′-H), 2.07–2.17 (m, 1H, 2′-H); ESI-MS m/z: 283.8 [M + H]+.
4.3. Proliferation Inhibitory Effect Assay
Cells were plated in a 96-well plate (4–5 × 103 cells per well) in the medium supplemented with 10% fetal bovine serum (FBS) and Dulbecco’s modified Eagle medium (DMEM) at 37 °C with a humidified 5% CO2 atmosphere. After 24 h, different concentrations of synthesized compounds (as positive control; 100 μmol/L to 0.01 nmol/L) were added and the cells were exposed to compounds for 72 h. Cell proliferation was evaluated by MTT assay according to the manufacturer’s instructions and incubated for 4 h. The optical density was determined at 570 nm by Varioskan Flash Multimode Reader (Thermo Electron, Finland). The IC50 value, which refers to the concentration (μM) of the compound to cause 50% cell death with respect to the control, was calculated according to the inhibition ratios. The experiments were performed in triplicates to obtain the mean cell viability.
5. Conclusions
The 8-aza-7-deaza-purine bases A, B, and C were glycosylated with l-O-acetyl-2,3,5-tri-O-benzoyl-d-ribofuranose (ribose I) and 2-deoxy-3,5-di-O-(p-toluoyl)-α-D-erythro-pentafuranosyl chloride (ribose II), and further deprotection provided the corresponding key intermediates 1, 2, 3, and 19. These intermediates were further substituted with NH2, OH, NHNH2 and NHOH to furnish final products 5–13 and 20–23. The iodinated intermediate 3 was cross-coupled with aromatic and alkynyl reagents to provide final products 14–17. The newly synthesized compounds were all well characterized. The structures of N9- and N8-glycosylated products were assigned. HMBC analysis of 2D NMR spectra further verified the N9- and N8-products. X-ray crystallographic studies unambiguously confirmed the conclusion. Antitumor inhibitory test resulted in some good hits to guide further studies. The protocols discovered herein can be utilized to expand further research in related fields.
Supplementary Materials
Supplementary materials are available online. Crystal data, HRMS, 1H-NMR, and 13C-NMR spectra of the target products.
Author Contributions
Conceptualization, H.A.; Data curation, H.R.; Formal analysis, J.T.; Funding acquisition, H.A.; Investigation, H.R.; Project administration, H.A.; Supervision, H.A. and J.T.; Writing—original draft, H.R.; Writing—review & editing, H.A.
Funding
Overseas Chinese Affairs Office of the State Council.
Acknowledgments
The authors thank Granlen’s teams for their effort.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cottam, H.B.; Wasson, D.B.; Shih, H.C.; Raychaudhuri, A.; Di Pasquale, G.; Carson, D.A. New adenosine kinase inhibitors with oral anti-inflammatory activity: Synthesis and biological evaluation. J. Med. Chem. 1993, 36, 3424–3430. [Google Scholar] [CrossRef] [PubMed]
- Charles, R.P., III; Howard, B.C.; Patricia, A.M.; Roland, K.R.; Ganapathi, R.R. Synthesis and biological activity of 6-azacadeguomycin and certain 3,4,6-trisubstituted pyrazolo[3,4-d]pyrimidine ribonucleosides. J. Med. Chem. 1985, 28, 1010–1016. [Google Scholar] [CrossRef]
- Michael, A.P.; Marr, J.J. Antileishmanial effect of allopurinol. Antimicrob. Agents Chemother. 1974, 5, 469–472. [Google Scholar] [CrossRef]
- Garaeva, L.D.; Korbukh, I.A.; Dobrynin, Y.V.; Nikolaeva, T.G.; Preobrazhenskaya, M.N. Synthesis and cytotoxic activity of 4,6-diaminopyrazolo[3,4-d] pyrimidine riboside and its 3-carbamoyl derivative. Pharm. Chem. J. 1988, 22, 523–526. [Google Scholar] [CrossRef]
- Junlin, H.; Mikhailopulo, I.; Seela, F. 3-Bromopyrazolo[3,4-d]pyrimidine 2′-Deoxy-2′-fluoro-β-D-arabinonucleosides: Modified DNA constituents with an unusually rigid sugar N-conformation. J. Org. Chem. 2003, 68, 5519–5524. [Google Scholar] [CrossRef]
- Brian, C.K.; Lisa, L.E.; Justin, D.B.; David, O.M.; Kevan, M.S. Inhibitor scaffolds as new allele specific kinase substrates. J. Am. Chem. Soc. 2002, 124, 12118–12128. [Google Scholar] [CrossRef]
- Ilja, V.F.; Maria, I.K.; Konstantin, V.A.; Igor, A.M. Recognition of artificial nucleobases by E. coli purine nucleoside phosphorylase versus its Ser90Ala mutant in the Synthesis of base-modified nucleosides. Chem. Eur. J. 2015, 21, 13401–13419. [Google Scholar] [CrossRef]
- Wenqing, L.; Kuiying, X.; Seela, F. 7-Substituted 8-aza-7-deazapurines and 2,8-diaza-7-deaza-purines: Synthesis of nucleosides and oligonucleotides. Nucleosides Nucleotides Nucleic Acids 2005, 24, 869–873. [Google Scholar] [CrossRef]
- Junlin, H.; Seela, F. Oligonucleotides incorporating 8-aza-7-deazapurines: synthesis and base pairing of nucleosides with nitrogen-8 as a glycosylation position. Org. Biomol. Chem. 2003, 1, 1873–1883. [Google Scholar] [CrossRef]
- Seela, F.; Kuiying, X. Pyrazolo[3,4-d]pyrimidine ribonucleosides related to 2-aminoadenosine and isoguanosine: synthesis, deamination and tautomerism. Org. Biomol. Chem. 2007, 5, 3034–3045. [Google Scholar] [CrossRef] [PubMed]
- Tiannan, H.; Sutera, S.R.; Mumbleaua, M.M.; Beal, P.A. TLR8 activation and inhibition by guanosine analogs in RNA: Importance of functional groups and chain length. Bioorg. Med. Chem. 2018, 26, 77–83. [Google Scholar] [CrossRef]
- Seela, F.; Suresh, S.P. Hydrogelation and spontaneous fiber formation of 8-aza-7-deazaadenine nucleoside ‘click’ conjugates. Tetrahedron 2011, 67, 7418–7425. [Google Scholar] [CrossRef]
- Seela, F.; Pujari, S.S. Azide-alkyne “Click” conjugation of 8-aza-7-deazaadenine-DNA: Synthesis, duplex stability, and fluorogenic dye labeling. Bioconjugate Chem. 2010, 21, 1629–1641. [Google Scholar] [CrossRef] [PubMed]
- Yuxuan, Z.; Beal, P.A. Synthesis and evaluation of an alkyne-modified ATP analog for enzymatic incorporation into RNA. Bioorg. Med. Chem. Lett. 2016, 26, 1799–1802. [Google Scholar] [CrossRef]
- Yang, L.; Zhiwen, L.; Gaofeng, L.; Keliang, L.; Junlin, H. Breaking the conservation of guanine residues in the catalytic loop of 10–23 DNAzyme by position-specific nucleobase modifications for rate enhancement. Chem. Commun. 2013, 49, 5037–5039. [Google Scholar] [CrossRef]
- Seela, F.; Steker, H. Synthesis of 2′-deoxyribofuranosides of 8-aza-7-deazaguanine and related pyrazolo[3,4-d]pyrimidines. Helv. Chim. Acta 1986, 69, 1602–1613. [Google Scholar] [CrossRef]
- Seela, F.; Becher, G. Synthesis of 7-halogenated 8-aza-7-deaza-2′-deoxyguanosines and related pyrazolo[3,4-d]pyrimidine 2′-deoxyribonucleosides. Synthesis 1998, 207–214. [Google Scholar] [CrossRef]
- Bontems, R.J.; Anderson, J.D.; Cottam, H.B. Guanosine analogues. Synthesis ofnucleosides of certain 3-substituted 6-aminopyrazolo[3,4-d]pyrimidin-4(5H)-ones as potential immunotherapeutic agents. J. Med. Chem 1990, 33, 2174–2178. [Google Scholar] [CrossRef] [PubMed]
- Seela, F.; Xiaohua, P. 7-Functionalized 7-deazapurine ribonucleosides related to 2-aminoadenosine, guanosine, and xanthosine: Glycosylation of pyrrolo[2,3-d]pyrimidines with 1-O-acetyl-2,3,5-tri-O-benzoyl-D-ribofuranose. J. Org. Chem. 2006, 71, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Haixin, D.; Yanhui, D.; Ruchun, Y.; Qi, S.; Qiang, X.; Yong, J. Efficient and practical synthesis of 5′-deoxytubercidin and its analogues via vorbrüggen glycosylation. Synthesis 2011, 9, 1442–1446. [Google Scholar] [CrossRef]
- Seela, F.; Becher, G.; Rosemeyer, H.; Reuter, H.; Kastner, G.; Igor, A.M. The high-anti conformation of 7-halogenated 8-aza-7-deaza-2′-deoxyguanosines: A study of the influence of modified bases on the sugar structure of nucleosides. Helv. Chim. Acta 1999, 82, 105–124. [Google Scholar] [CrossRef]
- Becher, G.; He, J.; Seela, F. Major-Groove-Halogenated DNA: The effects of bromo and iodo substituents replacing H-C(7) of 8-aza-7-deazapurine-2,6-diamine or H-C(5) of uracil residues. Helv. Chim. Acta 2001, 84, 1048–1065. [Google Scholar] [CrossRef]
- Yoshinaga, Y.; Kenzo, F. Ultrafast reversible photo-cross-linking reaction: Toward in situ DNA manipulation. Org. Lett. 2008, 10, 3227–3230. [Google Scholar] [CrossRef]
- Hall, L.M.; Gerowska, M.; Brown, T. A highly fluorescent DNA toolkit: Synthesis and properties of oligonucleotides containing new Cy3, Cy5 and Cy3B monomers. Nucleic Acids Res. 2012, 40, e108. [Google Scholar] [CrossRef] [PubMed]
- Ilirian, D.; John, S.J. Efficient preparation of 2-deoxy-3,5-di-O-p-toluoyl-α-D-ribofuranosyl chloride. Synlett 2004, 335–337. [Google Scholar] [CrossRef]
- He, Z.; Yuqi, C.; Yafen, W.; Yuhao, D.; Xiang, Z. A rapidly photo-activatable light-up fluorescent nucleoside and its application in DNA base variation sensing. Chem. Commun. 2016, 52, 8545–8548. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–23 are available from the authors. |
Figure 1.
8-Aza-7-deaza purine ribonucleosides.

Figure 2.
8-Aza-7-deaza-2′-deoxy purine nucleosides.

Scheme 1.
Synthesis of compounds 1–4 and 24–27. Reagents and conditions: (a) 1) purine base A, HMDS, (NH4)2SO4, reflux; 2) TMSOTf, 1,2-dichloroethane, rt, overnight; (b) BF3·Et2O, acetonitrile, reflux, 15 min; (c) purine base B, BF3·Et2O, acetonitrile, rt, 5 h; (d) NaOMe, MeOH, rt; (e) NH3/MeOH, 120 °C, overnight.
Scheme 1.
Synthesis of compounds 1–4 and 24–27. Reagents and conditions: (a) 1) purine base A, HMDS, (NH4)2SO4, reflux; 2) TMSOTf, 1,2-dichloroethane, rt, overnight; (b) BF3·Et2O, acetonitrile, reflux, 15 min; (c) purine base B, BF3·Et2O, acetonitrile, rt, 5 h; (d) NaOMe, MeOH, rt; (e) NH3/MeOH, 120 °C, overnight.

Scheme 2.
Synthesis of compounds 5–9 and 10–13. Reagents and conditions: (a) 25% NH4OH, 80 °C, overnight; (b) 80% NH2NH2, 70 °C, 3 h; (c) 2N KOH, rt, overnight; (d) 50% NH2OH, 60 °C.
Scheme 2.
Synthesis of compounds 5–9 and 10–13. Reagents and conditions: (a) 25% NH4OH, 80 °C, overnight; (b) 80% NH2NH2, 70 °C, 3 h; (c) 2N KOH, rt, overnight; (d) 50% NH2OH, 60 °C.

Figure 3.
UV spectral comparison of N8- and N9-products.

Figure 4.
HMBC spectra of compounds 7 (a) and 10 (b).

Figure 5.
Molecular structure of compound 7 taken from the X-ray analysis. Crystal Data for C10H13N5O5 (M = 283.25 g/mol): orthorhombic, space group P212121 (no. 19), a = 4.7669 (2) Å, b = 11.0409 (5) Å, c = 21.6334 (9) Å, V = 1138.59 (8) Å3, Z = 4, T = 293 (2) K, μ (CuKα) = 1.157 mm−1, Dcalc = 1.652 g/cm3, 4100 reflections measured (8.174° ≤ 2Θ ≤ 134.138°), 2032 unique (Rint = 0.0304, Rsigma = 0.0405) which were used in all calculations. The final R1 was 0.0366 (I > 2σ (I)) and wR2 was 0.0924 (all data).
Figure 5.
Molecular structure of compound 7 taken from the X-ray analysis. Crystal Data for C10H13N5O5 (M = 283.25 g/mol): orthorhombic, space group P212121 (no. 19), a = 4.7669 (2) Å, b = 11.0409 (5) Å, c = 21.6334 (9) Å, V = 1138.59 (8) Å3, Z = 4, T = 293 (2) K, μ (CuKα) = 1.157 mm−1, Dcalc = 1.652 g/cm3, 4100 reflections measured (8.174° ≤ 2Θ ≤ 134.138°), 2032 unique (Rint = 0.0304, Rsigma = 0.0405) which were used in all calculations. The final R1 was 0.0366 (I > 2σ (I)) and wR2 was 0.0924 (all data).

Figure 6.
Molecular structure of compound 8 taken from the X-ray analysis. Crystal Data for C10H15IN6O5 (M = 426.18 g/mol): monoclinic, space group P21 (no. 4), a = 5.08845 (13) Å, b = 12.7514 (2) Å, c = 11.3200 (2) Å, β = 91.4146 (19)°, V = 734.28 (3) Å3, Z = 2, T = 293 (2) K, μ (CuKα) = 17.478 mm−1, Dcalc = 1.928 g/cm3, 5309 reflections measured (7.812° ≤ 2Θ ≤ 134.094°), 2616 unique (Rint = 0.0300, Rsigma = 0.0391) which were used in all calculations. The final R1 was 0.0286 (I > 2σ (I)) and wR2 was 0.0711 (all data).
Figure 6.
Molecular structure of compound 8 taken from the X-ray analysis. Crystal Data for C10H15IN6O5 (M = 426.18 g/mol): monoclinic, space group P21 (no. 4), a = 5.08845 (13) Å, b = 12.7514 (2) Å, c = 11.3200 (2) Å, β = 91.4146 (19)°, V = 734.28 (3) Å3, Z = 2, T = 293 (2) K, μ (CuKα) = 17.478 mm−1, Dcalc = 1.928 g/cm3, 5309 reflections measured (7.812° ≤ 2Θ ≤ 134.094°), 2616 unique (Rint = 0.0300, Rsigma = 0.0391) which were used in all calculations. The final R1 was 0.0286 (I > 2σ (I)) and wR2 was 0.0711 (all data).

Scheme 3.
Synthesis of compounds 14–17. Reagents and conditions: (a) bis(triphenylphosphine) -palladium (II) chloride, 2-(tributylstannyl)furan, DMF, 90 °C, 18 h; (b) phenylboronic acid, K2CO3, Pd(PPh3)4, DME–water (2:1), 90 °C, 4 h; (c) trimethylsilylacetylene, Pd (PPh3)4, CuI, Et3N, DMF, rt, overnight; (d) K2CO3, MeOH, rt, 2 h; (e) Pd (PPh3)4, CuI, N-(2-propynyl)-2,2,2-trifluoroacetamide, Et3N, DMF, rt, overnight.
Scheme 3.
Synthesis of compounds 14–17. Reagents and conditions: (a) bis(triphenylphosphine) -palladium (II) chloride, 2-(tributylstannyl)furan, DMF, 90 °C, 18 h; (b) phenylboronic acid, K2CO3, Pd(PPh3)4, DME–water (2:1), 90 °C, 4 h; (c) trimethylsilylacetylene, Pd (PPh3)4, CuI, Et3N, DMF, rt, overnight; (d) K2CO3, MeOH, rt, 2 h; (e) Pd (PPh3)4, CuI, N-(2-propynyl)-2,2,2-trifluoroacetamide, Et3N, DMF, rt, overnight.

Scheme 4.
Synthesis of 8-aza-7-deaza-2′-deoxy purine ribonucleosides 18–23. Reagents and conditions: (a) TDA-1, KOH, CH3CN, rt; (b) NaOMe, MeOH, rt; (c) 2 N KOH, rt, for 20; 25% NH4OH, 80 °C, for 21; 80% NH2NH2, 70 °C, for 22; 50% NH2OH, 60 °C, for 23.
Scheme 4.
Synthesis of 8-aza-7-deaza-2′-deoxy purine ribonucleosides 18–23. Reagents and conditions: (a) TDA-1, KOH, CH3CN, rt; (b) NaOMe, MeOH, rt; (c) 2 N KOH, rt, for 20; 25% NH4OH, 80 °C, for 21; 80% NH2NH2, 70 °C, for 22; 50% NH2OH, 60 °C, for 23.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
UV absorption maxima of bases and corresponding nucleosides in methanol.
| Compound | λmax/nm (Methanol) |
|---|---|
| Purine base A | 223, 250, 280 |
| Compound 1 | 222, 252, 276 |
| Purine base B | 231, 278 |
| Compound 3 | 232, 278 |
Table 2.
Antitumor activity of newly synthesized compounds.
| Compound | A549 | MDA-MB-231 | ||
|---|---|---|---|---|
| Cytotoxicity IC50 (μM) | 100 μM IC% a | Cytotoxicity IC50 (μM) | 100 μM IC% a | |
| 2 | >100 | 41.42 | >100 | 2.69 |
| 6 | >100 | 17.56 | >100 | 17.81 |
| 8 | 7.68 | 61.44 | >100 | 14.04 |
| 9 | >100 | 43.78 | >100 | 14.14 |
| 10 | >100 | 26.76 | >100 | 10.32 |
| 11 | >100 | 39.18 | >100 | 10.69 |
| 12 | >100 | 21.98 | >100 | 5.55 |
| 13 | >100 | 27.25 | >100 | 11.59 |
| 14 | 100 | 56.18 | >100 | 6.41 |
| 16 | >100 | 39.50 | 81.72 | 54.70 |
| 18 | >100 | 37.18 | >100 | 25.67 |
| 19 | >100 | 27.31 | >100 | 3.04 |
| 22 | >100 | 18.64 | >100 | 7.64 |
| 23 | >100 | 19.63 | >100 | 10.37 |
| DOX | 0.019 | 84.32 | 0.001 | 95.94 |
a Inhibitory concentration percentage in 100 μM testing compounds.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ren, H.; An, H.; Tao, J. Investigation of 8-Aza-7-Deaza Purine Nucleoside Derivatives. Molecules 2019, 24, 983. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050983
AMA Style
Ren H, An H, Tao J. Investigation of 8-Aza-7-Deaza Purine Nucleoside Derivatives. Molecules. 2019; 24(5):983. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050983
Chicago/Turabian StyleRen, Hang, Haoyun An, and Jingchao Tao. 2019. "Investigation of 8-Aza-7-Deaza Purine Nucleoside Derivatives" Molecules 24, no. 5: 983. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050983