3.1. General Information
All melting points were determined with a Stuart Digital Melting Point apparatus SMP10 and are uncorrected. Elemental analyses were performed on a Perkin-Elmer 240 microanalyser, PE 2400 Series II CHNS/O Analyzer, carried out at the regional center for mycology and biotechnology, Al-Azhar University, cairo, Egypt. IR spectra were determined as KBr pellets on a Thermo Nicolet apparatus (Thermo Scientific, Madison, WI, USA) at Postgraduate campus for Girls at Lassan, King Khalid University, Abha, Saudi Arabia. The NMR spectra were recorded on a Bruker NMR spectrometer (Bruker, Billerica, MA, USA) in DMSO-
d6, as solvent at 300, 500 MHz for
1H-NMR and 75, 125 MHz for
13C-NMR at Faculty of Science, Cairo University, Egypt and King Khalid University, Abha, respectively. The chemical shifts (δ) are reported in parts per million (ppm). Mass spectra were measured on GC/MS-QP5 spectrometer at regional center for mycology and biotechnology, Al-Azhar University, Egypt. Antimicrobial activity was measured at the regional center for mycology and biotechnology, Al-Azhar University, Egypt. Follow-up of the reactions and checking the purity of the compounds were carried out using TLC on silica gel-precoated aluminum sheets (Fluorescent indicator 254 nm, Fluka, Germany) and the spots were detected by exposure to UV lamp at λ 254/366 nm for a few seconds or under iodine vapor. Compounds 2-(4-fluorphenylazo)malononitrile
2a and 3,5-diamino-4-(4-fluorophenylazo)-1
H-pyrazole
4b were prepared according to the reported procedure [
21,
22,
23].
3.1.1. General Procedure for the Synthesis of 2-Arylazomalononitrile 2a–c:
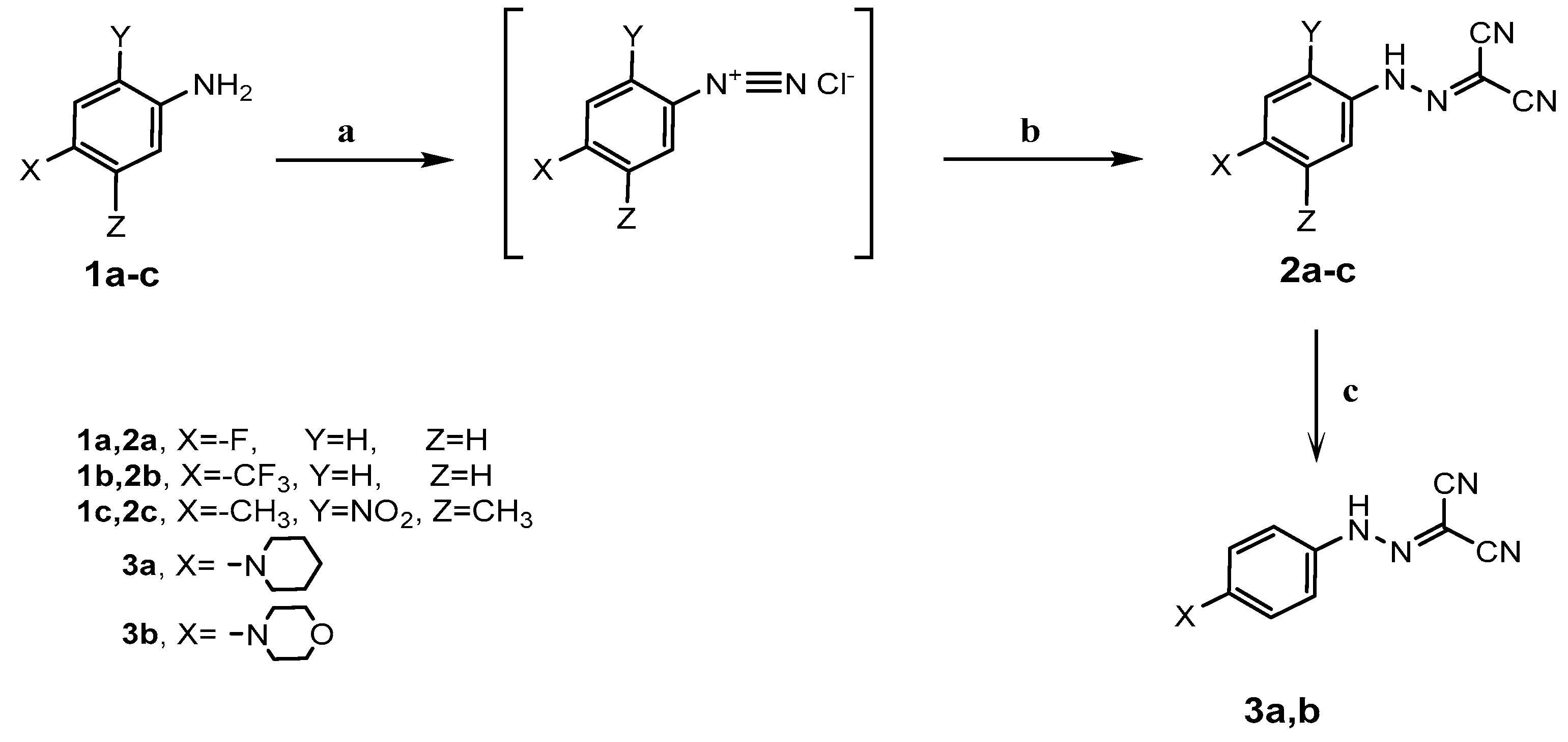
To a solution of aniline derivatives 1a–c (0.01 mol) in hydrochloric acid (6 mL), a solution of sodium nitrite (0.72 g, 0.0105 mol) in water (3 mL) was added portion wise with stirring at 0–5 °C for 1 h. The clear diazonium salt was added to a stirred solution of malononitrile (0.66 g, 0.01 mol) and sodium acetate (4.6 g) in aqueous ethanol 50% (50 mL) with continued stirring at 0–5 °C for 2 h. The reaction mixture was allowed to stand overnight at room temperature, then the precipitate that formed was filtered off, washed several times with water, dried, and recrystallized from ethanol to afford 2-arylazomalononitrile 2a–c.
2-[4-(Trifluoromethyl)phenylazo]malononitrile (2b): Yield: 93%; (orange powder): mp 179–180 °C; IR (KBr) νmax in cm−1: 3141 (NH), 2227 (C≡N), 1618 (C=N) 1H-NMR (500 MHz, DMSO-d6): δ 7.64 (d, 2H, Ar–H, J = 6.9 Hz), 7.76 (d, 2H, Ar–H, J = 7.1 Hz), 10.36 (s, br, 1H, NH); 13C-NMR (125 MHz, DMSO-d6): δ 86.98 (C(CN)2), 109.52 (C≡N), 113.88, 116.69 (Ar–C), 123.03, 125.19 (q, C–F3, J = 270 Hz), 126.72, 127.35, 144.52 (Ar–C); MS (m/z), 239 (M+ + 1; 6%), 238 (M+; 10%), 94 (95%), 67 (100%). Anal. Calcd. for C10H5F3N4 (238.17): C, 50.43; H, 2.12; N, 23.52%. Found C, 50.47; H, 2.34; N, 23.64%.
2-(4,5-Dimethyl-2-nitrophenylazo)malononitrile (2c): Yield: 45%; (yellow powder): mp 160–162 °C; IR (KBr) νmax in cm−1: 3231 (NH), 2989, 2954 (C-H aliphatic), 2215 (C≡N), 1617 (C=N); 1H-NMR (500 MHz, DMSO-d6): δ 2.30, 2.36 (2s, 6H, 2CH3), 7.60 (s, 1H, Ar–H), 7.98 (s, 1H, Ar–H), 9.88 (s, br, 1H, NH); 13C-NMR (125 MHz, DMSO-d6): δ 18.52, 19.69 (CH3), 89.05 (C(CN)2), 109.05 (C≡N), 113.34, 118.97, 125.61, 134.62, 135.11, 146.70 (Ar–C); MS (m/z), 244 (M+ + 1; 11%), 243 (M+; 25%), 61 (100%). Anal. Calcd. for C11H9N5O2 (243.23): C, 54.32; H, 3.73; N, 28.79%. Found C, 54.19; H, 3.59; N, 28.86%.
3.1.2. General Procedures for the Synthesis of 2-[(4-Substituted)arylazo] malononitrile 3a,b:
A mixture of 2-arylazomalononitrile 2a (0.01 mol) and piperidine or morpholine (0.01 mol) in ethanol (30 mL) was refluxed for 1 h and then left to cool to room temperature. The reaction mixture was poured onto cooled water with continuous stirring. The precipitate that formed was filtered, dried, and recrystallized from toluene to afford 2-[(4-substituted)arylazo] malononitrile 3a,b.
2-[4-(Piperidino)phenylazo]malononitrile (3a): Yield: 81%; (yellow plates): mp 197–199 °C; IR (KBr) νmax in cm−1: 3301 (NH), 2942, 2856 (C-H aliphatic), 2184 (C≡N), 1621 (C=N); 1H-NMR (300 MHz, DMSO-d6): δ 1.64 (m, 6H, 3CH2), 3.55 (m, 4H, 2N–CH2), 7.13 (d, 2H, Ar–H, J = 6.9 Hz), 7.50 (m, 3H, 2H Ar–H + NH); 13C-NMR (75 MHz, DMSO-d6): δ 23.61, 25.62 (CH2), 49.31 (N–CH2), 92.58 (C(CN)2), 115.12, 115.41 (C≡N), 116.95, 121.75, 121.86 (Ar–C); MS (m/z), 253 (M+; 7%), 242 (88%), 72 (100%). Anal. Calcd. for C14H15N5 (253.31): C, 66.38; H, 5.97; N, 27.65%. Found C, 66.33; H, 5.80; N, 27.54%.
2-(4-Morpholinophenylazo)malononitrile (3b): Yield: 81%; (yellow crystals): mp 166–167 °C; IR (KBr) νmax in cm−1: 3303 (NH), 2934, 2861 (C–H aliphatic), 2179 (C≡N), 1618 (C=N); 1H-NMR (300 MHz, DMSO-d6): δ 3.60 (t, 4H, 2N–CH2), 3.70 (t, 4H, 2O–CH2), 7.15 (d, 2H, 2 Ar–H, J = 4.8 Hz), 7.51(d, 2H, 2 Ar–H, J = 5.4 Hz), 7.69 (s, 1H, NH); 13C-NMR (75 MHz, DMSO-d6): δ 48.75 (N–CH2), 65.99 (O–CH2), 92.64 (C(CN)2), 115.19, 115.48 (C≡N), 116.87, 121.91, 122.02 (Ar–C); MS (m/z), 255 (M+; 21%), 111 (56%), 43 (100%). Anal. Calcd. for C13H13N5O (255.28): C, 61.17; H, 5.13; N, 27.43%. Found C, 61.28; H, 5.22; N, 27.48%.
3.1.3. General Procedure for the Synthesis of 4-Arylazo-3,5-diaminopyrazole 4a,b
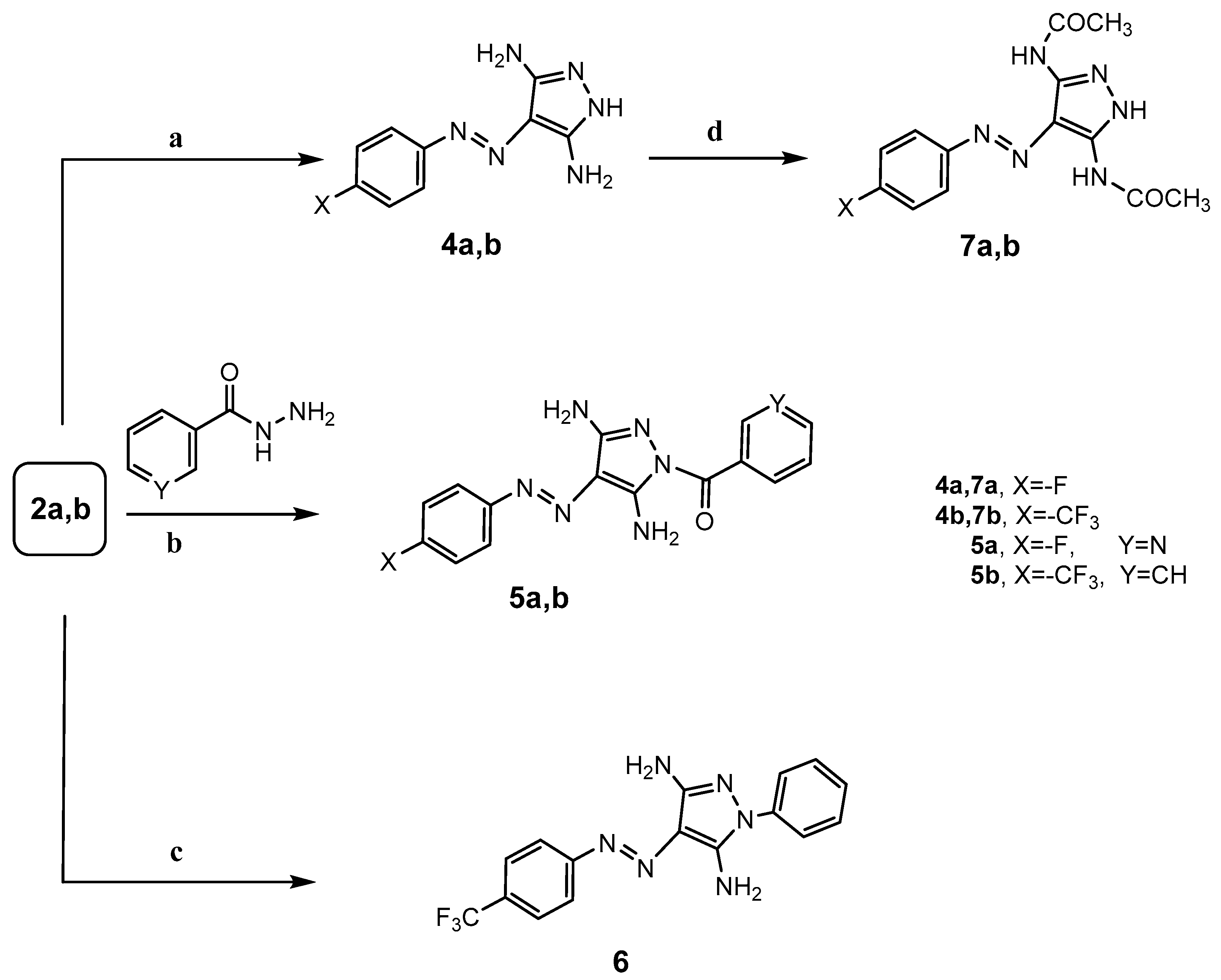
To a solution of 2-arylazomalononitrile 2a,b (0.01 mol) in ethanol (30 mL), hydrazine hydrate (0.01 mol) and pyridine (0.5 mL) were added. The reaction mixture was heated under reflux for 2 h (monitored by TLC). After completion of the reaction, the precipitate that formed was filtered off, dried, and recrystallized from ethanol to afford 3,5-diamino-4-(arylazo)pyrazole 4a,b.
3,5-Diamino-4-[4-(trifluoromethyl)phenylazo]-1H-pyrazole (4b): Yield: 98%; (orange crystals): mp 231–232 °C; IR (KBr) νmax in cm−1: 3465, 3389, 3296 (NH2,NH), 1622 (C=N), 1421 (N=N); 1H-NMR (500 MHz, DMSO-d6): δ 6.29 (br, 4H, 2NH2), 7.71 (d, 2H, Ar–H, J = 8.5 Hz), 7.83 (d, 2H, Ar–H, J = 8.0 Hz), 10.87 (br, 1H, NH); 13C-NMR (125 MHz, DMSO-d6): δ 115.61, 120.62, 120.94 (Ar–C), 123.47, 125.63 (q, C–F3, J = 270 Hz), 125.80, 125.83, 127.79 (Ar–C), 156.37 (C=N); MS (m/z), 271 (M+ + 1; 14%), 270 (M+; 100%). Anal. Calcd. for C10H9F3N6 (270.22): C, 44.45; H, 3.36; N, 31.10%. Found C, 44.39; H, 3.23; N, 31.19%.
3.1.4. General Procedure for the Synthesis of 4-Arylazo-3,5-diamino-N-substitutedpyrazole 5a,b
To a solution of 2-arylazomalononitrile 2a,b (0.01 mol) in ethanol (30 mL), hydrazide derivatives, namely nicotinohydrazide or benzohydrazide (0.01 mol) and pyridine (0.5 mL), were added. The reaction mixture was heated under reflux for 15 h (monitored by TLC). After completion of the reaction, the precipitate that formed was filtered off, dried, and recrystallized from ethanol to afford N-substituted-3,5-diamino-4-(arylazo)pyrazole 5a,b.
{3,5-Diamino-4-[(4-fluorophenyl)azo]-1H-pyrazol-1-yl}(pyridin-3-yl)methanone (5a): Yield: 69%; (orange plates): mp >3 00 °C; IR (KBr) νmax in cm−1: 3477, 3274, 3127 (NH2), 1672 (C=O), 1593 (C=N), 1423 (N=N); 1H-NMR (500 MHz, DMSO-d6): δ 6.92 (s, 2H, NH2), 7.28–7.32 (m, 4H, Ar–H), 7.72–7.75 (m, 2H, Ar–H), 7.95–7.98 (m, 2H, Ar–H), 8.08 (br, s, 2H, NH2); 13C-NMR (125 MHz, DMSO-d6): δ 107.82, 112.32, 120.54, 121.24, 125.82, 125.89, 126.92, 127.16, 128.35, 129.02, 131.04 (Ar–C), 153.15, 157.93 (C=N), 160.52 (C–F), 166.38 (C=O); MS (m/z), 327 (M+ + 2; 3.3%), 325 (M+; 12%), 95 (100%). Anal. Calcd. for C15H12FN7O (325.31): C, 55.38; H, 3.72; N, 30.14%. Found C, 55.28; H, 3.68; N, 30.24%.
{3,5-Diamino-4-[(4-(trifluoromethyl)phenylazo]-1H-pyrazol-1-yl}(phenyl)methanone (5b): Yield: 71%; (orange powder): mp 284–286 °C; IR (KBr) νmax in cm−1: 3506, 3353, (NH2), 1689 (C=O), 1618 (C=N), 1411 (N=N); 1H-NMR (500 MHz, DMSO-d6): δ 7.02 (s, 2H, NH2), 7.67–7.73 (m, 4H, Ar–H), 7.77–7.86 (m, 7H, Ar–H+NH2); 13C-NMR (125 MHz, DMSO-d6): δ 107.46, 121.02, 121.16, 121.65 (Ar–C), 123.63, 125.81 (q, C–F3, J = 272.5 Hz), 126.15, 127.02, 127.68, 149.08 (Ar–C), 150.11 (C=N), 167.31 (C=O); MS (m/z), 375 (M+ + 1; 5%), 374 (M+; 12%), 145 (100%). Anal. Calcd. for C17H13F3N6O (374.33): C, 54.55; H, 3.50; N, 22.45%. Found C, 54.75; H, 3.58; N, 22.35%.
3.1.5. Synthesis of 3,5-Diamino-N-phenyl-4-[4-(trifluoro-methyl)phenylazo]-1H-pyrazole (6)
Phenylhydrazine (0.98 mL, 0.01 mol) and pyridine (0.5 mL) were added to a solution of 2-arylazomalononitrile 2b (2.7 g, 0.01 mol) in ethanol (30 mL). The reaction mixture was heated under reflux for 3 h (monitored by TLC). After completion of the reaction, the precipitate that formed was filtered off, dried, and recrystallized from ethanol to give 3,5-diamino-1-phenyl-4-[4-(trifluoro-methyl)phenylazo]-1H-pyrazole 6. Yield: 71%; (yellow crystals): mp >300 °C; IR (KBr) νmax in cm−1: 3400, 3304, 3197 (NH2, NH), 1616 (C=N), 1423 (N=N); 1H-NMR (500 MHz, DMSO-d6): δ 6.99 (br, s, 2H, NH2), 7.33 (d, 2H, Ar–H, J = 7.5 Hz), 7.50 (d, 2H, Ar–H, J = 8.0 Hz), 7.53–7.76 (m, 5H, Ar–H), 7.95 (br, s, 2H, NH2); 13C-NMR (125 MHz, DMSO-d6): δ 101.00, 121.07, 121.26, 122.26 (Ar–C), 123.42, 125.59, 125.95 (q, C–F3, J = 270 Hz), 126.16, 126.41, 129.27, 149.07 (Ar–C), 156.07 (C=N). Anal. Calcd. for C16H13F3N6 (346.32): C, 55.49; H, 3.78; N, 24.27%. Found C, 55.68; H, 3.91; N, 24.22%.
3.1.6. Synthesis of 4-Arylazo-3,5-diacetamido-1H-pyrazole 7a,b
A solution of 3,5-diamine-4-arylazo-1H-pyrazole 4a,b (0.01 mol) in acetic acid (30 mL) was heated under reflux for 10 h (monitored by TLC). The reaction mixture was left to cool to room temperature, and then poured onto cooled water (50 mL) portion wise with continuous stirring for 1 h. The separated yellow compound was filtered off, washed with water, and, finally, dried and recrystallized from ethanol to afford 3,5-diacetamio-4-arylazo-1H-pyrazole 7a,b.
3,5-Diacetamido-4-[(4-fluorophenyl)azo]-1H-pyrazole (7a): Yield: 86%; (yellow powder): mp 234–236 °C; IR (KBr) νmax in cm−1: 3277, 3144 (NH), 1698 (C=O), 1605 (C=N), 1418 (N=N); 1H-NMR (300 MHz, DMSO-d6): δ 2.12, 2.17 (2s, 6H, 2CH3), 6.56 (br, 1H, NH), 7.77–7.92 (m, 5H, Ar–H+NH), 10.27 (br, 1H, NH); 13C-NMR (75 MHz, DMSO-d6): δ 23.12 (CH3), 115.82, 116.08, 120.56, 122.74, 122.85, 123.68, 123.80 (Ar–C), 149.12 (C=N), 161.04 (C–F), 169.24 (2C=O): MS (m/z), 305 (M+ + 1; 28%), 304 (M+; 100%), 42 (76%). Anal. Calcd. for C13H13FN6O2 (304.29): C, 51.31; H, 4.31; N, 27.62%. Found C, 51.38; H, 4.41; N, 27.53%.
3,5-Diacetamido-4-[4-(trifluoromethyl)phenylazo]-1H-pyrazole (7b): Yield: 80%; (Yellow plates): mp 236–238 °C; IR (KBr) νmax in cm−1: 3238 (NH), 1678 (C=O), 1595 (C=N), 1426 (N=N); 1H-NMR (300 MHz, DMSO-d6): δ 2.24 (s, 6H, 2CH3), 7.82–7.96 (m, 5H, Ar–H+NH), 8.40, 10.11 (2br, s, 2H, 2NH); 13C-NMR (125 MHz, DMSO-d6): δ 23.38 (CH3), 100.33, (Ar–C), 123.10, 125.26 (q, C–F3, J = 270 Hz), 126.07, 126.24, 126.26, 128.75, 129.00 (Ar–C), 155.61 (C=N), 167.33 (C=O): MS (m/z), 354 (M+; 19%), 312 (72%), 70 (100%). Anal. Calcd. for C14H13F3N6O2 (354.29): C, 47.46; H, 3.70; N, 23.72%. Found C, 47.57; H, 3.79; N, 23.76%.
3.1.7. General Procedure for the Synthesis of 3,6-Diarylazo-2,5,7-triaminopyrazolo[1,5-a]pyrimidine derivatives 8a,b
A. Under reflux Condition:
Method A: A mixture of 2-arylazomalononitrile 2a,b (0.02 mol) and hydrazine hydrate (0.01 mol) in ethanol (30 mL) in the presence of pyridine (0.5 mL) was heated under reflux for 5 h, (monitored by TLC). After completion of the reaction, the precipitates those formed were filtered off, dried, and recrystallized from 1,4-dioxane to afford 8a,b.
Method B: A mixture of 4-arylazo-3,5-diaminopyrazole 4a,b (0.01 mol) and 2-arylazomalononitrile 2a,b (0.01 mol) in ethanol (30 mL) and in the presence of pyridine (0.5 mL) was heated under reflux for 4–5 h, (monitored by TLC). After completion of the reaction, the precipitates those formed were filtered off, dried, and recrystallized from 1,4-dioxane to afford 8a and 8b, respectively.
B. Under Microwave Irradiation:
A mixture of 5-diamino-4-arylazopyrazole 4a,b (0.01 mol) and 2-arylazomalononitrile 2a,b (0.01 mol) was grinded carefully in a porcelain mortar using a pestle and transferred to a pyrex test tube; then, ethanol (4 mL) was added, followed by pyridine (0.5 mL). The reaction mixture was heated under microwave irradiation at 50% power for 2 min at 140 °C, monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and the precipitated solid was filtered off, washed with MeOH, and recrystallized from dioxane to afford 8a and 8b, respectively.
C-Under Ultrasound Condition:
To a solution of 4-arylazo-3,5-diaminopyrazole 4a,b (0.01 mol) in ethanol (30 mL), 2-arylazomalononitrile 2a,b (0.01 mol) and pyridine (0.5 mL) was added. The reaction mixture was sonicated for 1 h at room temperature, (monitored by TLC). After completion of the reaction, the precipitate product was filtered off, dried, and recrystallized from dioxane to afford 8a and 8b respectively.
3,6-Bis[(4-fluorophenyl)azo]-2,5,7-triaminopyrazolo[1,5-a]pyrimidine (8a): Yield: 74%; (orange plates): mp >300 °C; IR (KBr) νmax in cm−1: 3422, 3274 (NH2), 1615 (C=N), 1420 (N=N); 1H-NMR (500 MHz, DMSO-d6): δ 6.92 (s, 2H, NH2), 7.29–7.32 (m, 4H, Ar–H), 7.72–7.75 (m, 4H, Ar–H), 7.96 (s, br, 2H, NH2), 8.10 (s, 2H, NH2); 13C-NMR (125 MHz, DMSO-d6): δ 107.81, 115.11, 115.66, 115.70, 115.84, 115.86, 122.34, 122.40, 123.72, 123.79, 147.22, 149.08, 149.10 (Ar–C), 150.05, 152.31 (C=N), 160.52, 161.18 (C–F); MS (m/z), 409 (M+ + 1; 30%), 408 (M+; 100%), 363 (54%), Anal. Calcd. for C18H14F2N10 (408.38): C, 52.94; H, 3.46; N, 34.30%. Found C, 52.83; H, 3.52; N, 34.22%.
3,6-Bis[4-(trifluoromethyl)phenylazo]-2,5,7-triaminopyrazolo[1,5-a]pyrimidine (8b): Yield: 71%; (orange crystals): mp >300 °C; IR (KBr) νmax in cm−1: 3460, 3259, 3103 (NH2), 1602 (C=N), 1417 (N=N); 1H-NMR (500 MHz, DMSO-d6): δ 7.06 (s, 2H, NH2; exchangeable with D2O), 7.77–7.86 (m, 10H, Ar–H+NH2; exchangeable with D2O), 8.19 (s, 2H, NH2; exchangeable with D2O); 13C-NMR (125 MHz, DMSO-d6): δ 108.98, 116.57, 121.82, 122.25 (Ar–C), 123.19, 125.35 (q, C–F3, J = 287.5 Hz), 125.49, 125.99, 126.02, 126.17, 126.20, 126.83, 127.08, 127.86, 128.11, 148.05 (Ar–C), 152.38, 154.71 (C=N); MS (m/z), 509 (M+ + 1; 25%), 508 (M+; 100%). Anal. Calcd. for C20H14F6N10 (508.39): C, 47.25; H, 2.78; F, 22.42; N, 27.55%. Found C, 47.34; H, 2.71; N, 27.44%.
3.1.8. General Procedure for the Synthesis of 3,6-Diarylazo-2,5,7-triaminopyrazolo[1,5-a]pyrimidine Derivatives 9a,b
A. Under Reflux Condition:
To a solution of 4-arylazo-3,5-diaminopyrazole 4a,b (0.01 mol) in ethanol (30 mL), 2-arylazomalononitrile 2a,b (0.01 mol) and pyridine (0.5 mL) were added. The reaction mixture was heated under reflux for 6–7 h (monitored by TLC). After completion of the reaction, the formed precipitates were filtered off, dried, and recrystallized from ethanol to afford 9a,b.
B. Under Microwave Irradiation:
A mixture of 5-diamino-4-(arylazo)pyrazole 4a,b (0.01 mol) and 2-arylazomalononitrile 2a,b (0.01 mol) was grinded carefully in a porcelain mortar using a pestle and transferred to a pyrex test tube, and ethanol (4 mL) was added, followed by pyridine (0.5 mL). The reaction mixture was heated under microwave irradiation at 50% power for 2 min at 140 °C, monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and the precipitated solid was filtered off, washed with MeOH, and recrystallized from ethanol to afford 9a,b.
C-Under Ultrasound Condition:
To 5-diamino-4-(arylazo)pyrazole 4a,b (0.01 mol) in ethanol (30 mL), 2-arylazomalononitrile 2a,b (0.01 mol) and pyridine (0.5 mL) was added. The reaction mixture was sonicated for 1 h at room temperature (monitored by TLC). After completion of the reaction, the precipitate was filtered off, dried, and recrystallized from ethanol to afford 9a,b.
6-[(4-Trifluoromethyl)phenylazo]-3-[(4-fluorophenyl)azo]-2,5,7-triaminopyrazolo [1,5-a]pyrimidine (9a): Yield: 76%; (orange plates): mp >300 °C; IR (KBr) νmax in cm−1: 3474, 3276, 3123 (NH2), 1616 (C=N), 1423 (N=N); 1H-NMR (500 MHz, DMSO-d6): δ 6.98 (s, 2H, NH2), 7.31–7.35 (m, 4H, Ar–H), 7.76–7.82 (m, 6H, Ar–H+NH2), 8.22 (s, 2H, NH2); MS (m/z), 408 (45%), 458 (M+; 100%), 459 (M+ + 1; 22%). Anal. Calcd. for C19H14F4N10 (458.13): C, 49.79; H, 3.08; N, 30.56. Found C, 49.65; H, 3.19; N, 30.45%.
3-[(4-Trifluoromethyl)phenylazo]-6-[(4-fluorophenyl)azo]-2,5,7-triaminopyrazolo [1,5-a]pyrimidine (9b): Yield: 78%; (orange plates): mp >300 °C; IR (KBr) νmax in cm−1: 3468, 3266, 3124 (NH2), 1594 (C=N), 1419 (N=N); 1H-NMR (500 MHz, DMSO-d6): δ 7.06 (s, 2H, NH2), 7.28–7.32 (m, 4H, Ar–H), 7.80 (d, 2H, Ar–H, J = 7.5 Hz), 8.10 (s, br, 2H, NH2), 8.13 (s, 2H, NH2), 7.84 (d, 2H, Ar–H, J = 7.5 Hz); 13C-NMR (125 MHz, DMSO-d6): δ 108.98, 116.48, 121.82, 122.25 (Ar–C), 123.19, 125.35 (q, C–F3, J = 270 Hz), 125.52, 125.99, 126.02, 126.17, 126.20, 126.90, 127.68, 128.11, 148.05 (Ar–C), 152.83, 154.90 (C=N), 161.27 (C–F). Anal. Calcd. for C19H14F4N10 (458.13): C, 49.79; H, 3.08; N, 30.56%. Found C, 49.50; H, 3.21; N, 30.46%.
3.1.9. General Procedure for the Synthesis of 3,6-Diarylazo-2,5,7-triaminopyrazolo[1,5-a] pyrimidine Derivatives 10a,b
A. Under reflux Condition:
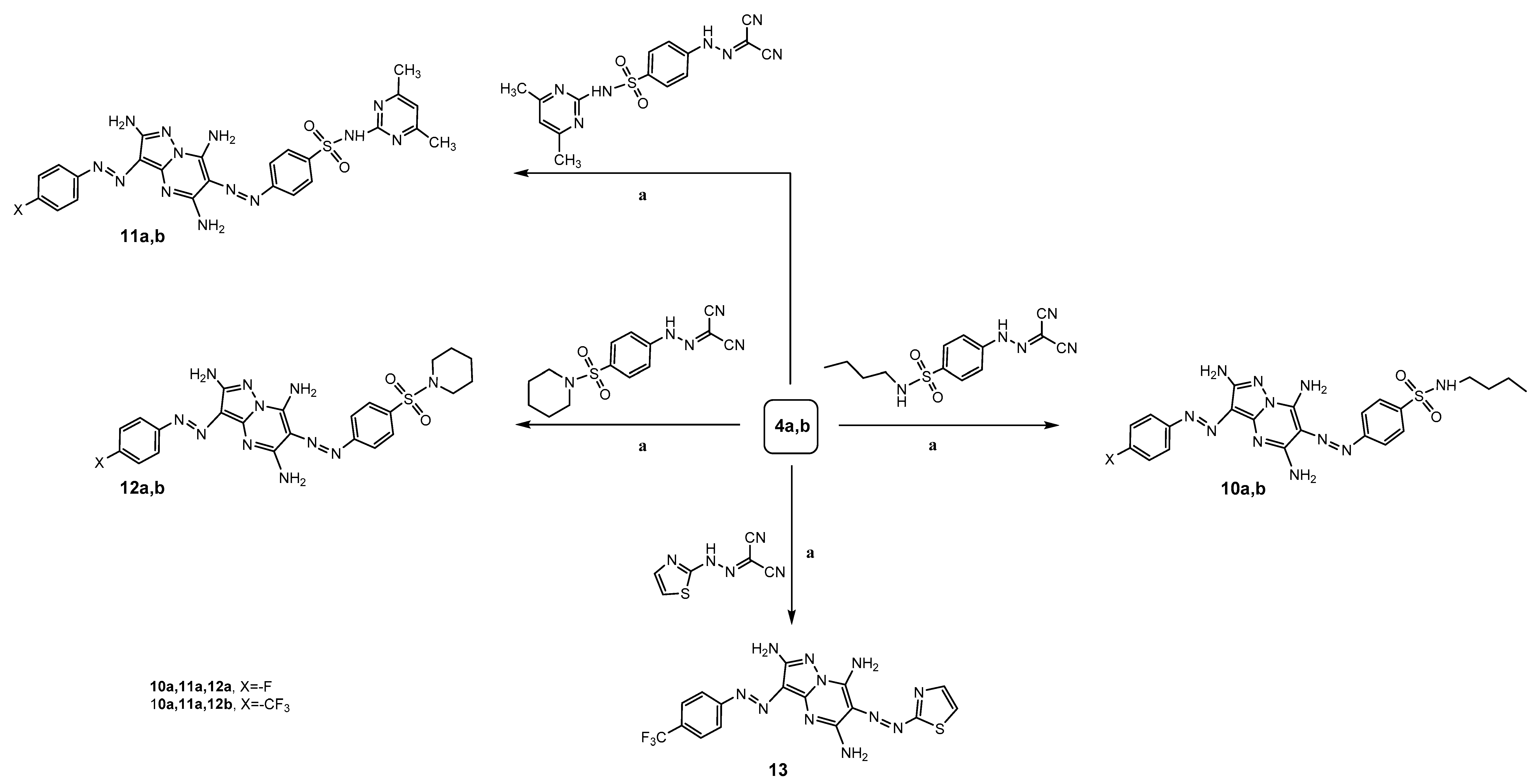
To a solution of 4-arylazo-3,5-diaminopyrazole 4a,b (0.01 mol) in ethanol (30 mL), N-[4-(N-butylsulfamoyl)phenyl]carbonohydrazonoyl dicyanide (3.05 g, 0.01 mol) and pyridine (0.5 mL) was added. The reaction mixture was heated under reflux for 8–9 h (monitored by TLC). After completion of the reaction, the precipitated product that formed was filtered off, dried, and recrystallized from toluene to afford 3-arylazo-2,5,7-triaminopyrazolo[1,5-a] pyrimidine 10a,b.
B. Under Microwave Irradiation:
A mixture of 5-diamino-4-(arylazo)pyrazole 4a,b (0.01 mol) and N-[4-(N-butylsulfamoyl)phenyl]carbonohydrazonoyl dicyanide (3.05 g, 0.01 mol) was grinded carefully in a porcelain mortar using a pestle and transferred to a pyrex test tube, and ethanol (4 mL) was added, followed by pyridine (0.5 mL). The reaction mixture was heated under microwave irradiation at 50% power for 5 min at 140 °C, monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and the precipitated solid was filtered off, washed with MeOH, and recrystallized from ethanol to afford 10a,b.
N-Butyl-4-{[2,5,7-triamino-3-(4-fluorophenylazo)pyrazolo[1,5-a]pyrimidin-6-yl]azo}benzenesulfonamide (10a): Yield: 76%; (orange plates): mp >300 °C; IR (KBr) νmax in cm−1 3474, 3279 (NH2), 2932, 2870 (C–H aliphatic), 1611 (C=N), 1420 (N=N), 1369 (SO2); 1H-NMR (500 MHz, DMSO-d6): δ 0.80 (t, 3H, CH3), 1.22–1.27 (m, 2H, CH2), 1.34–1.37 (m, 2H, CH2), 2.75–2.77 (t, 2H, CH2), 6.94 (s, 2H, NH2), 7.29–7.33 (m, 4H, Ar–H), 7.60 (br, s, 1H, NH), 7.74–7.76 (m, 4H, Ar–H), 7.83 (s, 2H, NH2), 8.18 (s, 2H, NH2); 13C-NMR (125 MHz, DMSO-d6): δ 13.43 (CH3), 19.19, 31.02 (CH2), 42.20 (N–CH2), 108.95, 115.28, 115.73, 115.91, 116.65, 120.81, 122.06, 122.43, 122.50, 123.91, 127.44, 127.68, 139.29, 147.51, 150.01 (Ar–C), 152.58, 154.48 (C=N), 160.63 (C–F); MS (m/z), 525 (M+; 11%), 526 (M+ + 1; 4%), 403 (100%). Anal. Calcd. for C22H24FN11O2S (525.57): C, 50.28; H, 4.60; N, 29.32; S, 6.10%. Found C, 50.39; H, 4.78; N, 29.39; S, 6.01%.
N-Butyl-4-{[2,5,7-triamino-3-((4-(trifluoromethyl)phenyl)azo)pyrazolo[1,5-a]pyrimidin-6-yl]azo}benzene-sulfonamide (10b): Yield: 84%; (reddish brown powder): mp 289–291 °C; IR (KBr) νmax in cm−1: 3446, 3315 (NH2) 2960, 2870 (C-H aliphatic), 1611 (C=N), 1412 (N=N), 1366 (SO2); 1H-NMR (500 MHz, DMSO-d6): δ 0.79 (t, 3H, CH3), 1.23–1.26 (m, 2H, CH2), 1.34–1.37 (m, 2H, CH2), 2.76–2.77 (t, 2H, CH2), 7.07 (s, 2H, NH2), 7.61 (br, s, 1H, NH), 7.82–7.89 (m, 8H, Ar–H), 8.21 (br, s, 4H, 2NH2); 13C-NMR (125 MHz, DMSO-d6): δ 13.40 (CH3), 19.19, 31.01 (2CH2), 42.19 (N–CH2), 109.06, 116.58, 120.72, 120.89, 121.03 (Ar–C), 123.32, 125.48 (q, C–F3, J = 270 Hz), 126.22, 126.85, 127.10, 127.45, 127.69, 139.47, 148.04 (Ar–C), 152.40, 154.41 (C=N); MS (m/z), 576 (M+ + 1; 3%), 575 (M+; 9%), 398 (100%). Anal. Calcd. for C23H24F3N11O2S (575.58): C, 48.00; H, 4.20; N, 26.77%. Found C, 48.24; H, 4.14; N, 26.86%.
3.1.10. General Procedure for the Synthesis of 3,6-Diarylazo-2,5,7-triaminopyrazolo[1,5-a] pyrimidine Derivatives 11a,b
A. Under Reflux Condition:
To a solution of 5-diamino-4-(arylazo)pyrazole 4a,b (0.01 mol) in ethanol (30 mL), N-{4-[N-(4,6-dimethylpyrimidin-2-yl)sulfamoyl]phenyl} carbonohydrazonoyl dicyanide (3.55 g, 0.01 mol) and pyridine (0.5 mL) were added. The reaction mixture was heated under reflux for 6–7 h, (monitored by TLC). After completion of the reaction, the precipitated product that formed was filtered off, dried, and recrystallized from toluene to afford 3-arylazo-2,5,7-triamino pyrazolo[1,5-a]pyrimidine 11a,b.
B. Under Microwave Irradiation:
A mixture of 5-diamino-4-(arylazo)pyrazole 4a,b (0.01 mol) and N-{4-[N-(4,6-dimethylpyrimidin-2-yl)sulfamoyl]phenyl}carbonohydrazonoyl dicyanide (3.55 g, 0.01 mol) was grinded carefully in a porcelain mortar using a pestle and transferred to a pyrex test tube, and ethanol (4 mL) was added, followed by pyridine (0.5 mL). The reaction mixture was heated under microwave irradiation at power 50% for 3 min at 140 °C, monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and the precipitated solid was filtered off, washed with MeOH, and recrystallized from ethanol to afford 11a,b.
N-(4,6-Dimethylpyrimidin-2-yl)-4-{[2,5,7-triamino-3-(4-fluorophenylazo)pyrazolo [1,5-a]pyrimidin-6-yl]azo}benzenesulfonamide (11a): Yield: 92%; (reddish-brown powder): mp > 300 °C; IR (KBr) νmax in cm−1: 3403, 3307 (NH2), 2959 (C–H aliphatic), 1616 (C=N), 1421 (N=N), 1347 (SO2); 1H-NMR (500 MHz, DMSO-d6): δ 2.26 (s, 6H, 2CH3), 6.75 (s, 1H, CH–pyrimidine), 6.94 (s, 2H, NH2), 7.20–7.32 (m, 5H, Ar–H+NH), 7.73–7.76 (m, 4H, Ar–H), 8.03, 8.13 (2br, s, 4H, 2NH2); MS (m/z), 575 (M+; 41%), 408 (100%). Anal. Calcd. for C24H22FN13O2S (575.59): C, 50.08; H, 3.85; N, 31.64. Found C, 50.27; H, 3.97; N, 31.51%.
N-(4,6-Dimethylpyrimidin-2-yl)-4-{[2,5,7-triamino-3-(4-(trifluoromethyl) phenylazo)pyrazolo[1,5-a]pyrimidin-6-yl]azo}benzenesulfonamide (11b): Yield: 78%; (reddish orange powder): mp >300 °C; IR (KBr) νmax in cm−1: 3418, 3324 (NH2), 1600 (C=N), 1418 (N=N), 1355 (SO2); 1H-NMR (500 MHz, DMSO-d6): δ 2.26 (s, 6H, 2CH3), 6.75 (s, 1H, CH–pyrimidine), 7.07 (s, 2H, NH2), 7.80 (d, 2H, Ar–H, J = 8.5 Hz), 7.85 (d, 2H, Ar–H, J = 8.5 Hz), 8.04 (d, 2H, Ar–H, J = 7.5 Hz), 8.14 (d, 2H, ArvH, J = 7.5 Hz), 8.24 (br, 4H, 2NH2), 9.5 (br, 1H, NH); 13C-NMR (125 MHz, DMSO-d6): δ 22.73 (CH3), 109.08, 113.25, 116.52, 120.70, 121.03, 121.36, 122.26 (Ar–C), 123.32, 125.48 (q, C–F3, J = 270 Hz), 126.20, 126.22, 126.86, 127.11, 127.64, 128.98, 139.73, 148.03 (Ar–C), 152.40, 154.50, 155.80, 156.08 (C=N). Anal. Calcd. C25H22F3N13O2S (625.60): C, 48.00; H, 3.54; N, 29.11. Found C, 48.18; H, 3.46; N, 29.08%.
3.1.11. General Procedure for the Synthesis of 3,6-Diarylazo-2,5,7-triaminopyrazolo[1,5-a]pyrimidine Derivatives 12a,b
A. Under Reflux Condition:
N-[4-(Piperidin-1-ylsulfonyl)phenyl]carbonhydrazonoyl dicyanide (3.17 g, 0.01 mol) and pyridine (0.5 mL) were added to a solution of 4-arylazo-3,5-diaminopyrazole 4a,b (0.01 mol) in ethanol (30 mL). The reaction mixture was heated under reflux for 7 h (monitored by TLC). After completion of the reaction, the precipitates which formed were filtered off, dried, and recrystallized from ethanol to afford 3-arylazo-2,5,7-triaminopyrazolo[1,5-a]pyrimidine 12a,b.
B. Under Microwave Irradiation:
A mixture of 4-arylazo-3,5-diaminopyrazole 4a,b (0.01 mol) and N-[4-(piperidin-1-ylsulfonyl)phenyl]carbonhydrazonoyl dicyanide (3.17 g, 0.01 mol) was grinded carefully in a porcelain mortar using a pestle and transferred to a pyrex test tube, and ethanol (4 mL) was added, followed by pyridine (0.5 mL). The reaction mixture was heated under microwave irradiation at power 50% for 3–7 min at 140 °C (monitored by TLC). After completion of the reaction, the reaction mixture was cooled to room temperature and the precipitated solid was filtered off, washed with MeOH, and recrystallized from ethanol to afford 12a,b.
3-[(4-Fluorophenyl)azo]-6-[4-(piperidin-1-ylsulfonyl)phenylazo]-2,5,7-triamino pyrazolo[1,5-a]pyrimidine (12a): Yield: 80%; (orange plates): mp >300 °C; IR (KBr) νmax in cm−1: 3404, 3266 (NH2), 2940, 2835 (C-H aliphatic), 1611 (C=N), 1420 (N=N), 1371 (SO2); 1H-NMR (500 MHz, DMSO-d6): δ 1.36–1.37 (m, 2H, CH2), 1.54–1.55 (m, 4H, 2CH2), 2.91–2.93 (m, 4H, 2NCH2), 6.95 (s, 2H, NH2), 7.29–7.33 (m, 4H, Ar–H), 7.74–7.77 (m, 6H, Ar–H+NH2), 8.21 (br, s, 2H, NH2); 13C-NMR (125 MHz, DMSO-d6): δ 22.85 (CH2), 24.67 (CH2), 46.58 (N–CH2), 109.17, 115.30, 115.74, 115.92, 120.88, 122.14, 122.45, 122.51, 128.38, 128.67, 134.07, 147.54, 149.99 (Ar–C), 152.62, 154.97 (C=N), 160.64 (C–F). Anal. Calcd. for C23H24FN11O2S (537.58): C, 51.39; H, 4.50; N, 28.66. Found C, 51.32; H, 4.73; N, 28.49%.
6-[(4-Piperidin-1-ylsulfonyl)phenylazo]-3-[4-(trifluoromethyl)phenylazo]-2,5,7-triaminopyrazolo[1,5-a]pyrimidine (12b): Yield: 81%; (reddish brown crystals): mp > 300 °C; IR (KBr) νmax in cm−1: 3439, 3273 (NH2), 2941, 2855 (C-H aliphatic), 1607 (C=N), 1409 (N=N), 1361 (SO2); 1H-NMR (500 MHz, DMSO-d6): δ 1.36 (m, 2H, CH2), 1.55 (m, 4H, 2CH2), 2.91 (m, 4H, 2N–CH2), 7.07 (s, 2H, NH2), 7.76–7.87 (m, 10H, Ar–H+NH2), 8.23 (br, s, 2H, NH2); 13C-NMR (125 MHz, DMSO-d6): δ 22.85 (CH2), 24.66 (CH2), 46.58 (N–CH2), 109.27, 116.59, 120.62, 120.97, 121.06, 122.24 (Ar–C), 123.33, 125.50, (q, C–F3, J = 271.2 Hz), 126.22, 126.25, 126.87, 127.13, 128.38, 128.66, 134.28, 148.10 (Ar–C), 152.43, 154.90 (C=N); MS (m/z), 588 (M+ + 1; 21%), 587 (M+; 44%), 442 (100%), 54 (99%). Anal. Calcd. for C24H24F3N11O2S (587.59): C, 49.06; H, 4.12; N, 26.22. Found C, 49.21; H, 4.32; N, 26.41%.
3.1.12. Synthesis of 6-(Thiazol-2-yldiazenyl)-3-(4-(trifluoro methyl)phenylazo)-2,5,7-triaminopyrazolo[1,5-a] pyrimidine (13)
A. Under reflux Condition:
N-(Thiazol-2-yl)carbonohydrazonoyl dicyanide (1.77 g, 0.01 mol) and pyridine (0.5 mL) were added to a solution of 3,5-diamino-4-[4-(trifluoromethyl)phenylazo]-1H-pyrazole (4b) (2.7 g, 0.01 mol) in ethanol (30 mL). The reaction mixture was heated under reflux for 12 h (monitored by TLC). After completion of the reaction, the precipitate which formed was filtered off, dried, and recrystallized from ethanol to afford 6-(thiazol-2-yldiazenyl)-3-(4-(trifluoromethyl)phenylazo)-2,5,7-triaminopyrazolo[1,5-a] pyrimidine (13).
B. Under Microwave Irradiation:
A mixture of 3,5-diamino-4-[4-(trifluoromethyl)phenylazo]-1H-pyrazole (4b) (2.7 g, 0.01 mol) and N-(thiazol-2-yl)carbonohydrazonoyl dicyanide (1.77 g, 0.01 mol) was grinded carefully in a porcelain mortar using a pestle and transferred to a pyrex test tube, and ethanol (4 mL) was added, followed by pyridine (0.5 mL). The reaction mixture was heated under microwave irradiation at 50% power for 8 min at 140 °C, monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and the precipitated solid was filtered off, washed with MeOH, and recrystallized from ethanol to give 13.
Yield: 78%; (brown powder): mp > 300 °C; IR (KBr) νmax in cm−1: 3415, 3309 (NH2), 1608 (C=N), 1419 (N=N); 1H-NMR (500 MHz, DMSO-d6): δ 7.11 (s, 2H, NH2), 7.46 (d, 1H, CH–thiazole, J = 3.5 Hz), 7.65 (d, 1H, CH–thiazol, J = 3.5 Hz), 7.77–7.89 (m, 6H, Ar–H+NH2), 8.24 (br, s, 2H, NH2); 13C-NMR (125 MHz, DMSO-d6): δ 109.10, 109.20, 116.78, 119.63, 121.15, 121.34, 122.43 (Ar–C), 123.30, 125.46 (q, C–F3, J = 270 Hz), 126.04, 126.24, 126.27, 142.47 (Ar–C), 152.76, 155.58, (2C=N), 167.33 (C=N thiazole); MS (m/z), 448 (M+ + 1; 24%), 447 (M+; 100%). Anal. Calcd. for C16H12F3N11S (447.40): C, 42.95; H, 2.70; N, 34.44. Found C, 42.81; H, 2.49; N, 34.38%.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}