A Complete Assessment of Dopamine Receptor- Ligand Interactions through Computational Methods

 , , and
, , and 
Abstract
:1. Introduction
1.1. Dopamine Receptors
1.2. Computer-Aided Drug Design
1.3. Aim
2. Results
2.1. Homology Models of Dopamine Receptors D1R-D5R Are Stable
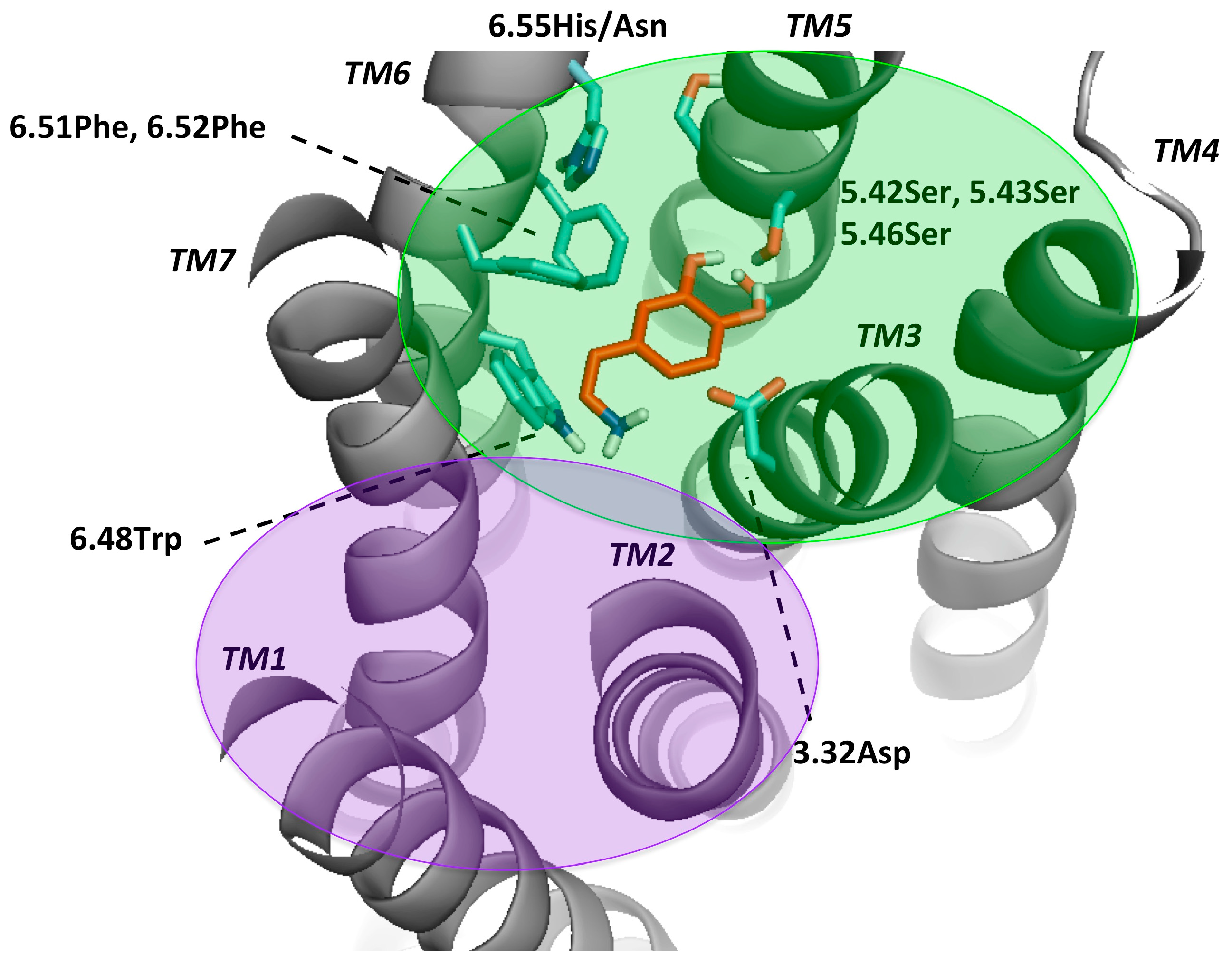
2.2. Dopamine Receptor Binding Pocket Definition
2.3. Proof-of-Concept of Molecular Docking Success
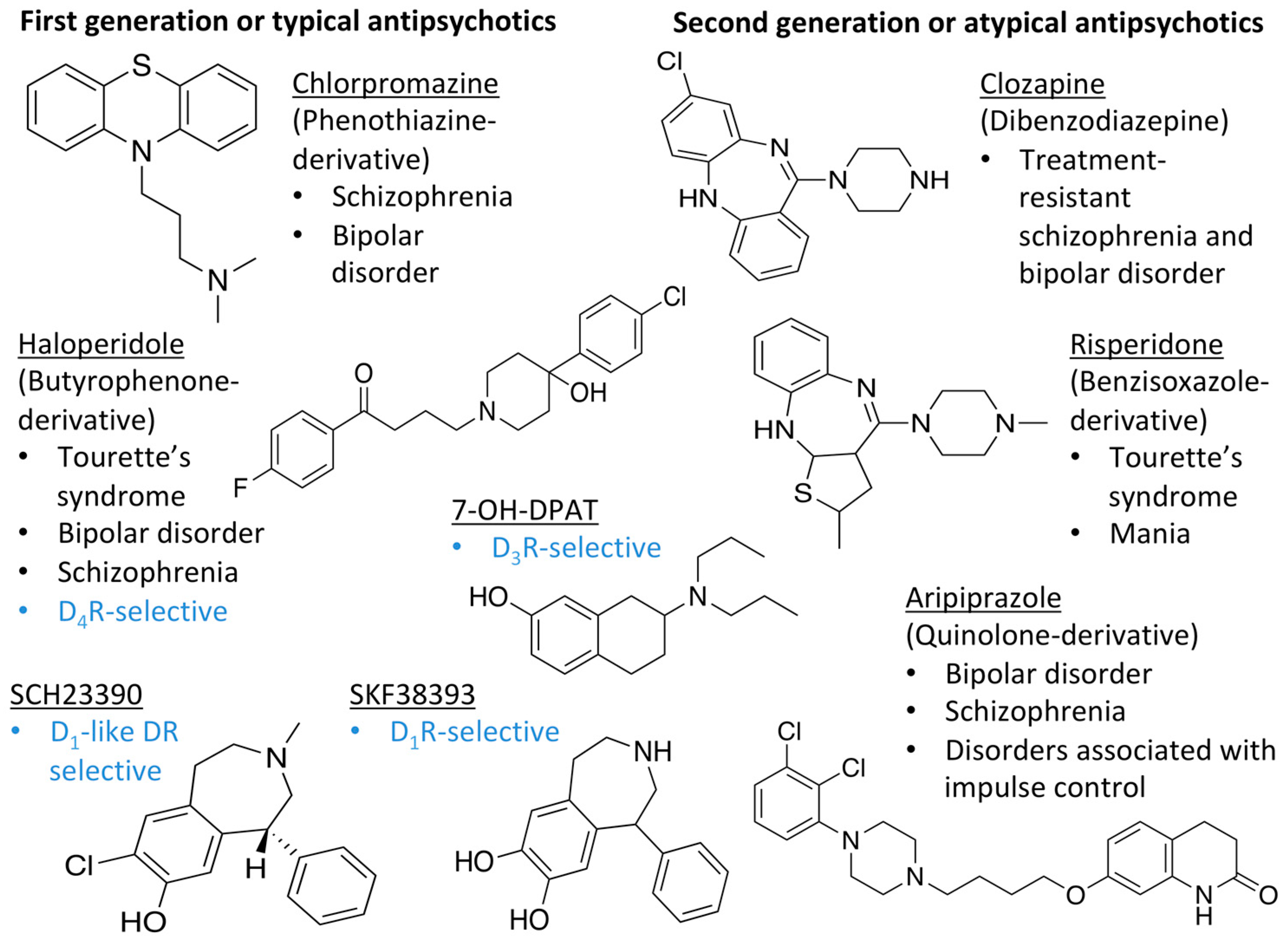
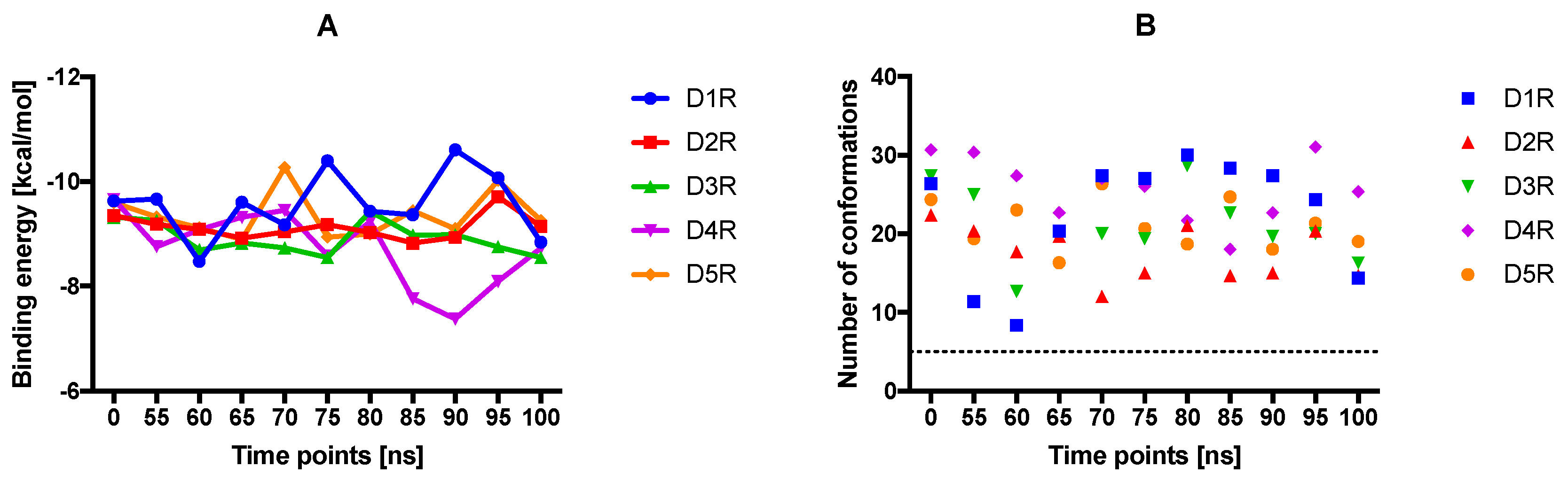
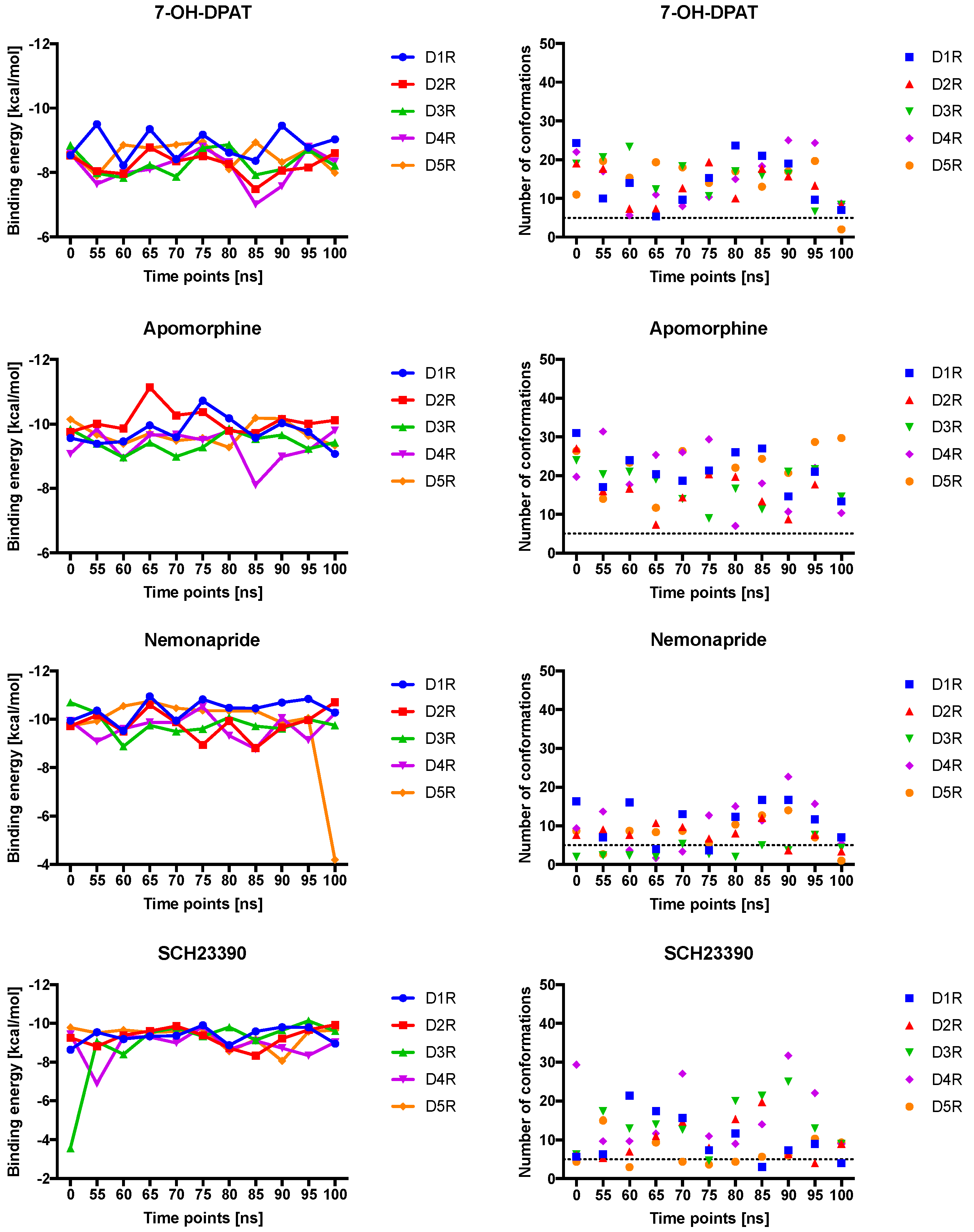
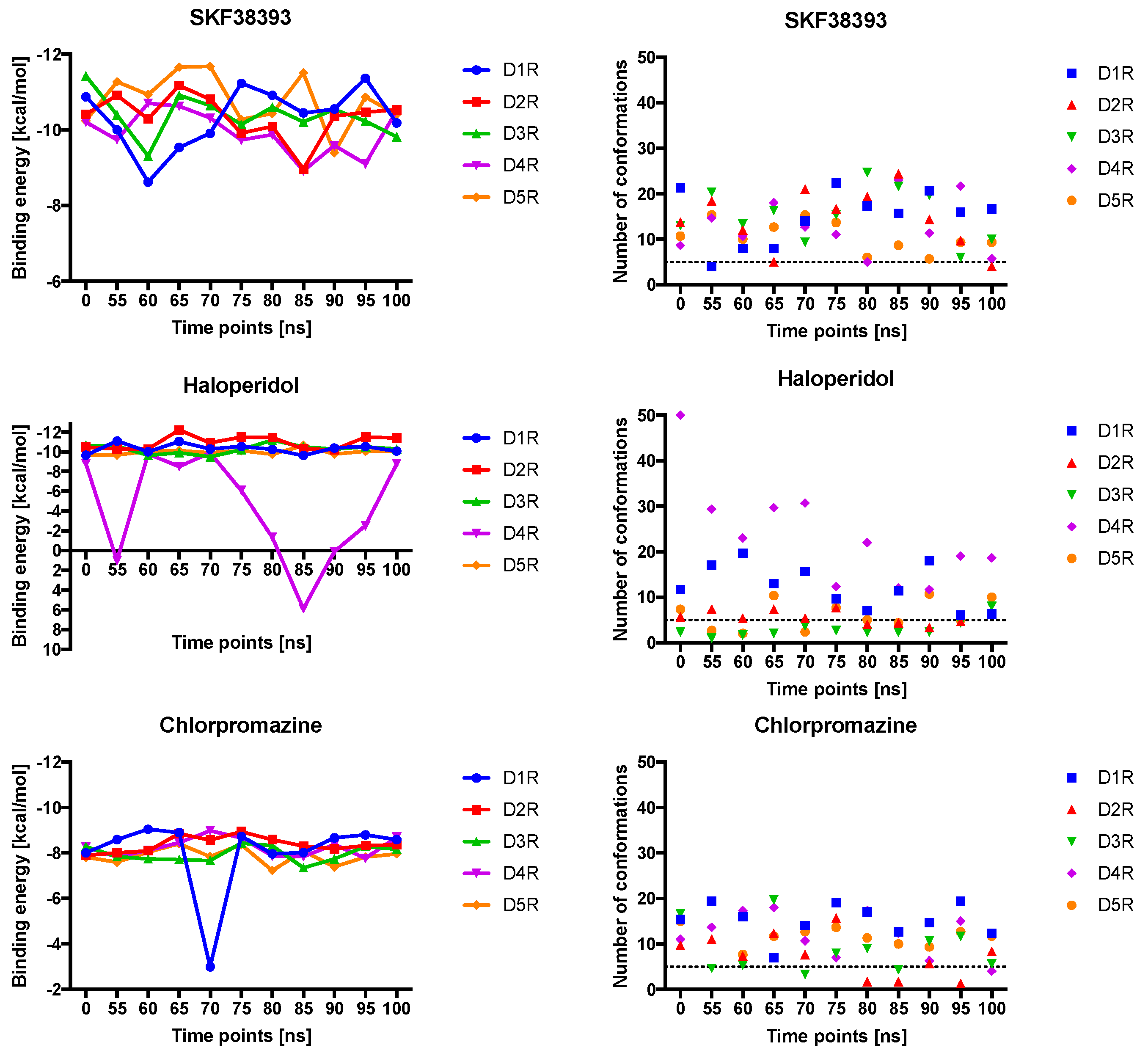
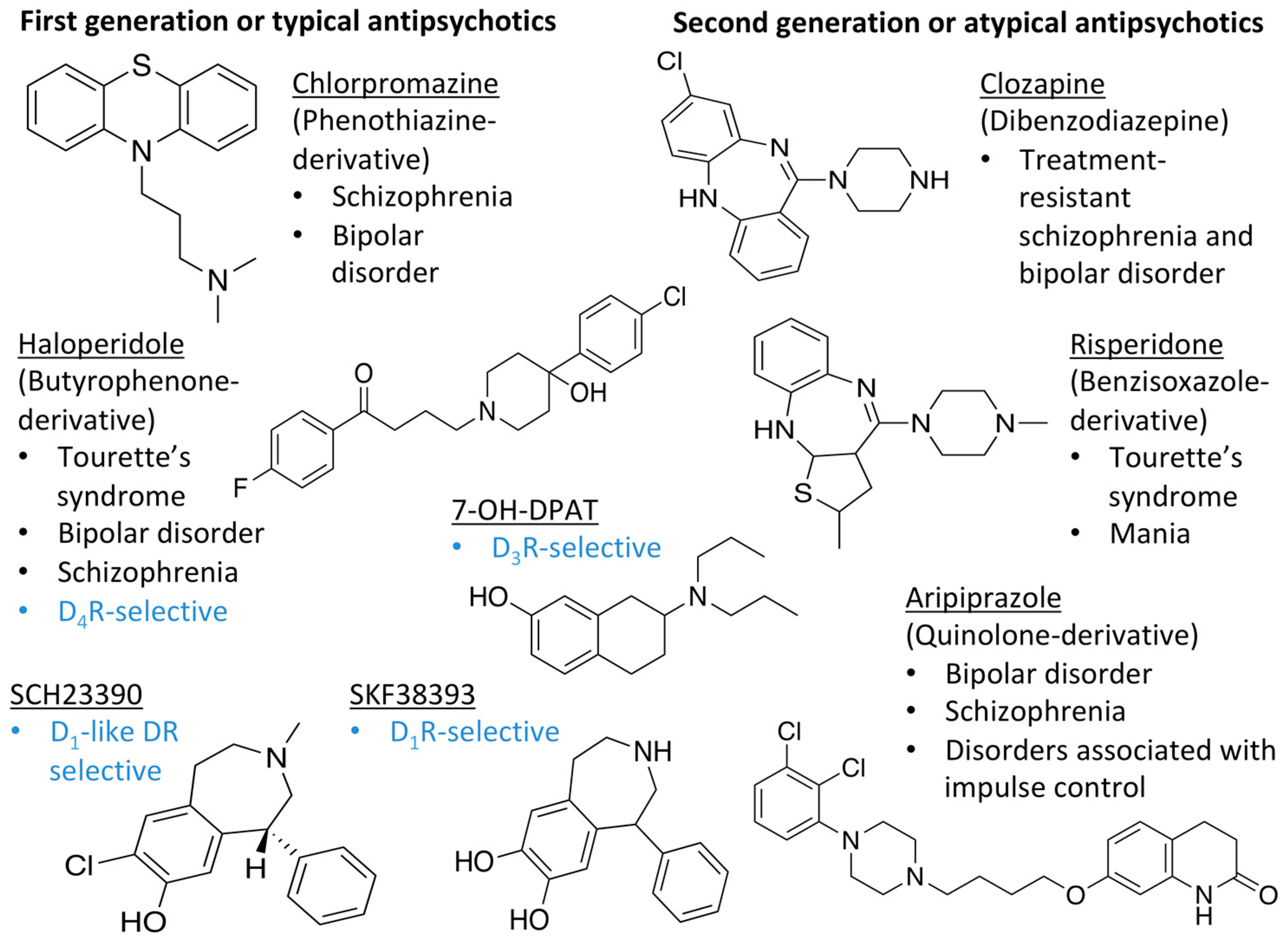
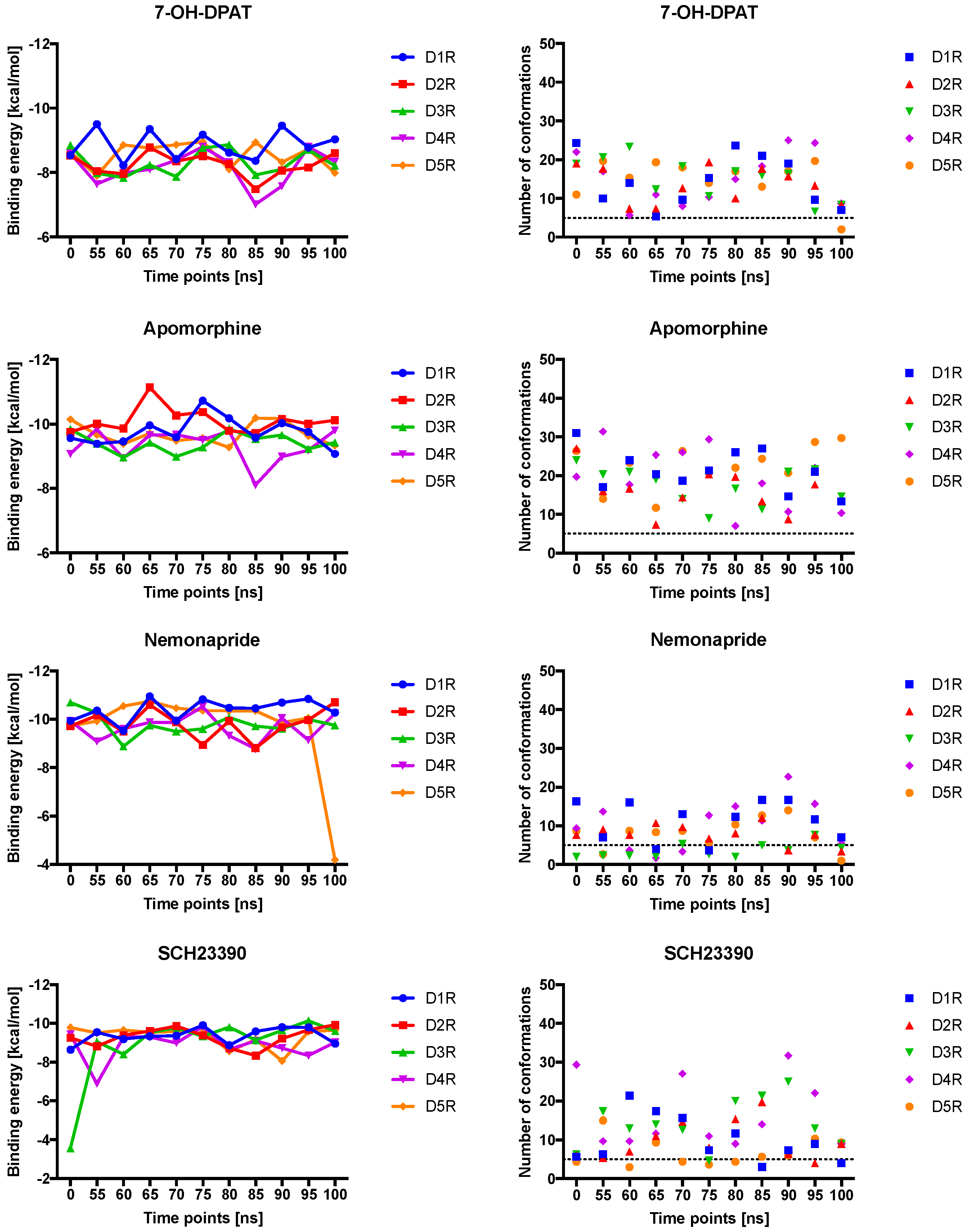
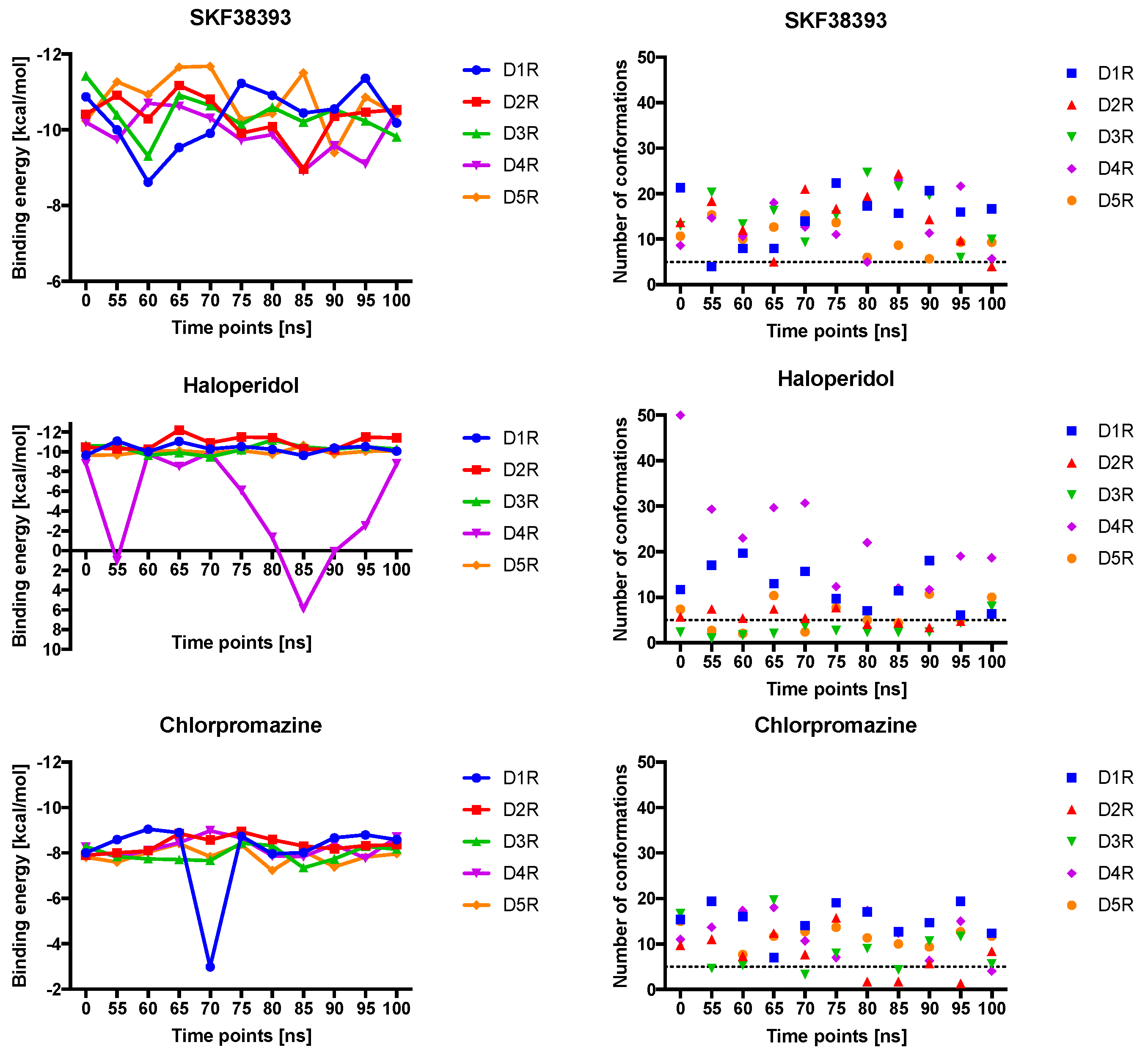
2.4. Docking of Various Ligands to DR Models
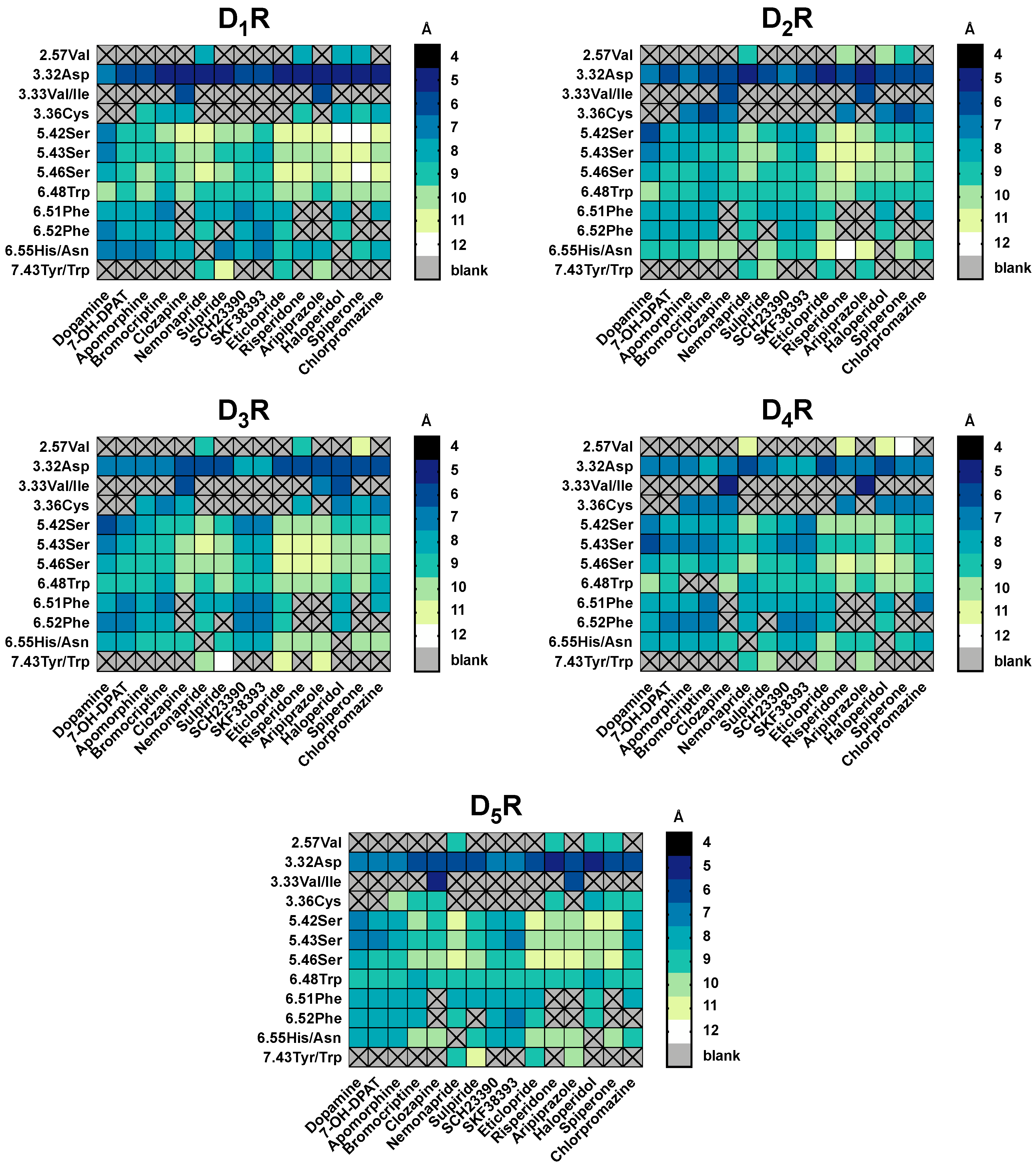
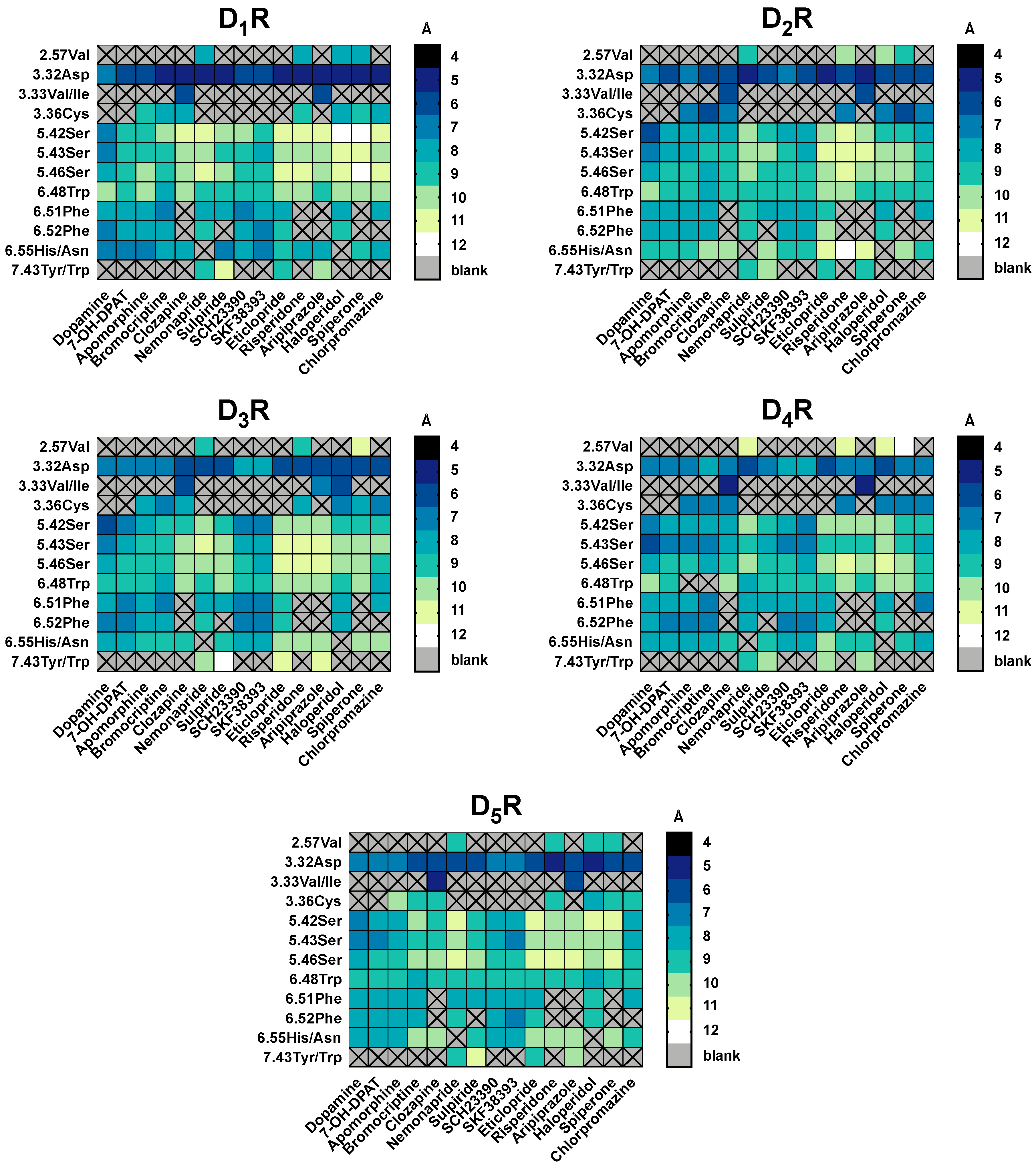
2.5. The Type of Pairwise Interactions Between Receptor Amino-Acids and Ligand is Relevant for Binding
2.6. 2.5 Å-Interactions
2.7. Hydrogen Bonds and Hydrophobic Contacts
2.8. Salt-Bridges
2.9. Cat-π- and π-π-Stacking Interactions
2.10. T-Stacking Interactions
3. Discussion
3.1. Validation of the In Silico Pipeline
3.2. Pairwise Interactions Analysis Was Able to Determine Key Amino-Acids and Types of Interaction
4. Materials and Methods
4.1. Homology Modeling
4.1.1. General Approach
4.1.2. Model Evaluation/Methods of Quality
4.2. Molecular Dynamics
4.2.1. System Setup
4.2.2. Molecular Dynamics Simulation Protocol
4.3. Molecular Docking
4.3.1. Ligand Dataset
4.3.2. Docking Procedure
4.3.3. Analysis of Molecular Docking
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Beaulieu, J.-M.; Gainetdinov, R.R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [Green Version]
- Platania, C.B.M.; Salomone, S.; Leggio, G.M.; Drago, F.; Bucolo, C. Homology Modeling of Dopamine D2 and D3 Receptors: Molecular Dynamics Refinement and Docking Evaluation. PLoS ONE 2012, 7, e44316. [Google Scholar] [CrossRef]
- Marsden, C.A. Dopamine: The rewarding years. Br. J. Pharmacol. 2006, 147 (Suppl. 1), 136–144. [Google Scholar] [CrossRef]
- Leggio, G.M.; Bucolo, C.; Platania, C.B.M.; Salomone, S.; Drago, F. Current drug treatments targeting dopamine D3 receptor. Pharmacol. Ther. 2016, 165, 164–177. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The structure and function of G protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef]
- Maurice, P.; Guillaume, J.L.; Benleulmi-Chaachoua, A.; Daulat, A.M.; Kamal, M.; Jockers, R. GPCR-Interacting Proteins, Major Players of GPCR Function. In GPCR—Interacting Proteins, Major Players of GPCR Function, 1st ed.; Elsevier Inc.: New York, NY, USA, 2011; Volume 62. [Google Scholar]
- Thimm, D.; Funke, M.; Meyer, A.; Müller, C.E. 6-Bromo-8-(4-methoxybenzamido)-4-oxo-4 H-chromene-2-carboxylic Acid: A powerful tool for studying orphan G protein-coupled receptor GPR35. J. Med. Chem. 2013, 56, 7084–7099. [Google Scholar] [CrossRef]
- Jaber, M.; Robinson, S.W.; Missale, C.; Caron, M.G. Dopamine receptors and brain function. Neuropharmacology 1997, 35, 1503–1519. [Google Scholar] [CrossRef]
- Seeman, P. Atypical Antipsychotics: Mechanism of Action. Focus 2002, 47, 27–38. [Google Scholar] [CrossRef]
- Rangel-Barajas, C.; Coronel, I.; Florán, B. Dopamine Receptors and Neurodegeneration. Aging Dis. 2015, 6, 349. [Google Scholar] [CrossRef]
- Amato, D.; Vernon, A.C.; Papaleo, F. Dopamine, the antipsychotic molecule: A perspective on mechanisms underlying antipsychotic response variability. Neurosci. Biobehav. Rev. 2017, 85, 146–159. [Google Scholar] [CrossRef]
- Noble, E.P. D2 dopamine receptor gene in psychiatric and neurologic disorders and its phenotypes. Am. J. Med. Genet. 2003, 116B, 103–125. [Google Scholar] [CrossRef]
- Zhang, A.; Neumeyer, J.L.; Baldessarini, R.J. Recent progress in development of dopamine receptor subtype-selective agents: Potential therapeutics for neurological and psychiatric disorders. Chem. Rev. 2007, 107, 274–302. [Google Scholar] [CrossRef]
- Miller, R. Mechanisms of action of antipsychotic drugs of different classes, refractoriness to therapeutic effects of classical neuroleptics, and individual variation in sensitivity to their actions: Part II. Curr. Neuropharmacol. 2009, 7, 315–330. [Google Scholar] [CrossRef]
- Mauri, M.C.; Paletta, S.; Maffini, M.; Colasanti, A.; Dragogna, F.; Di Pace, C.; Altamura, A.C. Clinical pharmacology of atypical antipsychotics: An update. EXCLI J. 2014, 13, 1163–1191. [Google Scholar]
- Sykes, D.A.; Moore, H.; Stott, L.; Holliday, N.; Javitch, J.A.; Lane, J.R.; Charlton, S.J. Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D2 receptors. Nat. Commun. 2017, 8, 763. [Google Scholar] [CrossRef]
- Salmas, R.; Serhat Is, Y.; Durdagi, S.; Stein, M.; Yurtsever, M. A QM protein–ligand investigation of antipsychotic drugs with the dopamine D2 Receptor (D2R). J. Biomol. Struct. Dyn. 2018, 36, 2668–2677. [Google Scholar] [CrossRef]
- Loebel, A.; Citrome, L.; Correll, C.U.; Xu, J.; Cucchiaro, J.; Kane, J.M. Treatment of early non-response in patients with schizophrenia: Assessing the efficacy of antipsychotic dose escalation. BMC Psychiatry 2015, 15, 1–7. [Google Scholar] [CrossRef]
- Behere, B.P.; Das, A.; Behere, A.P. Antipsychotics. In Clinical Psychopharmacology; Springer: Singapore, 2019; pp. 39–87. [Google Scholar]
- Moritz, A.E.; Benjamin Free, R.; Sibley, D.R. Advances and challenges in the search for D2 and D3 dopamine receptor-selective compounds. Cell. Signal. 2018, 41, 75–81. [Google Scholar] [CrossRef]
- Banala, A.K.; Levy, B.A.; Khatri, S.S.; Furman, C.A.; Roof, R.A.; Mishra, Y.; Gri, S.A.; Sibley, D.R.; Luedtke, R.R.; Newman, A.H. N-(3-Fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)arylcarboxamides as selective dopamine D3 receptor ligands: Critical role of the carboxamide linker for D3 recetpor selectivity. J. Med. Chem. 2011, 54, 3581–3594. [Google Scholar] [CrossRef]
- Newman, A.H.; Beuming, T.; Banala, A.K.; Donthamsetti, P.; Pongetti, K.; LaBounty, A.; Levy, B.A.; Cao, J.; Michino, M.; Luedtke, R.R.; et al. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem. 2012, 55, 6689–6699. [Google Scholar] [CrossRef]
- Damsma, G.; Bottema, T.; Westerink, B.H.C.; Tepper, P.G.; Dijkstra, D.; Pugsley, T.A.; MacKenzie, R.G.; Heffner, T.G.; Wikström, H. Pharmacological aspects of R-(+)-7-OH-DPAT, a putative dopamine D3 receptor ligand. Eur. J. Pharmacol. 1993, 249, 9–10. [Google Scholar] [CrossRef]
- Lévesque, D.; Diaz, J.; Pilon, C.; Martres, M.P.; Giros, B.; Souil, E.; Schott, D.; Morgat, J.L.; Schwartz, J.C.; Sokoloff, P. Identification, characterization, and localization of the dopamine D3 receptor in rat brain using 7-[3H]hydroxy-N,N-di-n-propyl-2-aminotetralin. Proc. Natl. Acad. Sci. USA 1992, 89, 8155–8159. [Google Scholar] [CrossRef]
- Sampson, D.; Zhu, X.Y.; Eyunni, S.V.K.; Etukala, J.R.; Ofori, E.; Bricker, B.; Lamango, N.S.; Setola, V.; Roth, B.L.; Ablordeppey, S.Y. Identification of a new selective dopamine D4receptor ligand. Bioorg. Med. Chem. 2014, 22, 3105–3114. [Google Scholar] [CrossRef]
- Zhang, J.; Xiong, B.; Zhen, X.; Zhang, A. Dopamine D1 receptor ligands: Where are we now and where are we going. Med. Res. Rev. 2009, 29, 272–294. [Google Scholar] [CrossRef]
- Conroy, J.L.; Free, R.B.; Sibley, D.R. Identification of G Protein-Biased Agonists That Fail to Recruit β-Arrestin or Promote Internalization of the D1 Dopamine Receptor. ACS Chem. Neurosci. 2015, 6, 681–692. [Google Scholar] [CrossRef]
- O’Sullivan, G.J.; Roth, B.L.; Kinsella, A.; Waddington, J.L. SK&F 83822 distinguishes adenylyl cyclase from phospholipase C-coupled dopamine D1-like receptors: Behavioural topography. Eur. J. Pharmacol. 2004, 486, 273–280. [Google Scholar]
- Butini, S.; Nikolic, K.; Kassel, S.; Brückmann, H.; Filipic, S.; Agbaba, D.; Gemma, S.; Brogi, S.; Brindisi, M.; Campiani, G.; et al. Polypharmacology of dopamine receptor ligands. Prog. Neurobiol. 2016, 142, 68–103. [Google Scholar] [CrossRef]
- Lee, S.M.; Kant, A.; Blake, D.; Murthy, V.; Boyd, K.; Wyrick, S.J.; Mailman, R.B. SKF-83959 is not a highly-biased functionally selective D1dopamine receptor ligand with activity at phospholipase C. Neuropharmacology 2014, 86, 145–154. [Google Scholar] [CrossRef]
- Arimitsu, E.; Ogasawara, T.; Takeda, H.; Sawasaki, T.; Ikeda, Y.; Hiasa, Y.; Maeyama, K. The ligand binding ability of dopamine D1 receptors synthesized using a wheat germ cell-free protein synthesis system with liposomes. Eur. J. Pharmacol. 2014, 745, 117–122. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Comput. Methods Drug Discov. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Jain, A. Computer Aided Drug Design & QSAR. J. Phys. Conf. Ser. 2017, 884, 012072. [Google Scholar]
- Lemos, A.; Melo, R.; Preto, A.J.; Almeida, J.G.; Moreira, I.S.; Natália, M.D.S. In silico studies targeting G-protein coupled receptors for drug research against Parkinson’s disease. Curr. Neuropharmacol. 2018, 16, 786–848. [Google Scholar] [CrossRef]
- Shin, W.H.; Christoffer, C.W.; Kihara, D. In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods 2017, 131, 22–32. [Google Scholar] [CrossRef]
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269. [Google Scholar] [CrossRef]
- Chien, E.Y.T.; Liu, W.; Zhao, Q.; Katritch, V.; Won Han, G.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the Human Dopamine D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef]
- Wang, S.; Wacker, D.; Levit, A.; Che, T.; Betz, R.M.; Mccorvy, J.D.; Venkatakrishnan, A.J.; Huang, X.-P.; Dror, R.O.; Shoichet, B.K.; et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 2017, 358, 381–386. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. BINANA: A novel algorithm for ligand-binding characterization. J. Mol. Graph. Model. 2011, 29, 888–893. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Shen, M.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Charron, C.L.; Fowkes, M.M.; Luyt, L.G. Bridging computational modeling with amino acid replacements to investigate GHS-R1a-peptidomimetic recognition. Eur. J. Med. Chem. 2016, 123, 822–833. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, 407–410. [Google Scholar] [CrossRef]
- Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Sci. 2003, 12, 1073–1086. [Google Scholar] [CrossRef] [Green Version]
- Wallner, B.; Elofsson, A. Identification of correct regions in protein models using structural, alignment, and consensus information. Protein Sci. 2006, 15, 900–913. [Google Scholar] [CrossRef] [Green Version]
- Floresca, C.Z.; Schetz, J.A. Dopamine Receptor Microdomains Involved in Molecular Recognition and the Regulation of Drug Affinity and Function. J. Recept. Signal Transduct. 2004, 24, 207–239. [Google Scholar] [CrossRef]
- Cummings, D.F.; Ericksen, S.S.; Goetz, A.; Schetz, J.A. Transmembrane Segment Five Serines of the D4 Dopamine Receptor Uniquely Influence the Interactions of Dopamine, Norepinephrine, and Ro10-4548. J. Pharmacol. Exp. Ther. 2010, 333, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Ericksen, S.S.; Cummings, D.F.; Teer, M.E.; Amdani, S.; Schetz, J.A. Ring Substituents on Substituted Benzamide Ligands Indirectly Mediate Interactions with Position 7.39 of Transmembrane Helix 7 of the D4 Dopamine Receptor. J. Pharmacol. Exp. Ther. 2012, 342, 472–485. [Google Scholar] [CrossRef]
- Bueschbell, B.; Preto, A.J.; Barreto, C.A.V.; Schiedel, A.C.; Moreira, I.S. Creating a valid in silico Dopamine D2-receptor model for small molecular docking studies. In MOL2NET, International Conference Series on Multidisciplinary Sciences; SCIFORUM: Basel, Switzerland, 2017; Volume 3, pp. 1–6. [Google Scholar]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Salmas, R.E.; Yurtsever, M.; Stein, M.; Durdagi, S. Modeling and protein engineering studies of active and inactive states of human dopamine D2 receptor (D2R) and investigation of drug/receptor interactions. Mol. Divers. 2015, 19, 321–332. [Google Scholar] [CrossRef]
- Moreira, I.S.; Shi, L.; Freyberg, Z.; Ericksen, S.S.; Weinstein, H.; Javitch, J.A. Structural Basis of Dopamine Receptor Activation. In The Dopamine Receptors; Neve, K.A., Ed.; Humana/Springer: Berlin, Germany, 2010; pp. 47–73. [Google Scholar]
- Huang, E.S. Construction of a sequence motif characteristic of aminergic G protein-coupled receptors. Protein Sci. 2003, 12, 1360–1367. [Google Scholar] [CrossRef]
- Tschammer, N.; Dörfler, M.; Hübner, H.; Gmeiner, P. Engineering a GPCR-ligand pair that simulates the activation of D 2L by dopamine. ACS Chem. Neurosci. 2010, 1, 25–35. [Google Scholar] [CrossRef]
- Kling, R.C.; Tschammer, N.; Lanig, H.; Clark, T.; Gmeiner, P. Active-state model of a dopamine D2 receptor—Galpha-i complex stabilized by aripiprazole-type partial agonists. PLoS ONE 2014, 9, e100069. [Google Scholar] [CrossRef]
- Kalani, M.Y.S.; Vaidehi, N.; Hall, S.E.; Trabanino, R.J.; Freddolino, P.L.; Kalani, M.A.; Floriano, W.B.; Kam, V.W.T.; Goddard, W.A. The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists. Proc. Natl. Acad. Sci. USA 2004, 101, 3815–3820. [Google Scholar] [CrossRef]
- Holst, B.; Nygaard, R.; Valentin-Hansen, L.; Bach, A.; Engelstoft, M.S.; Petersen, P.S.; Frimurer, T.M.; Schwartz, T.W. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J. Biol. Chem. 2010, 285, 3973–3985. [Google Scholar] [CrossRef]
- Männel, B.; Jaiteh, M.; Zeifman, A.; Randakova, A.; Möller, D.; Hübner, H.; Gmeiner, P.; Carlsson, J. Structure-guided screening for functionally selective D2 dopamine receptor ligands from a virtual chemical library. ACS Chem. Biol. 2017, 12, 2652–2661. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; Salmas, R.E.; Stein, M.; Yurtsever, M.; Seeman, P. Binding Interactions of Dopamine and Apomorphine in D2High and D2Low States of Human Dopamine D2 Receptor Using Computational and Experimental Techniques. ACS Chem. Neurosci. 2016, 7, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.N.; Mailman, R.B. Dopamine receptor signaling and cirrent and future antipyschotic drugs. Handb. Exp. Pharmacol. 2012, 212, 53–86. [Google Scholar]
- Bergman, J.; Madras, B.K.; Spealman, R.D. Behavioral effects of D1 and D2 dopamine receptor antagonists in squirrel monkeys. J. Pharmacol. Exp. Ther. 1991, 258, 910–917. [Google Scholar] [PubMed]
- Chen, T.; Hu, Y.; Lin, X.; Huang, X.; Liu, B.; Leung, P.; Chan, S.O.; Guo, D.; Jin, G. Dopamine signaling regulates the projection patterns in the mouse chiasm. Brain Res. 2015, 1625, 324–336. [Google Scholar] [CrossRef]
- Hidaka, K.; Matsumoto, M.; Tada, S.; Tasaki, Y.; Yamaguchi, T. Differential effects of [3H]nemonapride and [3H]spiperone binding on human dopamine D4 receptors. Neurosci. Lett. 1995, 186, 145–148. [Google Scholar] [CrossRef]
- Seeman, P.; Van Tol, H.H. Dopamine receptor pharmacology. Trends Pharmacol. Sci. 1994, 15, 264–270. [Google Scholar] [CrossRef]
- Lawler, C.P.; Prioleau, C.; Lewis, M.M.; Mak, C.; Jiang, D.; Schetz, J.A.; Gonzalez, A.M.; Sibley, D.R.; Mailman, R.B. Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology 1999, 20, 612–627. [Google Scholar] [CrossRef]
- Lindsley, C.W.; Hopkins, C.R. Return of D4 Dopamine Receptor Antagonists in Drug Discovery. J. Med. Chem. 2017, 60, 7233–7243. [Google Scholar] [CrossRef]
- Newton, C.L.; Wood, M.D.; Strange, P.G. Examining the effects of sodium ions on the binding of antagonists to dopamine D2 and D3 receptors. PLoS ONE 2016, 11, e0158808. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, X.; Tiberi, M. Functional importance of two conserved residues in intracellular loop 1 and transmembrane region 2 of Family A GPCRs: Insights from ligand binding and signal transduction responses of D1 and D5 dopaminergic receptor mutants. Cell. Signal. 2015, 27, 2014–2025. [Google Scholar] [CrossRef]
- Andringa, G.; Drukarch, B.; Leysen, J.E.; Cools, A.R.; Stoof, J.C. The alleged dopamine D1 receptor agonist SKF 83959 is a dopamine D1 receptor antagonist in primate cells and interacts with other receptors. Eur. J. Pharmacol. 1999, 364, 33–41. [Google Scholar] [CrossRef]
- Burris, K.D.; Molski, T.F.; Xu, C.; Ryan, E.; Tottori, K.; Kikuchi, T.; Yocca, F.D.; Molinoff, P.B. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J. Pharmacol. Exp. Ther. 2002, 302, 381–389. [Google Scholar] [CrossRef]
- López-Muñoz, F.; Alamo, C. The consolidation of neuroleptic therapy: Janssen, the discovery of haloperidol and its introduction into clinical practice. Brain Res. Bull. 2009, 79, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Madras, B.K. History of the discovery of the antipsychotic dopamine D2 receptor: A basis for the dopamine hypothesis of schizophrenia. J. Hist. Neurosci. 2013, 22, 62–78. [Google Scholar] [CrossRef]
- Abhijnhan, A.; Adams, C.E.; David, A.; Ozbilen, M. Depot fluspirilene for schizophrenia. Cochrane Database Syst. Rev. 2007, 1, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Valizade Hasanloei, M.A.; Sheikhpour, R.; Sarram, M.A.; Sheikhpour, E.; Sharifi, H. A combined Fisher and Laplacian score for feature selection in QSAR based drug design using compounds with known and unknown activities. J. Comput. Aided Mol. Des. 2018, 32, 375–384. [Google Scholar] [CrossRef]
- Kumar, S.P. PLHINT: A knowledge-driven computational approach based on the intermolecular H bond interactions at the protein-ligand interface from docking solutions. J. Mol. Graph. Model. 2018, 79, 194–212. [Google Scholar] [CrossRef]
- Trisciuzzi, D.; Nicolotti, O.; Miteva, M.A.; Villoutreix, B.O. Analysis of solvent-exposed and buried co-crystallized ligands: A case study to support the design of novel protein–protein interaction inhibitors. Drug Discov. Today 2018, 24, 551–559. [Google Scholar] [CrossRef]
- Tanina, A.; Wohlkönig, A.; Soror, S.H.; Flipo, M.; Villemagne, B.; Prevet, H.; Déprez, B.; Moune, M.; Perée, H.; Meyer, F.; et al. A comprehensive analysis of the protein-ligand interactions in crystal structures of Mycobacterium tuberculosis EthR. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 248–258. [Google Scholar] [CrossRef]
- Davis, A.M.; Teague, S.J. Hydrogen bonding, hydrophobic interactions, and failure of the rigid receptor hypothesis. Angew. Chem. Int. Ed. 1999, 38, 736–749. [Google Scholar] [CrossRef]
- Bosch, E.; Barnes, C.L.; Brennan, N.L.; Eakins, G.L.; Breyfogle, B.E. Cation-Induced π-stacking. J. Org. Chem. 2008, 73, 3931–3934. [Google Scholar] [CrossRef]
- Frontera, A.; Quiñonero, D.; Deyà, P.M. Cation-π and anion-π interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 440–459. [Google Scholar] [CrossRef]
- Wang, Q.; MacH, R.H.; Luedtke, R.R.; Reichert, D.E. Subtype selectivity of dopamine receptor ligands: Insights from structure and ligand-based methods. J. Chem. Inf. Model. 2010, 50, 1970–1985. [Google Scholar] [CrossRef]
- Simpson, M.M.; Ballesteros, J.A.; Chiappa, V.; Chen, J.; Suehiro, M.; Hartman, D.S.; Godel, T.; Snyder, L.A.; Sakmar, T.P.; Javitch, J.A. Dopamine D4/D2 receptor selectivity is determined by A divergent aromatic microdomain contained within the second, third, and seventh membrane-spanning segments. Mol. Pharmacol. 1999, 56, 1116–1126. [Google Scholar] [CrossRef]
- Salmas, R.E.; Yurtsever, M.; Durdagi, S. Atomistic molecular dynamics simulations of typical and atypical antipsychotic drugs at the dopamine D2 receptor (D2R) elucidates their inhibition mechanism. J. Biomol. Struct. Dyn. 2016, 35, 1–17. [Google Scholar] [CrossRef]
- Sukalovic, V.; Soskic, V.; Sencanski, M.; Andric, D.; Kostic-Rajacic, S. Determination of key receptor-ligand interactions of dopaminergic arylpiperazines and the dopamine D2 receptor homology model. J. Mol. Model. 2013, 19, 1751–1762. [Google Scholar] [CrossRef]
- Moreira, I.S. Structural features of the G-protein/GPCR interactions. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 16–33. [Google Scholar] [CrossRef]
- Scarselli, M.; Novi, F.; Schallmach, E.; Lin, R.; Baragli, A.; Colzi, A.; Griffon, N.; Corsini, G.U.; Sokoloff, P.; Levenson, R.; et al. D2/D3 Dopamine Receptor Heterodimers Exhibit Unique Functional Properties. J. Biol. Chem. 2001, 276, 30308–30314. [Google Scholar] [CrossRef]
- Bourne, J.A. SCH23390: The First Selective Dopamine D1-Like Receptor Antagonist. CNS Drug Rev. 2006, 7, 399–414. [Google Scholar] [CrossRef]
- Ferreira De Freitas, R.; Schapira, M. A systematic analysis of atomic protein-ligand interactions in the PDB. Medchemcomm 2017, 8, 1970–1981. [Google Scholar] [CrossRef]
- Hugo, E.A.; Cassels, B.K.; Fierro, A. Functional roles of T3.37 and S5.46 in the activation mechanism of the dopamine D1 receptor. J. Mol. Model. 2017, 23, 142. [Google Scholar] [CrossRef]
- Zarrindast, M.R.; Honardar, Z.; Sanea, F.; Owji, A.A. SKF 38393 and SCH 23390 inhibit reuptake of serotonin by rat hypothalamic synaptosomes. Pharmacology 2011, 87, 85–89. [Google Scholar] [CrossRef]
- Pettersson, I.; Gundertofte, K.; Palm, J.; Liljefors, T. A Study on the Contribution of the 1-Phenyl Substituent to the Molecular Electrostatic Potentials of Some Benzazepines in Relation to Selective Dopamine D-1 Receptor Activity. J. Med. Chem. 1992, 35, 502–507. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A.; Francisco, S. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinforma 2017, 54, 1–55. [Google Scholar]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Hege, S.B.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Cristobal, S.; Zemla, A.; Fischer, D.; Rychlewski, L.; Elofsson, A. A study of quality measures for protein threading models. BMC Bioinform. 2001, 2, 5. [Google Scholar] [CrossRef]
- Siew, N.; Elofsson, A.; Rychlewski, L.; Fischer, D. MaxSub: An automated measure for the assessment of protein structure prediction quality. Bioinformatics 2000, 16, 776–785. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [Green Version]
- Gaete-Eastman, C.; Morales-Quintana, L.; Herrera, R.; Moya-León, M.A. In-silico analysis of the structure and binding site features of an α-expansin protein from mountain papaya fruit (VpEXPA2), through molecular modeling, docking, and dynamics simulation studies. J. Mol. Model. 2015, 21, 1–12. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, 370–376. [Google Scholar] [CrossRef]
- Lomize, A.L.; Pogozheva, I.D.; Lomize, M.A.; Mosberg, H.I. Positioning of proteins in membranes: A computational approach. Protein Sci. 2006, 15, 1318–1333. [Google Scholar] [CrossRef] [Green Version]
- Lomize, A.L.; Pogozheva, I.D.; Lomize, M.A.; Mosberg, H.I. The role of hydrophobic interactions in positioning of peripheral proteins in membranes. BMC Struct. Biol. 2007, 7, 1–30. [Google Scholar] [CrossRef]
- Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. Anisotropic solvent model of the lipid bilayer. 2. Energetics of insertion of small molecules, peptides and proteins in membranes Andrei. J. Chem. Inf. Model. 2011, 51, 930–946. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, 665–667. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N*log(N) method for Ewald sums in large systems Tom. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schultern, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
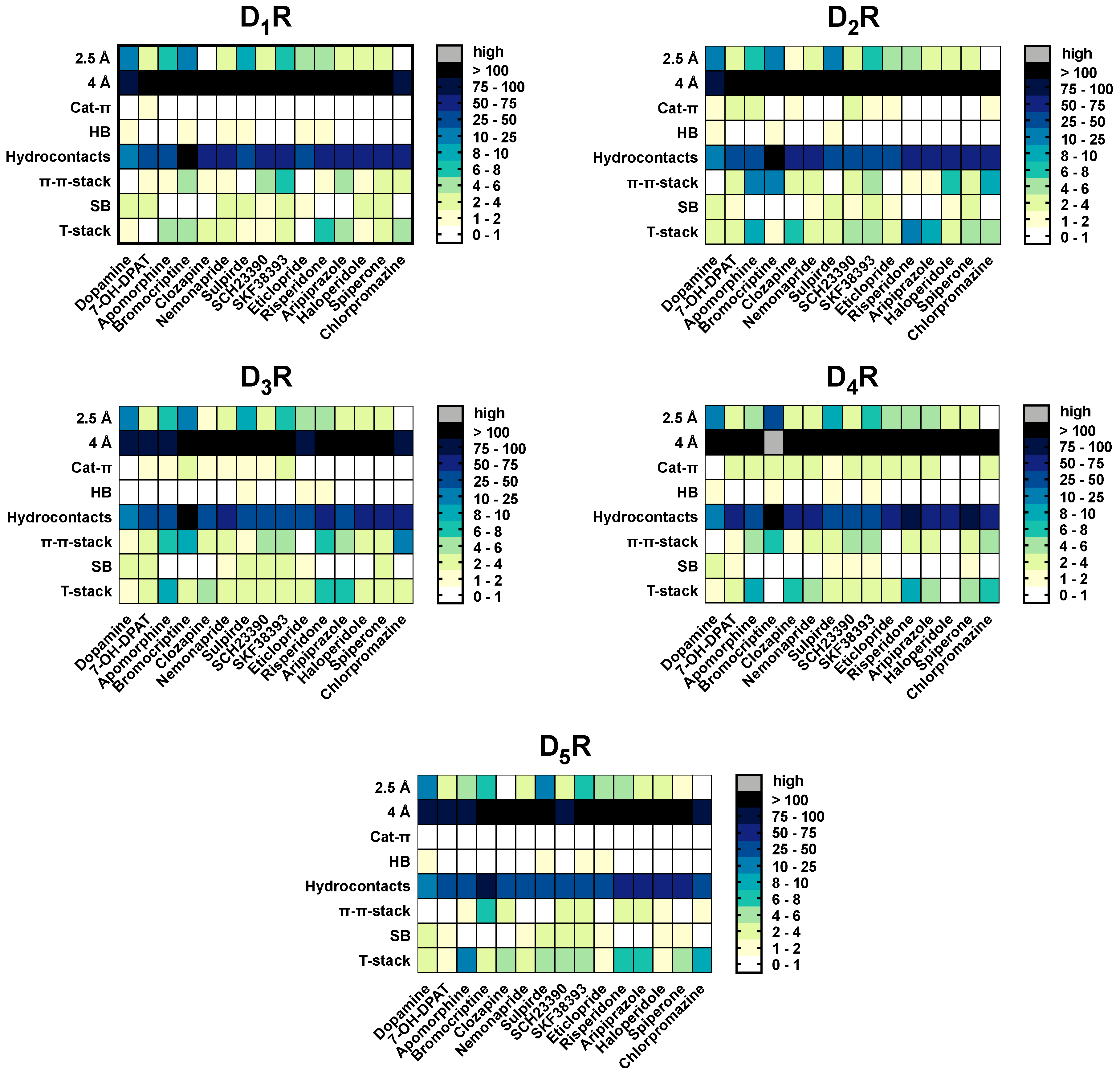

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
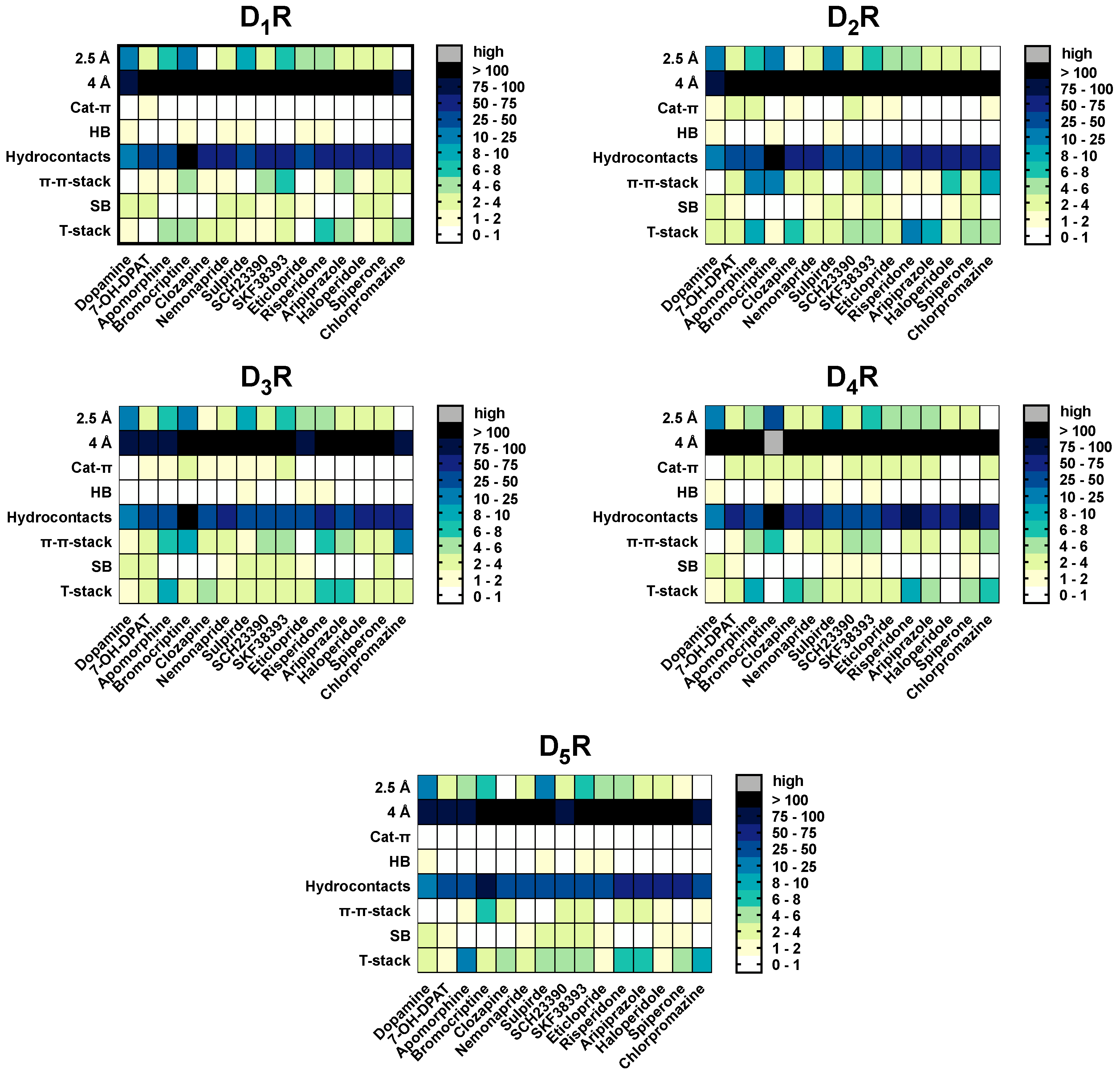{kind=link}
| LIGAND | FUNCTION | BP | REFERENCES | |
|---|---|---|---|---|
| DOPAMINE | 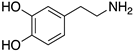 | Endogenous agonist of all DR | OBP | [47,52,65] |
| 7-OH-DPAT |  | Synthetic D3R selective agonist | OBP | [47,65,66] |
| APOMORPHINE | 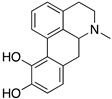 | D2R selective agonist | OBP | [47,52,65] |
| BROMOCRIPTINE |  | D2R selective agonist | OBP | [47,65] |
| CLOZAPINE | 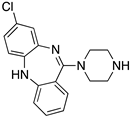 | “Dirty drug”, multiple receptor binding | OBP | [47,65,67,68] |
| NEMONAPRIDE | 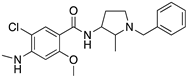 | D2R/D3R selective antagonist | OBP + SBP | [38,47,55,65] |
| SULPIRIDE | 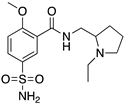 | “Dirty drug”, multiple receptor binding | OBP + SBP | [47,65,66] |
| SCH23390 | 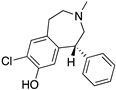 | D1R antagonist | OBP | [31,47,65,69] |
| SKF38393 | 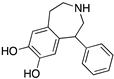 | D1R selective agonist | OBP | [31,47,65,70] |
| ETICLOPRIDE | 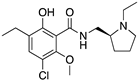 | D2R/D3R selective antagonist | OBP + SBP | [37,66] |
| RISPERIDONE | 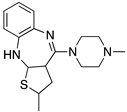 | “Dirty drug”, multiple receptor binding | OBP+SBP | [36,47] |
| ARIPIPRAZOLE | 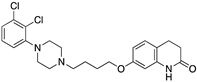 | Partial D2R agonist, D2R/D3R heterodimer antagonist | OBP + SBP | [66,71] |
| HALOPERIDOLE | 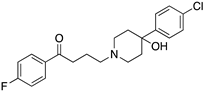 | D2R selective antagonist, D4R antagonist | OBP+SBP | [47,65,67,72,73] |
| SPIPERONE | 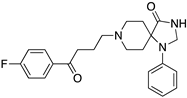 | Affinity for all DR | OBP + SBP | [47,65,66] |
| CHLORPROMAZINE | 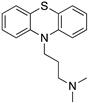 | Antagonist on all DR | OBP | [47,65,74] |
| DOPAMINE RECEPTOR | TEMPLATE | BLAST [%] | CLUSTALOMEGA [%] |
|---|---|---|---|
| D1R | 3PBL | 35.0 | 39.5 |
| D2R | 6CM4 | 97.0 | 100.0 |
| D3R | 3PBL | 93.0 | 99.3 |
| D4R | 5WIU | 93.0 | 100.0 |
| D5R | 5WIU | 35.0 | 39.1 |
| DR | DOPE | LGscore | LGscore + PSIPRED | MaxSub | MaxSub + PSIPRED | z-Score |
|---|---|---|---|---|---|---|
| D1R | −39070.82 | 2.53 | 4.26 | 0.18 | 0.53 | −2.14 |
| D2R | −39284.66 | 2.52 | 4.22 | 0.21 | 0.52 | −2.22 |
| D3R | −39458.37 | 3.14 | 4.19 | 0.27 | 0.55 | −3.12 |
| D4R | −36738.05 | 3.33 | 4.25 | 0.25 | 0.59 | −3.90 |
| D5R | −38356.05 | 2.60 | 4.14 | 0.15 | 0.57 | −1.49 |
| LIGAND | FLEXIBLE RESIDUES IN B&W NUMBERING |
|---|---|
| DOPAMINE | 3.32Asp, 5.42Ser, 5.43Ser, 5.46Ser, 6.48Trp, 6.51Phe, 6.52Phe, 6.55His/Asn |
| 7-OH-DPAT | |
| APOMORPHINE | 3.32Asp, 3.36/3.35Cys, 5.42Ser, 5.43Ser, 5.46Ser, 6.48Trp, 6.51Phe, 6.52Phe, 6.55His/Asn |
| BROMOCRIPTINE | |
| CLOZAPINE | 3.32Asp, 3.33Val, 3.36Cys, 5.42Ser, 5.43Ser, 5.46Ser, 6.48Trp, 6.55His/Asn |
| NEMONAPRIDE | 2.57Val, 3.32Asp, 5.42Ser, 5.43Ser, 5.46Ser, 6.48Trp, 6.51Phe, 6.52Phe, 7.43Tyr |
| SULPIRIDE | 3.32Asp, 6.48Trp, 5.42Ser, 5.43Ser, 5.46Ser, 6.55His/Asn, 7.43Tyr, 6.51Phe |
| SCH23390 | 3.32Asp, 6.48Trp, 5.42Ser, 5.43Ser, 5.46Ser, 6.55His/Asn, 6.51Phe, 6.52Phe |
| SKF38393 | |
| ETICLOPRIDE | 3.32Asp, 6.48Trp, 5.42Ser, 5.43Ser, 5.46Ser, 6.55His/Asn, 7.43Tyr, 6.51Phe, 6.52Phe |
| RISPERIDONE | 3.32Asp, 6.48Trp, 3.36Cys, 6.55His/Asn, 2.57Val, 5.42Ser, 5.43Ser, 5.46Ser |
| ARIPIPRAZOLE | 3.32Asp, 6.48Trp, 3.33Val, 5.42Ser, 5.43Ser, 5.46Ser, 7.43Tyr, 6.55His/Asn |
| HALOPERIDOLE | 3.32Asp, 6.48Trp, 6.51Phe, 6.52Phe, 3.36Cys, 2.57Val, 5.42Ser, 5.43Ser, 5.46Ser |
| SPIPERONE | 3.32Asp, 6.48Trp, 5.42Ser, 5.43Ser, 5.46Ser, 3.36Cys, 6.55His/Asn, 2.57Val |
| CHLORPROMAZINE | 3.32Asp, 6.48Trp, 5.42Ser, 5.43Ser, 5.46Ser, 6.55His/Asn, 3.36Cys, 6.51Phe |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bueschbell, B.; Barreto, C.A.V.; Preto, A.J.; Schiedel, A.C.; Moreira, I.S. A Complete Assessment of Dopamine Receptor- Ligand Interactions through Computational Methods. Molecules 2019, 24, 1196. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24071196
Bueschbell B, Barreto CAV, Preto AJ, Schiedel AC, Moreira IS. A Complete Assessment of Dopamine Receptor- Ligand Interactions through Computational Methods. Molecules. 2019; 24(7):1196. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24071196
Chicago/Turabian StyleBueschbell, Beatriz, Carlos A. V. Barreto, António J. Preto, Anke C. Schiedel, and Irina S. Moreira. 2019. "A Complete Assessment of Dopamine Receptor- Ligand Interactions through Computational Methods" Molecules 24, no. 7: 1196. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24071196