
Field-Based Affinity Optimization of a Novel Azabicyclohexane Scaffold HIV-1 Entry Inhibitor

 , , and
, , and
Abstract
:1. Introduction
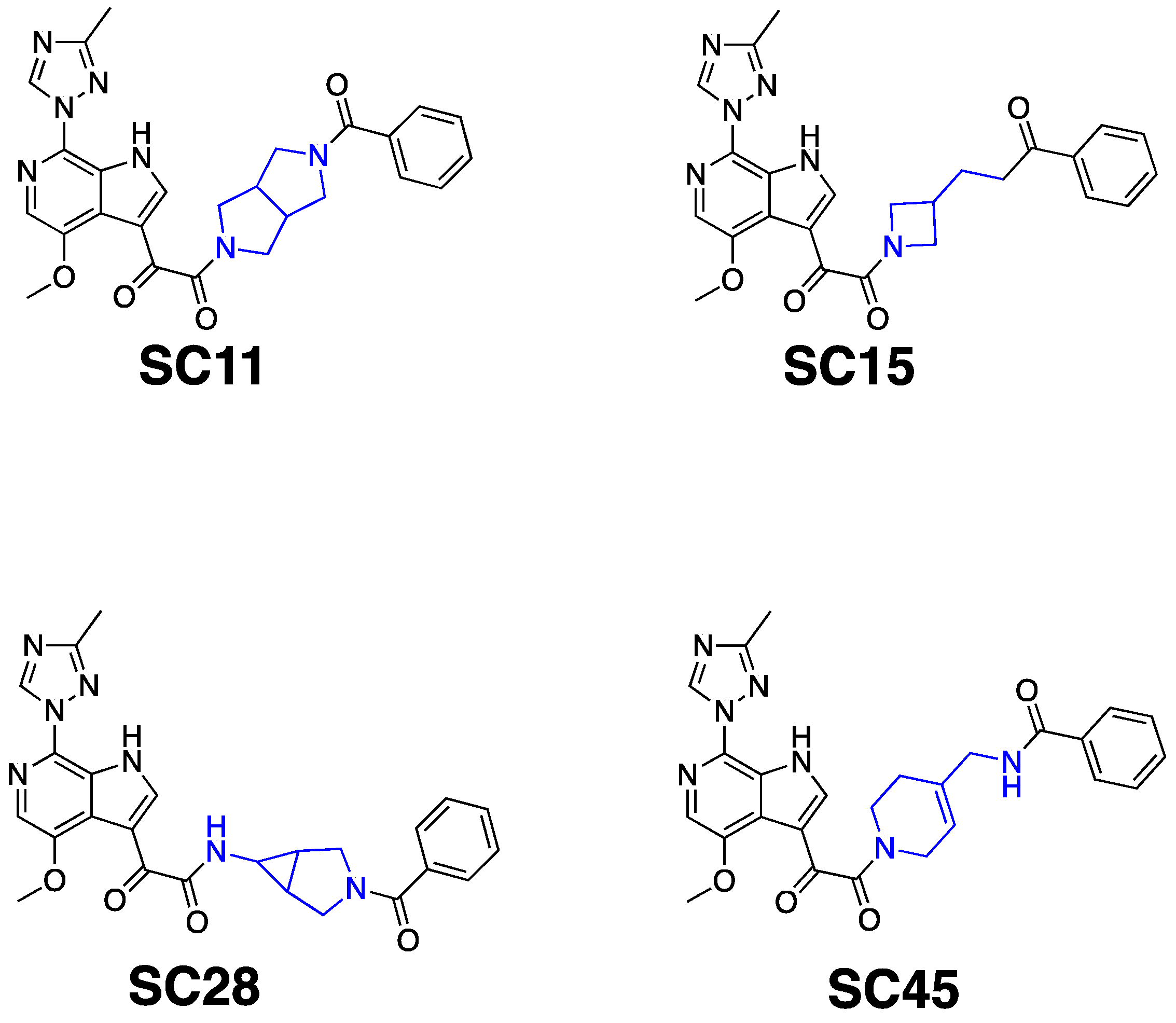
2. Results and Discussion
3. Material and Methods
3.1. General Information
3.2. Chemistry
3.2.1. Synthesis of SC49
General Procedure for the Preparation of 2

General Procedure for the Preparation of 3

General Procedure for the Preparation of 4

General Procedure for the Preparation of 5

General Procedure for the Preparation of 6

General Procedure for the Preparation of 7

General Procedure for the Preparation of 8

General Procedure for the Preparation of SC49

3.2.2. The Synthesis of SC50
General Procedure for the Preparation of 9

General Procedure for the Preparation of 10

General Procedure for the Preparation of 11

General Procedure for the Preparation of 12

General Procedure for the Preparation of 13

General Procedure for the Preparation of SC50

3.2.3. Synthesis of SC52
Preparation of tert-Butyl (3-benzoyl-3-azabicyclo [3.1.0]hexan-6-yl) Carbamate (14)

Preparation of (6-Amino-3-azabicyclo[3.1.0]hexan-3-yl)(phenyl) Methanone (15)

Preparation of 6-Allylpyrazolo[1,5-a]pyrimidine-5,7-diol (17)

Preparation of 6-Allyl-5,7-dichloropyrazolo[1,5-a]pyrimidine (18)

Preparation of 6-Allyl-5-chloro-N-(4-methoxybenzyl)pyrazolo[1,5-a] pyrimidin-7-amine (19)

Preparation of 5-Chloro-8-(4-methoxybenzyl)-8H-pyrazolo[1,5-a]pyrrolo[3,2-e]pyrimidine (20)

Preparation of 8-(4-Methoxybenzyl)-5-(3-methyl-1H-1,2,4-triazol-1-yl)-8H-pyrazolo[1,5-a]-pyrrolo[3,2-e]pyrimidine (21)

Preparation of 5-(3-Methyl-1H-1,2,4-triazol-1-yl)-8H-pyrazolo[1,5-a] pyrrolo[3,2-e]-pyrimidine (22)

Preparation of Ethyl 2-(5-(3-methyl-1H-1,2,4-triazol-1-yl)-8H-pyrazolo [1,5-a]pyrrolo[3,2-e]-pyrimidin-8-yl)acrylate (23)

Preparation of 2-(5-(3-Methyl-1H-1,2,4-triazol-1-yl)-8H-pyrazolo [1,5-a]pyrrolo[3,2-e]-pyrimidin-8-yl)acrylic acid (24)

Preparation of N-(3-benzoyl-3-azabicyclo[3.1.0]hexan-6-yl)-2-(5-(3- methyl-1H-1,2,4-triazol-1-yl)-8H-pyrazolo[1,5-a]pyrrolo[3,2-e]pyrimidin-8-yl)acrylamide (SC52)

3.2.4. The Synthesis of SC55
Preparation of 7-Bromo-4-fluoro-1H-indole (26)

Preparation of 4-Fluoro-1H-indole-7-carbonitrile (27)

Preparation of Methyl 2-(7-cyano-4-fluoro-1H-indol-3-yl)-2-oxoacetate (28)

Preparation of 2-(7-Cyano-4-fluoro-1H-indol-3-yl)-2-oxoacetic Acid (29)

Preparation of N-(3-benzoyl-3-azabicyclo[3.1.0]hexan-6-yl)-2-(7-cyano-4-fluoro-1H-indol-3-yl)-2-oxoacetamide (30)

Preparation of N-(3-benzoyl-3-azabicyclo[3.1.0]hexan-6-yl)-2-(4-fluoro-7-(N-hydroxy-carbamimidoyl)-1H-indol-3-yl)-2-oxoacetamide (31).

Preparation of N-(3-benzoyl-3-azabicyclo[3.1.0]hexan-6-yl)-2-(4-fluoro-7-(5-(trichloro-methyl)-1,2,4-oxadiazol-3-yl)-1H-indol-3-yl)-2-oxoacetamide (32)

Preparation of 2-(7-Nitro-1H-indazol-3-yl)acetic acid SC55

3.2.5. Synthesis of SC56
Preparation of 7-Bromo-4-methoxy-1H-indole (34)

Preparation of 4-Methoxy-1H-indole-7-carbonitrile (35)

Preparation of Methyl 2-(7-cyano-4-methoxy-1H-indol-3-yl)-2-oxoacetate (36)

Preparation of 2-(7-Cyano-4-methoxy-1H-indol-3-yl)-2-oxoacetic Acid (37)

Preparation of N-(3-Benzoyl-3-azabicyclo[3.1.0]hexan-6-yl)-2-(7-cyano-4-methoxy-1H-indol-3-yl)-2-oxoacetamide (38)

Preparation of N-(3-benzoyl-3-azabicyclo[3.1.0]hexan-6-yl)-2-(7-(N-hydroxycarbamimidoyl)-4-methoxy-1H-indol-3-yl)-2-oxoacetamide (39)

Preparation of N-(3-benzoyl-3-azabicyclo[3.1.0]hexan-6-yl)-2-(4-methoxy-7-(5-(trichloro-methyl)-1,2,4-oxadiazol-3-yl)-1H-indol-3-yl)-2-oxoacetamide (40)

Preparation of 2-(7-(5-Amino-1,2,4-oxadiazol-3-yl)-4-methoxy-1H-indol-3-yl)-N-(3-benzoyl-3-azabicyclo[3.1.0]hexan-6-yl)-2-oxoacetamide (SC56)

3.3. Cells
3.4. Proteins
3.5. Production of Pseudotyped Viruses
3.6. ELISA-Based Quantification of p24 Content
3.7. Single-Round Infection Assay
3.8. Cellular Toxicity
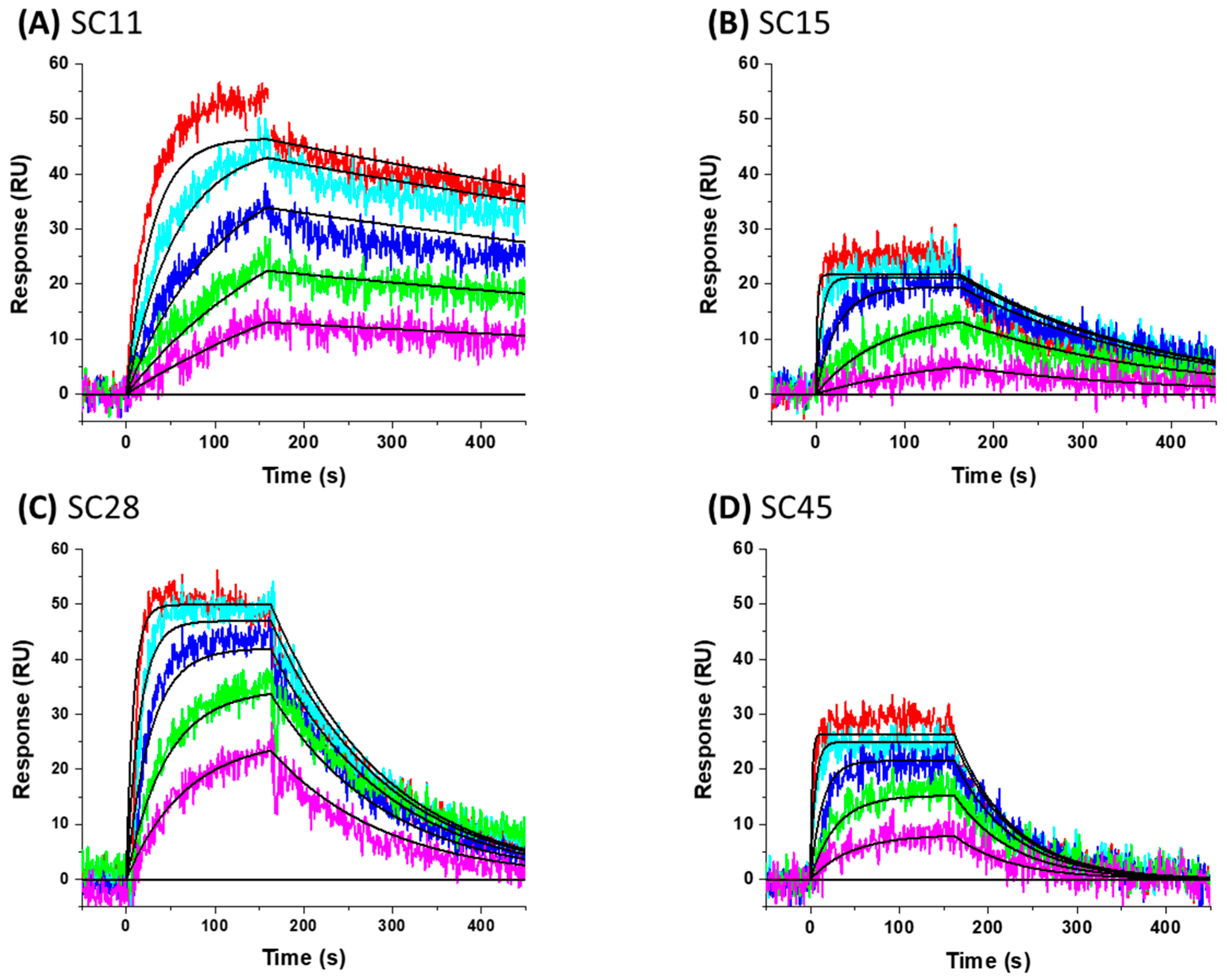
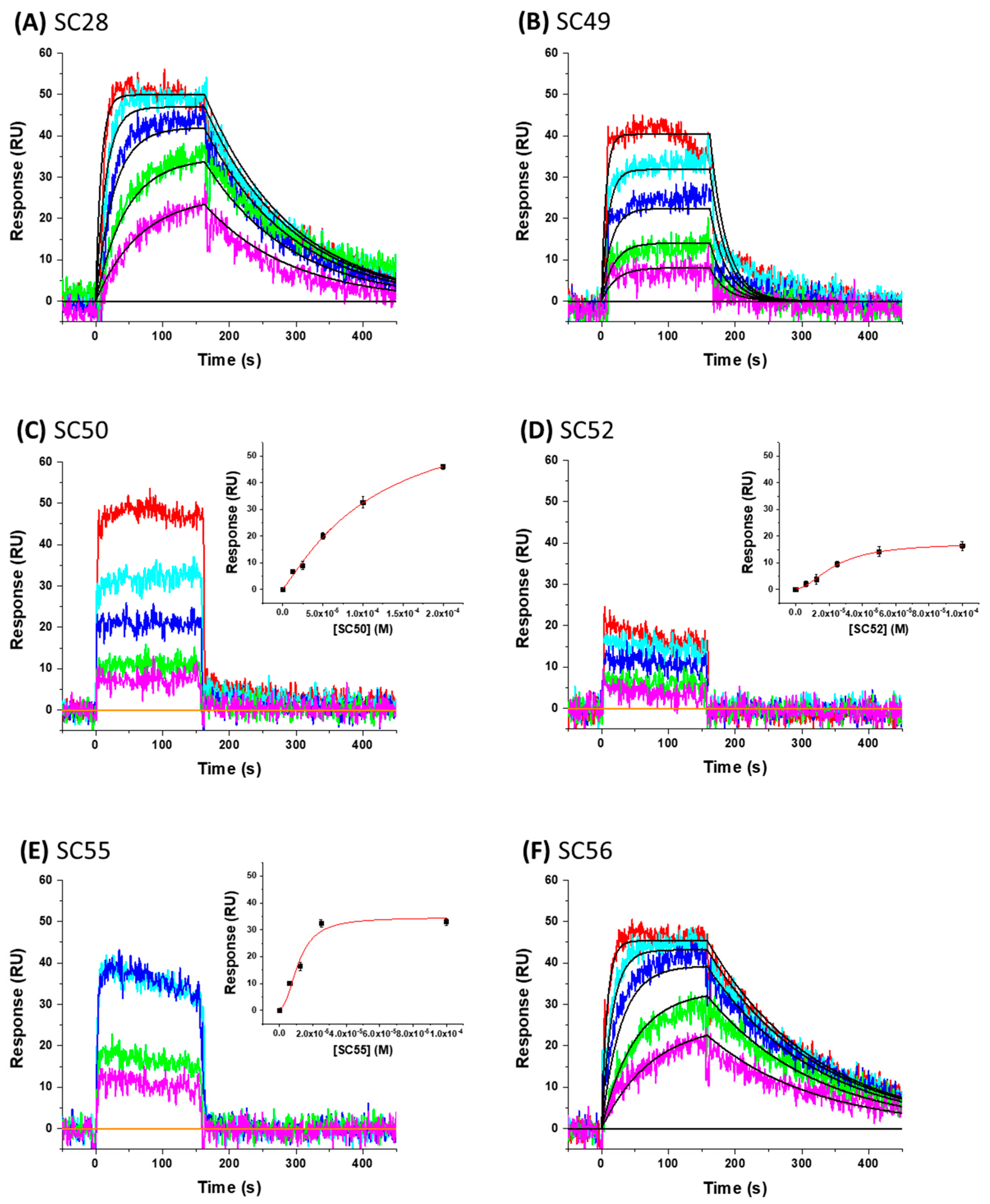
3.9. SPR Direct Interaction Analysis
Immobilization of Env Constructs
3.10. Direct Binding Analysis
3.11. Negative Stain Electron Microscopy (EM)
3.12. Differential Scanning Aalorimetry (DSC)
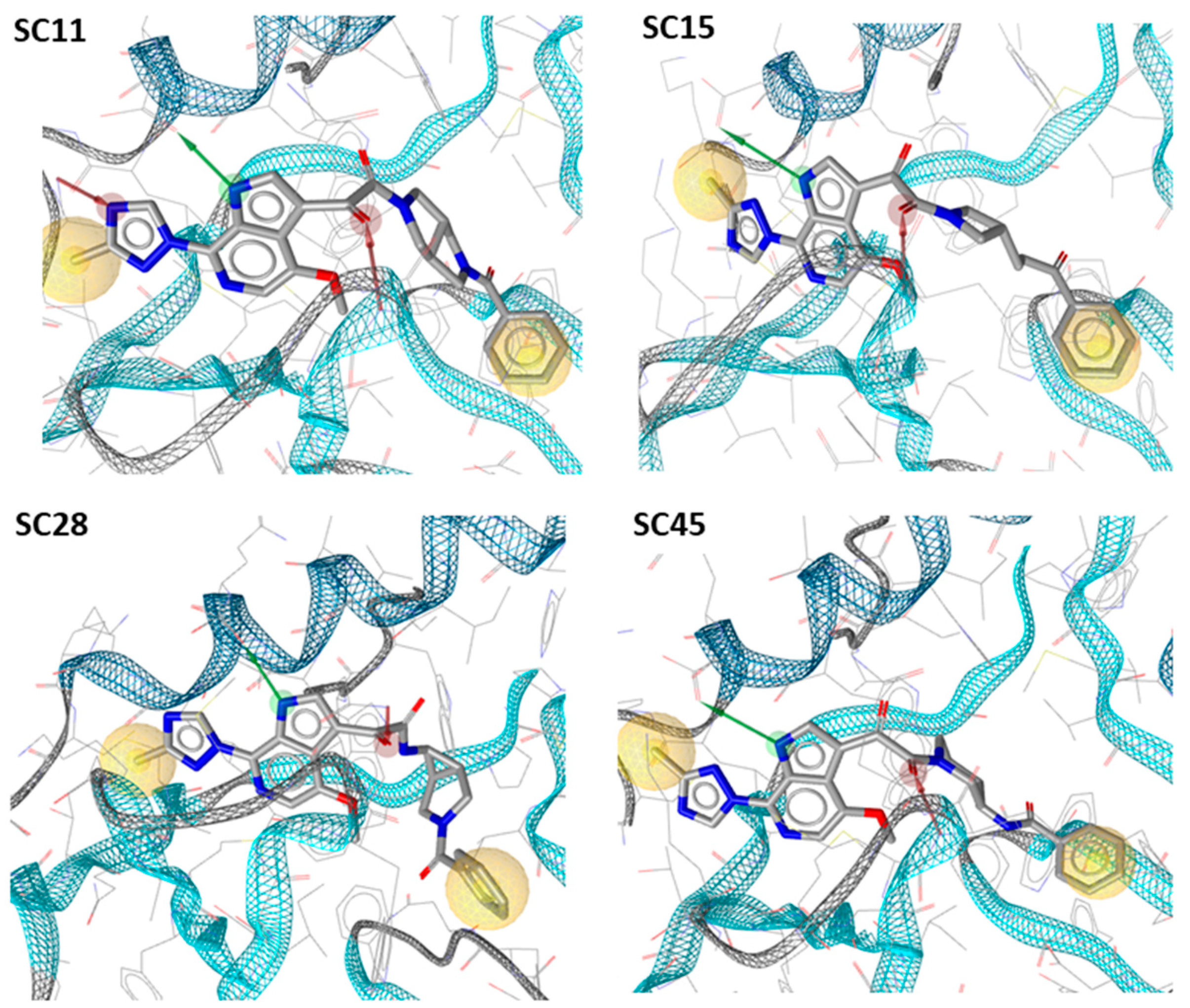
3.13. Molecular Modeling
3.13.1. Docking of Compounds SC11, SC15 and SC45
3.13.2. Docking of Compounds SC28, SC49, SC50, SC52, SC55 and SC56
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ward, A.B.; A Wilson, I. The HIV-1 envelope glycoprotein structure: Nailing down a moving target. Immunol. Rev. 2017, 275, 21–32. [Google Scholar]
- Wang, H.; Barnes, C.O.; Yang, Z.; Nussenzweig, M.C.; Bjorkman, P.J. Partially Open HIV-1 Envelope Structures Exhibit Conformational Changes Relevant for Coreceptor Binding and Fusion. Cell Host Microbe. 2018, 24, 579–592.e4. [Google Scholar] [CrossRef]
- Wagh, K.; Kreider, E.F.; Li, Y.; Barbian, H.J.; Learn, G.H.; Giorgi, E.; Hraber, P.T.; Decker, T.G.; Smith, A.G.; Gondim, M.V.; et al. Completeness of HIV-1 Envelope Glycan Shield at Transmission Determines Neutralization Breadth. Cell Rep. 2018, 25, 893–908.e7. [Google Scholar] [CrossRef]
- Schiffner, T.; Pallesen, J.; Russell, R.A.; Dodd, J.; De Val, N.; Labranche, C.C.; Montefiori, D.; Tomaras, G.D.; Shen, X.; Harris, S.L.; et al. Structural and immunologic correlates of chemically stabilized HIV-1 envelope glycoproteins. PLOS Pathog. 2018, 14, e1006986. [Google Scholar] [CrossRef]
- Wibmer, C.K.; Gorman, J.; Ozorowski, G.; Bhiman, J.N.; Sheward, D.J.; Elliott, D.H.; Rouelle, J.; Smira, A.; Joyce, M.G.; Ndabambi, N.; et al. Structure and Recognition of a Novel HIV-1 gp120-gp41 Interface Antibody that Caused MPER Exposure through Viral Escape. PLOS Pathog. 2017, 13, e1006074. [Google Scholar] [CrossRef]
- Pancera, M.; Lai, Y.-T.; Bylund, T.; Druz, A.; Narpala, S.; O’Dell, S.; Schön, A.; Bailer, R.T.; Chuang, G.-Y.; Geng, H.; et al. Crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nat. Methods 2017, 13, 1115–1122. [Google Scholar] [Green Version]
- Medina-Ramirez, M.; Garcés, F.; Escolano, A.; Skog, P.; De Taeye, S.W.; Del Moral-Sanchez, I.; McGuire, A.T.; Yasmeen, A.; Behrens, A.-J.; Ozorowski, G.; et al. Design and crystal structure of a native-like HIV-1 envelope trimer that engages multiple broadly neutralizing antibody precursors in vivo. J. Exp. Med. 2017, 214, 2573–2590. [Google Scholar] [CrossRef] [Green Version]
- Kulp, D.W.; Steichen, J.M.; Pauthner, M.; Hu, X.; Schiffner, T.; Liguori, A.; Cottrell, C.A.; Havenar-Daughton, C.; Ozorowski, G.; Georgeson, E.; et al. Structure-based design of native-like HIV-1 envelope trimers to silence non-neutralizing epitopes and eliminate CD4 binding. Nat. Commun. 2017, 8, 1655. [Google Scholar] [Green Version]
- Joyce, M.G.; Georgiev, I.S.; Yang, Y.; Druz, A.; Geng, H.; Chuang, G.-Y.; Kwon, Y.D.; Pancera, M.; Rawi, R.; Sastry, M.; et al. Soluble Prefusion Closed DS-SOSIP.664-Env Trimers of Diverse HIV-1 Strains. Cell Rep. 2017, 21, 2992–3002. [Google Scholar] [CrossRef]
- Wang, H.; A Cohen, A.; Galimidi, R.P.; Gristick, H.B.; Jensen, G.J.; Bjorkman, P.J. Cryo-EM structure of a CD4-bound open HIV-1 envelope trimer reveals structural rearrangements of the gp120 V1V2 loop. Proc. Acad. Sci. 2016, 113, E7151–E7158. [Google Scholar] [CrossRef]
- Lee, J.H.; Ozorowski, G.; Ward, A.B. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 2016, 351, 1043–1048. [Google Scholar] [CrossRef]
- Meanwell, N.A.; Krystal, M.R.; Nowicka-Sans, B.; Langley, D.R.; Conlon, D.A.; Eastgate, M.D.; Grasela, D.M.; Timmins, P.; Wang, T.; Kadow, J.F. Inhibitors of HIV-1 Attachment: The Discovery and Development of Temsavir and its Prodrug Fostemsavir. J. Med. Chem. 2018, 61, 62–80. [Google Scholar] [CrossRef]
- Kobayakawa, T.; Ohashi, N.; Hirota, Y.; Takahashi, K.; Yamada, Y.; Narumi, T.; Yoshimura, K.; Matsushita, S.; Harada, S.; Tamamura, H. Flexibility of small molecular CD4 mimics as HIV entry inhibitors. Bioorganic Med. Chem. 2018, 26, 5664–5671. [Google Scholar] [CrossRef]
- Li, W.; Lu, L.; Li, W.; Jiang, S. Small-molecule HIV-1 entry inhibitors targeting gp120 and gp41: A patent review (2010-2015). J. Expert Opin. Ther. Patents 2017, 27, 707–719. [Google Scholar] [CrossRef]
- Curreli, F.; Kwon, Y.D.; Belov, D.S.; Ramesh, R.R.; Kurkin, A.V.; Altieri, A.; Kwong, P.D.; Debnath, A.K. Synthesis, Antiviral Potency, in Vitro ADMET, and X-ray Structure of Potent CD4 Mimics as Entry Inhibitors That Target the Phe43 Cavity of HIV-1 gp120. J. Med. Chem. 2017, 60, 3124–3153. [Google Scholar] [CrossRef]
- Tuyishime, M.; Lawrence, R.; Cocklin, S. Core chemotype diversification in the HIV-1 entry inhibitor class using field-based bioisosteric replacement. Bioorganic Med. Chem. Lett. 2016, 26, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Dang, Z.; Zhu, L.; Lai, W.; Bogerd, H.; Lee, K.-H.; Huang, L.; Chen, C.-H. Aloperine and Its Derivatives as a New Class of HIV-1 Entry Inhibitors. ACS Med. Chem. Lett. 2016, 7, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Heredia, A.; Latinovic, O.S.; Barbault, F.; De Leeuw, E.P. A novel small-molecule inhibitor of HIV-1 entry. Drug Des. Dev. Ther. 2015, 9, 5469–5478. [Google Scholar] [CrossRef]
- Herschhorn, A.; Gu, C.; Espy, N.; Richard, J.; Finzi, A.; Sodroski, J.G. A broad HIV-1 inhibitor blocks envelope glycoprotein transitions critical for entry. Nat. Methods 2014, 10, 845–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuser, M.E.; Murphy, M.B.; Rashad, A.A.; Cocklin, S.; Narayan, M. Kinetic Characterization of Novel HIV-1 Entry Inhibitors: Discovery of a Relationship between Off-Rate and Potency. Molecules 2018, 23, 1940. [Google Scholar] [CrossRef]
- Tuyishime, M.; Danish, M.; Princiotto, A.; Mankowski, M.K.; Lawrence, R.; Lombart, H.-G.; Esikov, K.; Berniac, J.; Liang, K.; Ji, J.; et al. Discovery and optimization of novel small-molecule HIV-1 entry inhibitors using field-based virtual screening and bioisosteric replacement. Bioorganic Med. Chem. Lett. 2014, 24, 5439–5445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.-T.; Wang, T.; O’Dell, S.; Louder, M.K.; Schön, A.; Cheung, C.S.F.; Chuang, G.-Y.; Druz, A.; Lin, B.; McKee, K.; et al. Lattice engineering enables definition of molecular features allowing for potent small-molecule inhibition of HIV-1 entry. Nat. Commun. 2019, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Herschhorn, A.; Gu, C.; Moraca, F.; Ma, X.; Farrell, M.; Smith, A.B., 3rd; Pancera, M.; Kwong, P.D.; Schon, A.; Freire, E.; et al. The β20-β21 of gp120 is a regulatory switch for HIV-1 Env conformational transitions. Nat. Commun. 2017, 8, 1049. [Google Scholar] [CrossRef] [PubMed]
- Ozorowski, G.; Pallesen, J.; De Val, N.; Lyumkis, D.; Cottrell, C.A.; Torres, J.L.; Copps, J.; Stanfield, R.L.; Cupo, A.; Pugach, P.; et al. Open and closed structures reveal allostery and pliability in the HIV-1 envelope spike. Nat. Cell Boil. 2017, 547, 360–363. [Google Scholar] [CrossRef] [Green Version]
- Sanders, R.W.; van Gils, M.J.; Derking, R.; Sok, D.; Ketas, T.J.; Burger, J.A.; Ozorowski, G.; Cupo, A.; Simonich, C.; Goo, L.; et al. HIV-1 neutralizing antibodies induced by native-like envelope trimers. Science 2015, 349, aac4223. [Google Scholar] [CrossRef] [PubMed]
- Pugach, P.; Ozorowski, G.; Cupo, A.; Ringe, R.; Yasmeen, A.; de Val, N.; Derking, R.; Kim, H.J.; Korzun, J.; Golabek, M.; et al. A Native-Like SOSIP.664 Trimer Based on an HIV-1 Subtype B env Gene. J. Virol. 2015, 89, 3380–3395. [Google Scholar] [CrossRef]
- Munro, J.B.; Gorman, J.; Ma, X.; Zhou, Z.; Arthos, J.; Burton, D.R.; Koff, W.C.; Courter, J.R.; Smith, A.B., 3rd; Kwong, P.D.; et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 2014, 346, 759–763. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Yang, Z.; Zhang, Z.; Gong, Y.-F.; Riccardi, K.A.; Lin, P.-F.; Parker, D.D.; Rahematpura, S.; Mathew, M.; Zheng, M.; et al. Inhibitors of HIV-1 attachment. Part 10. The discovery and structure–activity relationships of 4-azaindole cores. Bioorganic Med. Chem. Lett. 2013, 23, 213–217. [Google Scholar] [CrossRef]
- Wang, T.; Yin, Z.; Zhang, Z.; Bender, J.A.; Yang, Z.; Johnson, G.; Yang, Z.; Zadjura, L.M.; D’Arienzo, C.J.; Parker, D.D.; et al. Inhibitors of Human Immunodeficiency Virus Type 1 (HIV-1) Attachment. 5. An Evolution from Indole to Azaindoles Leading to the Discovery of 1-(4-Benzoylpiperazin-1-yl)-2-(4,7-dimethoxy-1H-pyrrolo[2,3-c]pyridin-3-yl)ethane-1,2-dione (BMS-488043), a Drug Candidate That Demonstrates Antiviral Activity in HIV-1-Infected Subjects. J. Med. Chem. 2009, 52, 7778–7787. [Google Scholar]
- Meanwell, N.A.; Wallace, O.B.; Fang, H.; Wang, H.; Deshpande, M.; Wang, T.; Yin, Z.; Zhang, Z.; Pearce, B.C.; James, J.; et al. Inhibitors of HIV-1 attachment. Part 2: An initial survey of indole substitution patterns. Bioorganic Med. Chem. Lett. 2009, 19, 1977–1981. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-benzoyl-1-[(4-methoxy-1H- pyrrolo[2,3-b]pyridin-3-yl)oxoacetyl]-2-(R)-methylpiperazine (BMS-378806): A novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J. Med. Chem. 2003, 46, 4236–4239. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.P.; Francis, A.C.; Meuser, M.E.; Mankowski, M.; Ptak, R.G.; Rashad, A.A.; Melikyan, G.B.; Cocklin, S. Exploring Modifications of an HIV-1 Capsid Inhibitor: Design, Synthesis, and Mechanism of Action. J. Drug Des. Res. 2018, 5. [Google Scholar]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr Is Required for Efficient Replication of Human Immunodeficiency Virus Type-1 in Mononuclear Phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, J.; Coligan, J.; Barin, F.; McLane, M.; Sodroski, J.; Rosen, C.; Haseltine, W.; Lee, T.; Essex, M. Major glycoprotein antigens that induce antibodies in AIDS patients are encoded by HTLV-III. Science 1985, 228, 1091–1094. [Google Scholar] [CrossRef]
- Koyanagi, Y.; O’Brien, W.; Zhao, J.; Golde, D.; Gasson, J.; Chen, I. Cytokines alter production of HIV-1 from primary mononuclear phagocytes. Science 1988, 241, 1673–1675. [Google Scholar] [CrossRef]
- Koyanagi, Y.; Miles, S.; Mitsuyasu, R.; Merrill, J.; Vinters, H.; Chen, I. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science 1987, 236, 819–822. [Google Scholar] [CrossRef]
- O’Brien, W.A.; Koyanagi, Y.; Namazie, A.; Zhao, J.-Q.; Diagne, A.; Ldler, K.; A Zack, J.; Chen, I.S.Y. HIV-1 tropism for mononuclear phagocytes can be determined by regions of gp120 outside the CD4-binding domain. Nat. Cell Boil. 1990, 348, 69–73. [Google Scholar] [CrossRef]
- Li, Y.; Hui, H.; Burgess, C.J.; Price, R.W.; Sharp, P.M.; Hahn, B.H.; Shaw, G.M. Complete nucleotide sequence, genome organization, and biological properties of human immunodeficiency virus type 1 in vivo: Evidence for limited defectiveness and complementation. J. Virol. 1992, 66, 6587–6600. [Google Scholar] [PubMed]
- Li, Y.; Kappes, J.C.; A Conway, J.; Price, R.W.; Shaw, G.M.; Hahn, B.H. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: Identification of replication-competent and -defective viral genomes. J. Virol. 1991, 65, 3973–3985. [Google Scholar]
- Björndal, A.; Deng, H.; Jansson, M.; Fiore, J.R.; Colognesi, C.; Karlsson, A.; Albert, J.; Scarlatti, G.; Littman, D.R.; Fenyö, E.M. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 1997, 71, 7478–7487. [Google Scholar] [PubMed]
- Zentner, I.; Sierra, L.-J.; Fraser, A.K.; Maciunas, L.; Mankowski, M.K.; Vinnik, A.; Fedichev, P.; Ptak, R.G.; Martín-García, J.; Cocklin, S.; et al. Identification of a Small-Molecule Inhibitor of HIV-1 Assembly that Targets the Phosphatidylinositol (4,5)-bisphosphate Binding Site of the HIV-1 Matrix Protein. ChemMedChem 2013, 8, 426–432. [Google Scholar] [CrossRef]
- Kortagere, S.; Madani, N.; Mankowski, M.K.; Schon, A.; Zentner, I.; Swaminathan, G.; Princiotto, A.; Anthony, K.; Oza, A.; Sierra, L.J.; et al. Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J. Virol. 2012, 86, 8472–8481. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.; Schubert, D.; LaBonte, J.; Munson, L.; Gibson, S.; Scammell, J.; Ferrigno, P.; Sodroski, J. Species-Specific, Postentry Barriers to Primate Immunodeficiency Virus Infection. J. Virol. 1999, 73, 10020–10028. [Google Scholar]
- Marcon, L.; Choe, H.; A Martin, K.; Farzan, M.; Ponath, P.D.; Wu, L.; Newman, W.; Gerard, N.; Gerard, C.; Sodroski, J. Utilization of C-C chemokine receptor 5 by the envelope glycoproteins of a pathogenic simian immunodeficiency virus, SIVmac239. J. Virol. 1997, 71, 2522–2527. [Google Scholar]
- Ratner, L.; Haseltine, W.; Patarca, R.; Livak, K.J.; Starcich, B.; Josephs, S.F.; Doran, E.R.; Rafalski, J.A.; Whitehorn, E.A.; Baumeister, K.; et al. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nat. Cell Boil. 1985, 313, 277–284. [Google Scholar] [CrossRef]
- Cocklin, S.; Gopi, H.; Querido, B.; Nimmagadda, M.; Kuriakose, S.; Cicala, C.; Ajith, S.; Baxter, S.; Arthos, J.; Martín-García, J.; et al. Broad-Spectrum Anti-Human Immunodeficiency Virus (HIV) Potential of a Peptide HIV Type 1 Entry Inhibitor. J. Virol. 2007, 81, 3645–3648. [Google Scholar] [CrossRef]
- He, J.; Choe, S.; Walker, R.; Di Marzio, P.; O Morgan, D.; Landau, N.R. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995, 69, 6705–6711. [Google Scholar]
- Bravman, T.; Bronner, V.; Lavie, K.; Notcovich, A.; Papalia, G.A.; Myszka, D.G. Exploring “one-shot” kinetics and small molecule analysis using the ProteOn XPR36 array biosensor. Anal. Biochem. 2006, 358, 281–288. [Google Scholar] [CrossRef] [PubMed]
- De Taeye, S.W.; Ozorowski, G.; de la Pena, A.T.; Guttman, M.; Julien, J.P.; van den Kerkhof, T.L.; Burger, J.A.; Pritchard, L.K.; Pugach, P.; Yasmeen, A.; et al. Immunogenicity of Stabilized HIV-1 Envelope Trimers with Reduced Exposure of Non-neutralizing Epitopes. Cell 2015, 163, 1702–1715. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ka (M−1s−1) | kd (s−1) | KD |
|---|---|---|---|
| SC11 | 3.83 ± 1.12 × 103 | 5.02 ± 2.67 × 10−4 | 0.131 µM |
| SC15 | 3.01 ± 0.188 × 105 | 5.44 ± 0.677 × 10−3 | 0.0181 µM |
| SC28 | 1.39 ± 0.14 × 104 | 6.99 ± 0.43 × 10−3 | 0.511 µM |
| SC45 | 1.38 ± 0.045 × 104 | 1.52 ± 0.0294 × 10−2 | 1.10 µM |
| Compound | Calculated Overall Ligand-Protein Interaction Energy Kcal/Mol |
|---|---|
| SC28 | −16.13700 |
| SC49 | −6.58188 |
| SC50 | −12.87666 |
| SC52 | −2.25288 |
| SC55 | −10.12901 |
| SC56 | −18.37654 |
| Compound | ka (M−1s−1) | kd (s−1) | KD (µM) |
|---|---|---|---|
| SC28 | 1.39 ± 0.14 × 104 | 6.99 ± 0.43 × 10−3 | 0.504 µM |
| SC49 | 9.49 ± 1.6 × 102 | 3.43 ± 0.36 × 10−2 | 36.1 µM |
| SC50 | N.D. | N.D. | 114 ± 51 µM |
| SC52 | N.D. | N.D. | 23.7 ± 2.4 µM |
| SC55 | N.D. | N.D. | 11.4 ± 3.7 µM |
| SC56 | 1.22 ± 0.067 × 107 | 6.39 ± 0.31 × 10−3 | 0.526 nM |
| Compound | IC50 B41 (µM) | IC50 HxBc2 (µM) | IC50 JRCSF (µM) | IC50 JRFL (µM) | IC50 YU-2 (µM) | Median IC50 (µM) | CC50 (µM) | Therapeutic Index (CC50/IC50) |
|---|---|---|---|---|---|---|---|---|
SC28 (parental) | 0.035 ± 0.0062 | 0.021 ± 0.005 | 0.026 ± 0.009 | 0.17 ± 0.05 | 0.48 ± 0.05 | 0.15 ± 0.17 | 190 ± 54 | 1291.21 |
SC49 | 26 ± 19 | 62 ± 1.4 | NA | 33.32 ± 11.56 | 57.2 ± 26.5 | 45 ± 15 | 326 ± 81 | 7.31 |
SC50 | 7.1 ± 4.2 | 6.9 ± 5.2 | 28.92 ± 5.62 | 24.28 ± 6.2 | 22.6 ± 4.2 | 18 ± 9 | 120 ± 18 | 6.63 |
SC52 | 0.43 ± 0.1 | 0.44 ± 0.17 | 2.02 ± 0.25 | NA | 1.35 ± 0.44 | 1.06 ± 0.668 | 9.2 ± 1.1 | 8.65 |
SC55 | 2.02 ± 0.81 | 0.73 ± 0.56 | 0.45 ± 0.35 | 0.45 ± 0.33 | 1.15 ± 0.81 | 0.96 ± 0.59 | 73 ± 14 | 75.85 |
SC56 | 0.051 ± 0.008 | 0.011 ± 0.003 | 0.046 ± 0.009 | 0.13 ± 0.015 | 0.11 ± 0.049 | 0.0698 ± 0.045 | 94 ± 10 | 1344.71 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meuser, M.E.; Rashad, A.A.; Ozorowski, G.; Dick, A.; Ward, A.B.; Cocklin, S. Field-Based Affinity Optimization of a Novel Azabicyclohexane Scaffold HIV-1 Entry Inhibitor. Molecules 2019, 24, 1581. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24081581
Meuser ME, Rashad AA, Ozorowski G, Dick A, Ward AB, Cocklin S. Field-Based Affinity Optimization of a Novel Azabicyclohexane Scaffold HIV-1 Entry Inhibitor. Molecules. 2019; 24(8):1581. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24081581
Chicago/Turabian StyleMeuser, Megan E., Adel A. Rashad, Gabriel Ozorowski, Alexej Dick, Andrew B. Ward, and Simon Cocklin. 2019. "Field-Based Affinity Optimization of a Novel Azabicyclohexane Scaffold HIV-1 Entry Inhibitor" Molecules 24, no. 8: 1581. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24081581