
Super Secondary Structures of Proteins with Post-Translational Modifications in Colon Cancer

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
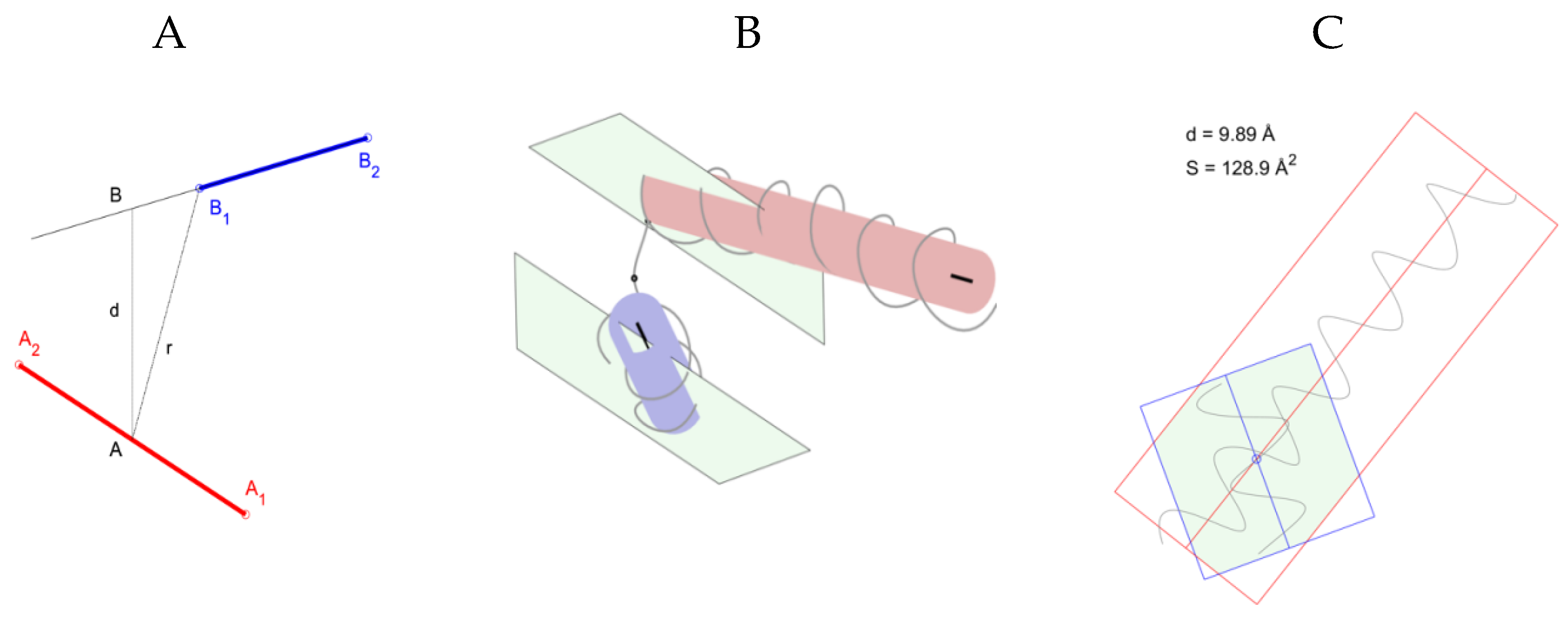
2.1. Identification of Helical Pairs in Protein Structures
2.2. Recognition and Selection of Protein Structures
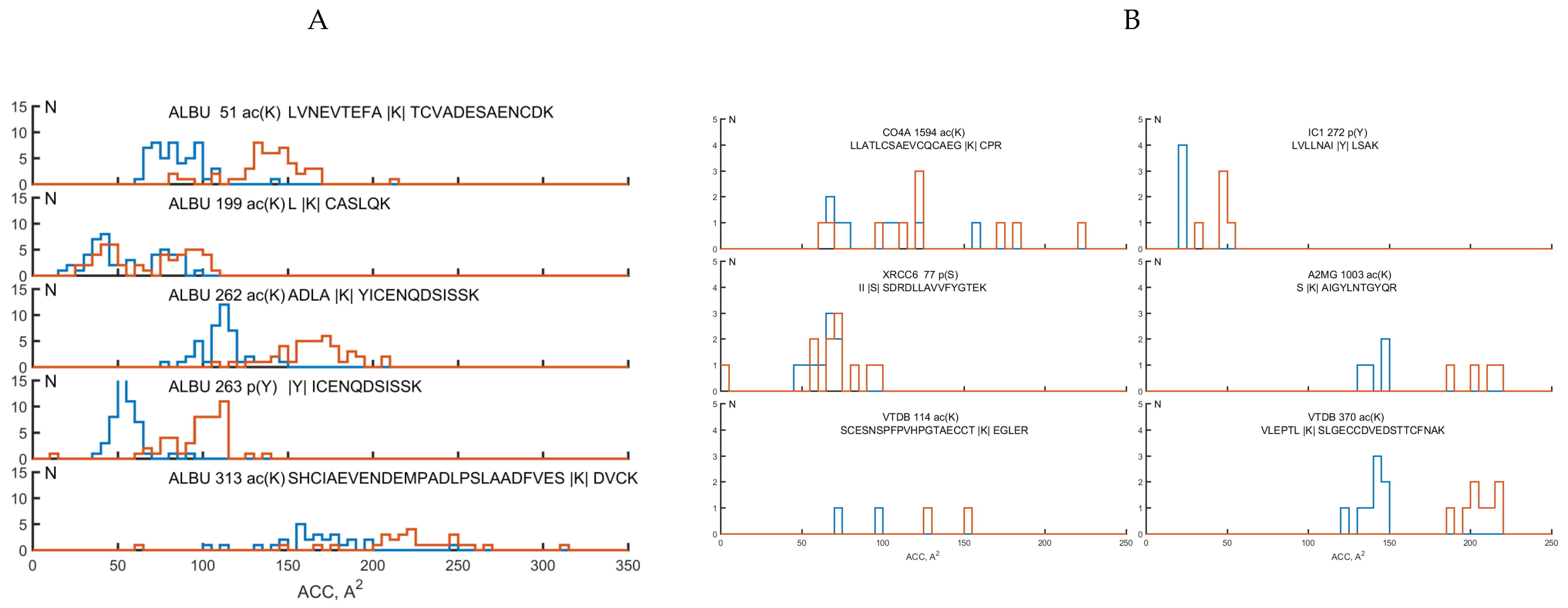
2.3. Analysis of Structural Motifs of Protein Molecules Containing PTM Associated with the Development of Colorectal Cancer
2.4. The Biological Significance of PTM in Target Proteins Detected in Samples of Patients with Colorectal Cancer
3. Materials and Methods
3.1. Demography
3.2. Sample Preparation for MS Analysis
3.3. Mass Spectrometry Protein Registration
3.4. Protein Identification and Criteria Selection for Post-translational Modifications
3.5. Analysis of Post-translational Protein Modifications
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharma, B.S.; Prabhakaran, V.; Desai, A.P.; Bajpai, J.; Verma, R.J.; Swain, P.K. Post-Translational Modifications (PTMs), from a Cancer Perspective: An Overview. Oncogen 2019, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Villanueva, J.F.; Díaz-Molina, R.; García-González, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karve, T.; Cheema, A. Small Changes Huge Impact: The Role of Protein Posttranslational Modifications in Cellular Homeostasis and Disease. J. Amino Acids 2011, 2011, 207691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Post-Translational Modifications in Health and Disease; Vidal, C.J. (Ed.) Springer: New York, NY, USA, 2011. [Google Scholar] [CrossRef]
- Virág, D.; Dalmadi-Kiss, B.; Vékey, K.; Drahos, L.; Klebovich, I.; Antal, I.; Ludányi, K. Current Trends in the Analysis of Post-Translational Modifications. Chromatographia 2020, 83, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bidlingmaier, S.; Liu, B. Identification of Posttranslational Modification-Dependent Protein Interactions Using Yeast Surface Displayed Human Proteome Libraries. Methods Mol. Biol. Clifton NJ 2015, 1319, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Sundararajan, N.; Mao, D.; Chan, S.; Koo, T.-W.; Su, X.; Sun, L.; Zhang, J.; Sung, K.; Yamakawa, M.; Gafken, P.R.; et al. Ultrasensitive Detection and Characterization of Posttranslational Modifications Using Surface-Enhanced Raman Spectroscopy. Anal. Chem. 2006, 78, 3543–3550. [Google Scholar] [CrossRef]
- Li, X.; Kapoor, T.M. Approach to Profile Proteins That Recognize Post-Translationally Modified Histone “Tails”. J. Am. Chem. Soc. 2010, 132, 2504–2505. [Google Scholar] [CrossRef] [Green Version]
- Khidekel, N.; Hsieh-Wilson, L.C. A ‘Molecular Switchboard’—Covalent Modifications to Proteins and Their Impact on Transcription. Org. Biomol. Chem. 2004, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- McDonald, W.H.; Yates, J.R. Shotgun Proteomics and Biomarker Discovery. Available online: https://www.hindawi.com/journals/dm/2002/505397/ (accessed on 18 June 2020). [CrossRef] [Green Version]
- Martín-Bernabé, A.; Balcells, C.; Tarragó-Celada, J.; Foguet, C.; Bourgoin-Voillard, S.; Seve, M.; Cascante, M. The Importance of Post-Translational Modifications in Systems Biology Approaches to Identify Therapeutic Targets in Cancer Metabolism. Curr. Opin. Syst. Biol. 2017, 3, 161–169. [Google Scholar] [CrossRef]
- Hitosugi, T.; Chen, J. Post-Translational Modifications and the Warburg Effect. Oncogene 2014, 33, 4279–4285. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of Colorectal Cancer: Incidence, Mortality, Survival, and Risk Factors. Przeglad Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Efimov, A.V. Standard Structures in Proteins. Prog. Biophys. Mol. Biol. 1993, 60, 201–239. [Google Scholar] [CrossRef]
- Brazhnikov, E.; Efimov, A. Structure of α-α-Hairpins with Short Connections in Globular Proteins. Mol. Biol. 2001, 35, 89–97. [Google Scholar] [CrossRef]
- Efimov, A.V. l-shaped structure from two alpha-helices with a proline residue between them. Mol. Biol. (Mosk.) 1992, 26, 1370–1376. [Google Scholar] [PubMed]
- Kopylov, A.T.; Stepanov, A.A.; Malsagova, K.A.; Soni, D.; Kushlinsky, N.E.; Enikeev, D.V.; Potoldykova, N.V.; Lisitsa, A.V.; Kaysheva, A.L. Revelation of Proteomic Indicators for Colorectal Cancer in Initial Stages of Development. Mol. Basel Switz. 2020, 25, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikhonov, D.A.; Kulikova, L.I.; Efimov, A.V. Statistical Analysis of the Internal Distances of Helical Pairs in Protein Molecules. Math. Biol. Bioinforma. 2019, 14, t18–t36. [Google Scholar] [CrossRef] [Green Version]
- Tikhonov, D.A. The Study of Interhelical Angles in the Structural Motifs Formed by Two Helices. Math. Biol. Bioinforma. 2019, 14, t1–t17. [Google Scholar] [CrossRef]
- Rudnev, V.R.; Pankratov, A.N.; Kulikova, L.I.; Dedus, F.F.; Tikhonov, D.A.E.; Efimov, A.V.E. Recognition and Stability Analysis of Structural Motifs of α-α-Corner Type in Globular Proteins. Mat. Biol. Bioinformatika 2013, 8, 398–406. [Google Scholar] [CrossRef]
- Ying, H.-Q.; Sun, H.-L.; He, B.-S.; Pan, Y.-Q.; Wang, F.; Deng, Q.-W.; Chen, J.; Liu, X.; Wang, S.-K. Circulating Vitamin D Binding Protein, Total, Free and Bioavailable 25-Hydroxyvitamin D and Risk of Colorectal Cancer. Sci. Rep. 2015, 5, 7956. [Google Scholar] [CrossRef]
- Andersen, S.W.; Shu, X.-O.; Cai, Q.; Khankari, N.K.; Steinwandel, M.D.; Jurutka, P.W.; Blot, W.J.; Zheng, W. Total and Free Circulating Vitamin D and Vitamin d–Binding Protein in Relation to Colorectal Cancer Risk in a Prospective Study of African Americans. Cancer Epidemiol. Prev. Biomark. 2017, 26, 1242–1247. [Google Scholar] [CrossRef] [Green Version]
- Buda, A.; Pignatelli, M. Cytoskeletal Network in Colon Cancer: From Genes to Clinical Application. Int. J. Biochem. Cell Biol. 2004, 36, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Darling, V.; Hauke, R.; Tarantolo, S.; Agrawal, D. Immunological Effects and Therapeutic Role of C5a in Cancer. Expert Rev. Clin. Immunol. 2014, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bau, D.-T.; Tsai, C.-W.; Wu, C.-N. Role of the XRCC5/XRCC6 Dimer in Carcinogenesis and Pharmacogenomics. Pharmacogenomics 2011, 12, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Lexa, K.W.; Dolghih, E.; Jacobson, M.P. A Structure-Based Model for Predicting Serum Albumin Binding. PLoS ONE 2014, 9, e93323. [Google Scholar] [CrossRef]
- Bikle, D.D.; Schwartz, J. Vitamin D Binding Protein, Total and Free Vitamin D Levels in Different Physiological and Pathophysiological Conditions. Front. Endocrinol. 2019, 10, 317. [Google Scholar] [CrossRef] [Green Version]
- Nazha, B.; Moussaly, E.; Zaarour, M.; Weerasinghe, C.; Azab, B. Hypoalbuminemia in Colorectal Cancer Prognosis: Nutritional Marker or Inflammatory Surrogate? World J. Gastrointest. Surg. 2015, 7, 370–377. [Google Scholar] [CrossRef]
- Read, J.A.; Choy, S.T.B.; Beale, P.J.; Clarke, S.J. Evaluation of Nutritional and Inflammatory Status of Advanced Colorectal Cancer Patients and Its Correlation with Survival. Nutr. Cancer 2006, 55, 78–85. [Google Scholar] [CrossRef]
- Boonpipattanapong, T.; Chewatanakornkul, S. Preoperative Carcinoembryonic Antigen and Albumin in Predicting Survival in Patients with Colon and Rectal Carcinomas. J. Clin. Gastroenterol. 2006, 40, 592–595. [Google Scholar] [CrossRef]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.-F.; Lim, H.-W.; Liu, S.; et al. Akt-Dependent Metabolic Reprogramming Regulates Tumor Cell Histone Acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Regulation of Cancer Cell Metabolism by Hypoxia-Inducible Factor 1. Semin. Cancer Biol. 2009, 19, 12–16. [Google Scholar] [CrossRef]
- Sutendra, G.; Dromparis, P.; Kinnaird, A.; Stenson, T.; Haromy, A.; Parker, J.; McMurtry, M.; Michelakis, E. Mitochondrial Activation by Inhibition of PDKII Suppresses HIF1a Signaling and Angiogenesis in Cancer. Oncogene 2012, 32, 1638–1650. [Google Scholar] [CrossRef] [Green Version]
- Liyasova, M.S.; Schopfer, L.M.; Lockridge, O. Reaction of Human Albumin with Aspirin in Vitro: Mass Spectrometric Identification of Acetylated Lysines 199, 402, 519, and 545. Biochem. Pharmacol. 2010, 79, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Lockridge, O.; Xue, W.; Mulvenon, A.; Grigoryan, H.; Ding, S.-J.; Schopfer, L.; Hinrichs, S.; Masson, P. Pseudo-Esterase Activity of Human Albumin. J. Biol. Chem. 2008, 283, 22582–22590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finamore, F.; Priego-Capote, F.; Gluck, F.; Zufferey, A.; Fontana, P.; Sanchez, J.-C. Impact of High Glucose Concentration on Aspirin-Induced Acetylation of Human Serum Albumin: An in Vitro Study. EuPA Open Proteomics 2014, 3, 100–113. [Google Scholar] [CrossRef]
- Martin, S.C.; Ekman, P. In Vitro Phosphorylation of Serum Albumin by Two Protein Kinases: A Potential Pitfall in Protein Phosphorylation Reactions. Anal. Biochem. 1986, 154, 395–399. [Google Scholar] [CrossRef]
- Parodi, A.; Miao, J.; Soond, S.M.; Rudzińska, M.; Zamyatnin, A.A. Albumin Nanovectors in Cancer Therapy and Imaging. Biomolecules 2019, 9, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in Cancer. Protein J. 2017, 36, 1–6. [Google Scholar] [CrossRef]
- Deutsch, E.; Lane, L.; Overall, C.; Bandeira, N.; Baker, M.; Pineau, C.; Moritz, R.; Corrales, F.; Orchard, S.; Eyk, J.; et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 3.0. J. Proteome Res. 2019, 18, 4108–4116. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Sayle, R.A.; Milner-White, E.J. RASMOL: Biomolecular Graphics for All. Trends Biochem. Sci. 1995, 20, 374. [Google Scholar] [CrossRef]
Sample Availability: The mass spectrometry proteomics data are available via ProteomeXchange repository with the dataset identifier PXD015163 (https://www.ebi.ac.uk/pride/archive/projects/PXD015163). |



{kind=link}
{kind=link}
| Protein Name | PTM SEQQ | Nprot | Npass | SS of PTM via DSSP | ACC, Å2 | std |
|---|---|---|---|---|---|---|
| VTDB | vleptl|Ac(k)||slgeccdvedsttcfnak | 6 | 8 | H:8 | 136.8 | 8.8 |
| VTDB | scesnspfpvhpgtaecct|Ac(k)||egler | 6 | 2 | S:2 | 82.0 | 17.0 |
| CO4A | llatlcsaevcqcaeg|Ac(k)||cpr | 6 | 10 | C:4;G:2;S:3;T:1 | 91.3 | 30.5 |
| XRCC6 | ii|P(S)|sdrdllavvfygtek | 6 | 11 | T:11 | 57.0 | 20.8 |
| IC1 | lvllnai|P(Y)|lsak | 3 | 5 | E:5 | 23.8 | 4.7 |
| A2MG | s|Ac(k)|aigylntgyqr | 1 | 4 | H:4 | 139.8 | 6.1 |
| ALBU | l|Ac(k)|caslqk | 107 | 157 | H:157 | 48.0 | 20.4 |
| ALBU | shciaevendempadlpslaadfves|Ac(k)|dvck | 106 | 154 | S:90;T:64 | 123.3 | 47.8 |
| ALBU | adla|Ac(k)|yicenqdsissk | 106 | 156 | H:155;T:1 | 99.8 | 27.0 |
| ALBU | |P(Y)|icenqdsissk | 106 | 156 | H:155;T:1 | 53.9 | 10.9 |
| ALBU | lvnevtefa|Ac(k)|tcvadesaencdk | 106 | 156 | G:1;H:148;T:7 | 82.2 | 17.5 |
| Protein Name | PDB AC | PTM SEQQ | Sequence of the Helical Pair | Locus | d | r | ϕ | θ | S | P | Np | Motif |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VTDB | 1J78 | VLEPTL|Ac(k)|SLGECCDVEDSTTCFNAK | NTkvmdkytfelsRRTHLPevflskvleptl|k|slgEC | 323–359 (354) | 11.4 | 11.4 | 36.3 | −35.9 | 148.1 | 48.1 | 6 | α-α-corner |
| LPevflskvleptl|k|slgECCDVEDsttcfnakgpllkkelssfidkgqelCA | 340–392 (354) | 9.0 | 9.3 | 13.5 | −20.3 | 159.9 | 59.9 | 7 | α-α-hairpin | |||
| SCESNSPFPVHPGTAECCT|Ac(k)|EGLER | PGtaecCT|K|EglerklcmaaLK | 106–127 (114) | 0.7 | 8.7 | 13.5 | −1.9 | 11.3 | 14.2 | 4 | α-α-hairpin | ||
| |K|EglerklcmaaLKHQPQEFPTYVEPTndeiceafrkDp | 114–152 (114) | 16.4 | 37.5 | 33.1 | 21.7 | 0 | 0 | 15 | ||||
| CO4A | 5JPN | LLATLCSAEVCQCAEG|Ac(K)|CPR | CSaevcqcaEG|K|CPRQRRALERGLQDEDGyrmkfacYY | 1583–1620 (1594) | 10.1 | 23.5 | 112.4 | 148.4 | 0 | 0 | 20 | helical pair |
| XRCC6 | 1JEQ | II|P(s)|SDRDLLAVVFYGTEK | SKamfESQSEDELTpfdmsiqciqsvyiskii|S|S | 45–78 (77) | 0.49 | 4.42 | 109.8 | 22.91 | 1.64 | 6.93 | 9 | helical pair |
| LTpfdmsiqciqsvyiskii|S|SDRDLLAVVFYGTEKDKNSVNFKNIYVLQELDNPGakrileldQF | 57–122 (77) | 8.95 | 9.69 | 12.69 | 11.23 | 73.06 | 35.18 | 36 | α-α-hairpin | |||
| IC1 | 5DU3 | LVLLNAI|P(y)|LSAK | NNsdanlelintwvaknTNNKISRLLDSLPSDTRLVLLNAI|Y|LSAKWKTTFDpkkTR | 231–287 (272) | 8.0 | 30.9 | 79.7 | −25.0 | 0 | 0 | 35 | helical pair |
| A2MG | 4ACQ | S|Ac(k)|AIGYLNTGYQR | XNmvlfapniyvldylneTQQLTpeiks|k|aigylntgyqrqln | 975–1017(1003) | 9.3 | 9.3 | 32.2 | 22.7 | 179.8 | 58.9 | 5 | α-α-hairpin |
| ALBU | 1AO6 | L|Aс(к)|CASLQK | FYapellffakrykaaftecCQAADkaacllpkldelrdegkassakqrl|k|caslqkfGe | 149–208 (199) | 7.8 | 9.8 | 1.8 | 1.6 | 139.1 | 61.9 | 5 | α-α-hairpin |
| ADkaacllpkldelrdegkassakqrl|k|caslqkfGerafkawavarlsqrFP | 172–224 (199) | 2.3 | 5.0 | 52.1 | −29.1 | 25.2 | 22.1 | 1 | V-structure | |||
| SHCIAEVENDEMPADLPSLAADFVES|Ac(k)|DVCK | SLaadfVES|K|DvcknyaeAk | 304–323 (313) | 0.6 | 8.1 | 105.4 | 38.9 | 0 | 0 | 5 | l-structure | ||
| |K|DvcknyaeAkdvflgmflyeyarRH | 313–338 (313) | 0.5 | 4.8 | 64.8 | −7.3 | 15.2 | 16.1 | 1 | V-structure | |||
| ADLA|Ac(k)|YICENQDSISSK | AEfaevsklvtdltkvhteccHGDllecaddradla|k|yiceNq | 226–268 (262) | 6.1 | 7.2 | 19.7 | 16.7 | 90.5 | 53.7 | 3 | α-α-hairpin | ||
| GDllecaddradla|k|yiceNqdsiSS | 248–273 (262) | 1.2 | 4.6 | 99.0 | 33.4 | 5.2 | 9.3 | 1 | l-structure | |||
| |P(y)|ICENQDSISSK | AEfaevsklvtdltkvhteccHGDllecaddradlak|y|iceNq | 226–268 (263) | 6.1 | 7.2 | 19.7 | 16.7 | 90.5 | 53.7 | 3 | α-α-hairpin | ||
| GDllecaddradlak|y|iceNqdsiSS | 248–273 (263) | 1.2 | 4.6 | 98.9 | 33.4 | 5.2 | 9.3 | 1 | l-structure | |||
| LVNEVTEFA|Ac(k)|TCVADESAENCDK | SevahrfkdlgeenfkalvliafaqyLQQCPfedhvklvnevtefa|k|tcvaDE | 5–57 (51) | 9.4 | 9.4 | 9.1 | −9.0 | 304.9 | 76.5 | 5 | α-α-corner | ||
| CPfedhvklvnevtefa|k|tcvaDESAENCDKSlhtlfgdklctvaTL | 34–80 (51) | 10.9 | 10.9 | 32.5 | −28.7 | 171.0 | 53.0 | 10 | α-α-corner |
| Protein Name | PTM Position* | Binding Partner | Binding Site Position, a.a. | Structural Localization of PTM and Binding Site | The Role of Partner in Oncogenesis | Estimated Role of PTM |
|---|---|---|---|---|---|---|
| Vitamin d-binding protein (VTDB) | K370-ac K114-ac | Vitamin d metabolites | 35–49 | Spatially removed | Epidemiological studies suggest that the risk of developing colorectal cancer (CRC) is associated with a decrease in the level of 25-hydroxyvitamin d (25 (OH) d) in the blood [21,22]. However, little is known regarding the role of VDBP in colorectal carcinogenesis [21]. | Acetylation of VTDB is a response to the processes accompanying oncogenesis, including an increase in the level of 25 (OH) d, actin, C5a |
| Actin | 373–403 | Spatially brought together | In the case of CRC, cells undergo changes in the cytoskeleton formed by actin, and cell adhesion decreases during invasion [23]. | |||
| C5a/C5a des Arg | 130–149 | Spatially removed | The complement factor C5a is involved in the processes of tumor formation and profiling of cells [24]. | |||
| Complement C4-A (CO4A) | K370-ac | Immune aggregates or protein antigens | 1125 | Spatially removed | CO4A is involved in the formation of immune complexes. | Regulation of binding of immune aggregates or protein antigens, the content of which in the blood increases in response to tumor growth. |
| X-ray repair cross-complementing protein 6 (XRCC6) | S77-p | XRCC5 and DNA | 31 | Spatially brought together | XRCC5/6 dimer is involved in DNA repair mechanisms. DNA modification and binding sites in the protein structure are in the same domain and in close proximity to each other (UniProtKB - P12956 (XRCC6_HUMAN) https://www.uniprot.org/uniprot/P12956) | One of the main etiologies of the onset and development of cancer are genetic polymorphisms, which are associated with the regulation of cell proliferation and differentiation. Cells are likely to react to genetic damage and their ability to maintain genomic stability using the DNA repair mechanism through the XRCC5/XRCC6 dimer [25] is of paramount importance. |
| Plasma protease C1 inhibitor (IC1) | Y297-p | Chymotrypsin | 465–467 | Spatially removed | IC1 is involved in the regulation of the activity of the C1r or C1s complex and can play an important role in the activation of the complement system. Very efficient inhibitor of FXIIa. Inhibits chymotrypsin and kallikrein | Tyrosine phosphorylation of IC1 is probably the body’s response to the processes that accompany oncogenesis. |
| Serum albumin (ALBU) | K223-ac K341-ac K286-ac K75-ac Y287-S82-p K257-glygly | Most anions, hormones, heme, and lipophilic xenobiotics [26] | 218–257 (Site I) | Spatially brought together | The albumin/ligand complexation plays an important role in transport, regulation of metabolite and xenobiotic activity. Albumin binds most anions, independent of the hydrophobic character of the ligand side group. | Modification of lysine residues in ALBU may be caused by their increased esterase activity in patients with CRC. Enhanced non-specific phosphorylation of ALBU, especially in warfarin-binding site, is an effect of the impaired cell signaling network significantly contributing to tumor growth and development. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tikhonov, D.; Kulikova, L.; Kopylov, A.; Malsagova, K.; Stepanov, A.; Rudnev, V.; Kaysheva, A. Super Secondary Structures of Proteins with Post-Translational Modifications in Colon Cancer. Molecules 2020, 25, 3144. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25143144
Tikhonov D, Kulikova L, Kopylov A, Malsagova K, Stepanov A, Rudnev V, Kaysheva A. Super Secondary Structures of Proteins with Post-Translational Modifications in Colon Cancer. Molecules. 2020; 25(14):3144. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25143144
Chicago/Turabian StyleTikhonov, Dmitry, Liudmila Kulikova, Arthur Kopylov, Kristina Malsagova, Alexander Stepanov, Vladimir Rudnev, and Anna Kaysheva. 2020. "Super Secondary Structures of Proteins with Post-Translational Modifications in Colon Cancer" Molecules 25, no. 14: 3144. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25143144