
Workflow for the Quantification of Soluble and Insoluble Carbohydrates in Soybean Seed


Abstract
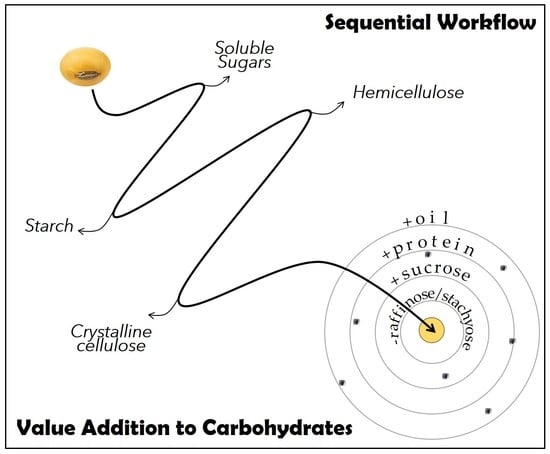
:
1. Introduction
2. Results and Discussion
2.1. Development of a Sequential Protocol for Quantifying Reserve and Structural Carbohydrates in Soybean Seeds
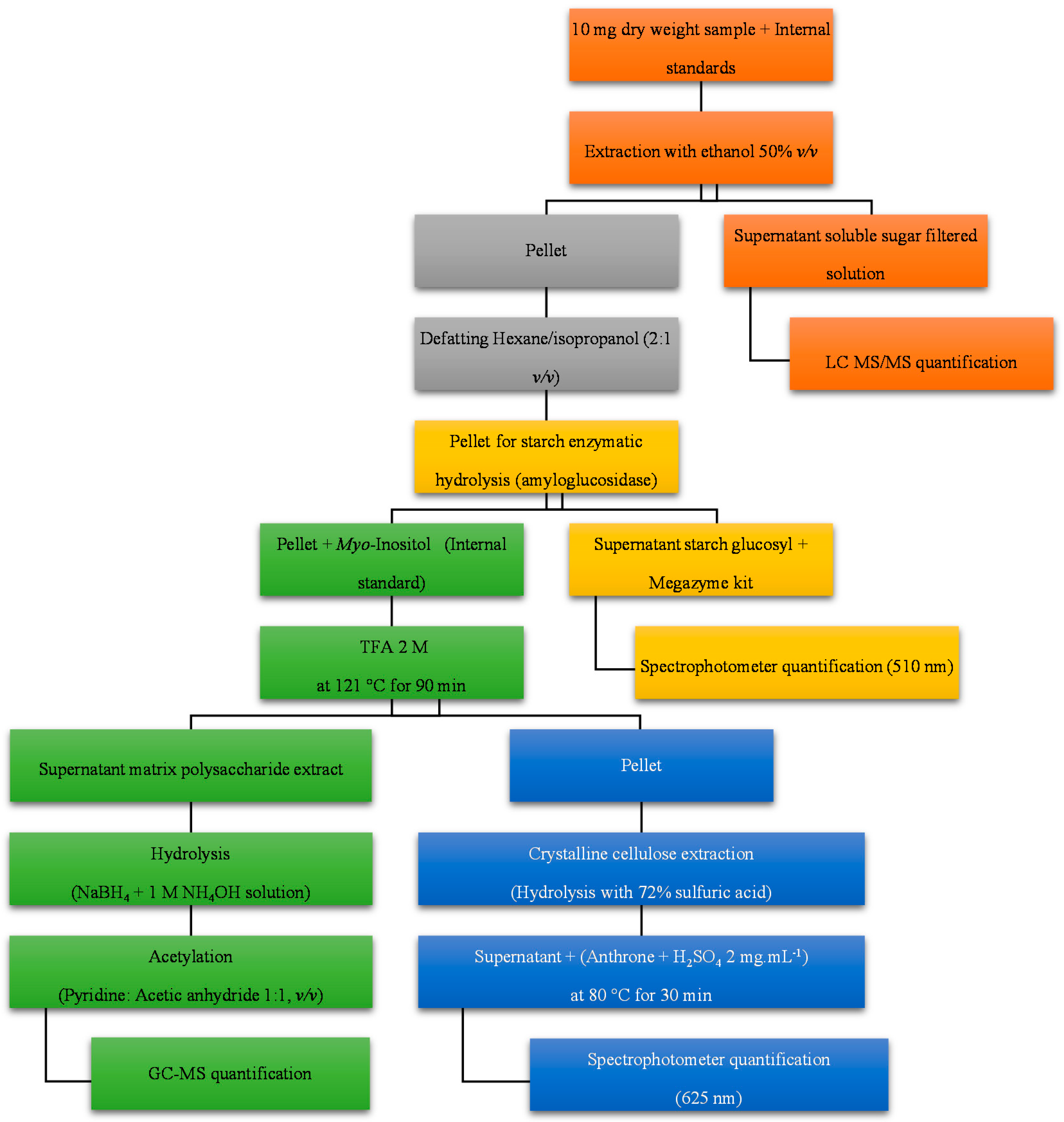
2.1.1. Biological Sample Preparation
2.1.2. Soluble Sugars
2.1.3. Starch
2.1.4. Hemicellulose
2.1.5. Crystalline Cellulose
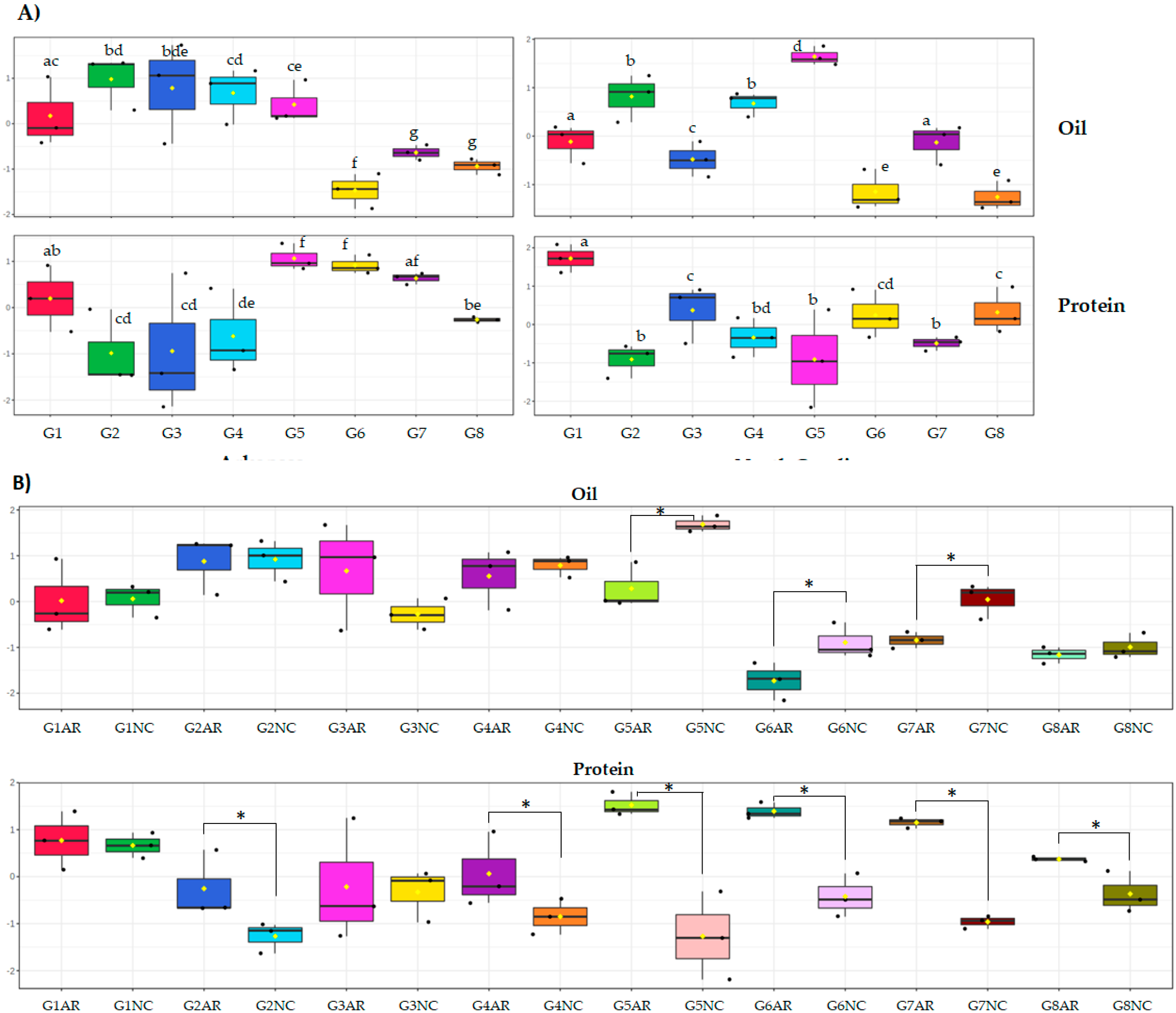
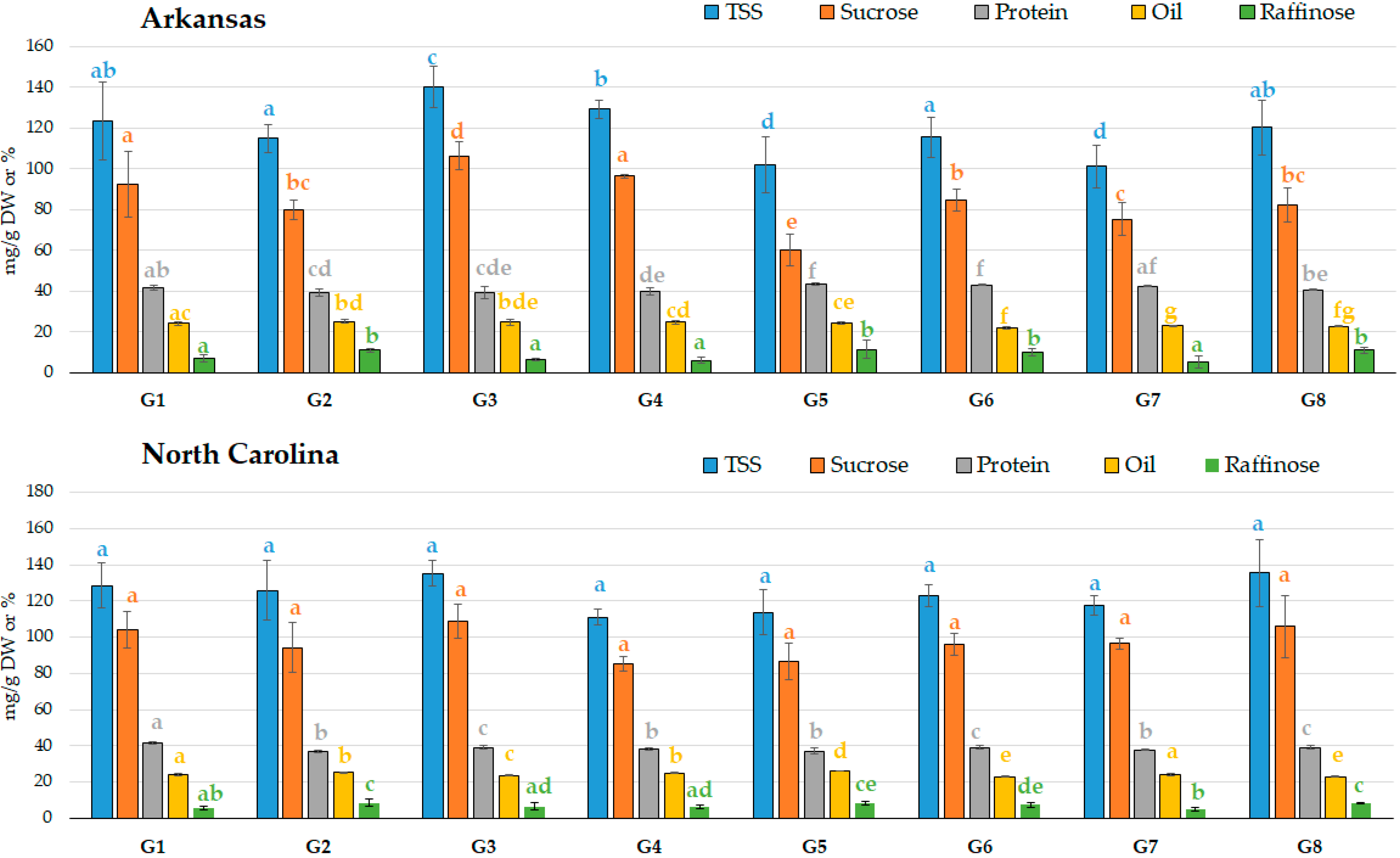
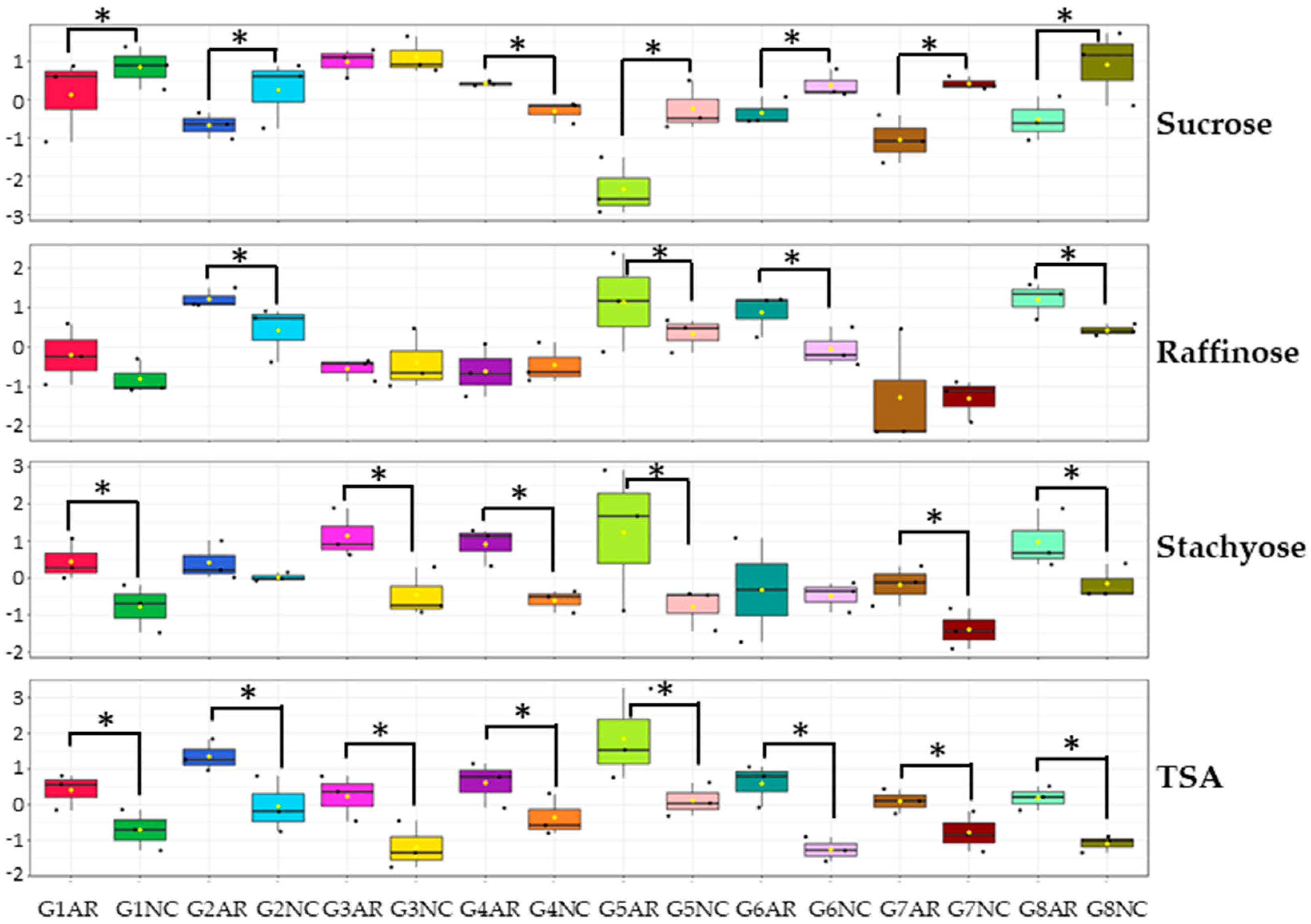
2.2. Application of the Novel Sequential Method to Eight Soybean Genotypes Grown in Two Different Locations
2.2.1. Carbohydrate Quantification in Eight Soybean Genotypes Grown in Two Different Locations
2.2.2. Line Variability and Location Effect
3. Materials and Methods
3.1. Reagents, Chemicals, and Enzymes
3.2. Standards Preparation
3.3. Plant Growing Locations and Conditions
3.4. Seed Oil and Protein Quantification
3.5. Carbohydrate Sequential Extraction
3.5.1. Biological Sample Preparation
3.5.2. Soluble Sugar and Sugar Alcohol Extraction
3.5.3. Defatting
3.5.4. Starch Extraction
3.5.5. Hemicellulose Extraction
3.5.6. Cellulose Extraction
3.6. Carbohydrate Quantification
3.6.1. Soluble Sugar and Sugar Alcohol Quantification by LC–MS/MS
3.6.2. Glucose from Starch Quantification by a Colorimetric Assay
3.6.3. Derivatization of Hemicellulose Components and Quantification by GC–MS
3.6.4. Cellulose-Derived Glucose Quantification by the Anthrone Method
3.7. Determination of Accuracy, Recovery, and Matrix Effect
3.7.1. Accuracy
3.7.2. Recovery
3.7.3. Matrix Effect
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- USDA World Agricultural Production. Available online: https://www.fas.usda.gov/data/world-agricultural-production (accessed on 29 June 2020).
- Voorhees, J.M.; Barnes, M.E.; Chipps, S.R.; Brown, M.L. Bioprocessed soybean meal replacement of fish meal in rainbow trout (Oncorhynchus mykiss) diets. Cogent Food Agric. 2019, 5. [Google Scholar] [CrossRef]
- Stein, H.H.; Berger, L.L.; Drackley, J.K.; Fahey, G.C.; Hernot, D.C.; Parsons, C.M. Nutritional Properties and Feeding Values of Soybeans and Their Coproducts. Soybeans Chem. Prod. Process. Util. 2008, 613–660. [Google Scholar] [CrossRef]
- Assefa, Y.; Purcell, L.C.; Salmeron, M.; Naeve, S.; Casteel, S.N.; Kovács, P.; Archontoulis, S.; Licht, M.; Below, F.; Kandel, H.; et al. Assessing variation in us soybean seed composition (protein and oil). Front. Plant Sci. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Clemente, T.E.; Cahoon, E.B. Soybean oil: Genetic approaches for modification of functionality and total content. Plant Physiol. 2009, 151, 1030–1040. [Google Scholar] [CrossRef] [Green Version]
- Pantalone, V.R. 11-Modern Breeding Approaches to Enhancing Soy Protein Quality; Wilson, R.F.B.T.-D.S., Ed.; AOCS Press: Urbana, IL, USA, 2012; pp. 197–226. ISBN 978-0-9830791-0-1. [Google Scholar]
- Medic, J.; Atkinson, C.; Hurburgh, C.R. Current knowledge in soybean composition. JAOCS, J. Am. Oil Chem. Soc. 2014, 91, 363–384. [Google Scholar] [CrossRef]
- Jo, H.; Lorenz, A.J.; Rainey, K.M.; Shannon, J.G.; Chen, P.; Bilyeu, K.D. Environmental stability study of soybeans with modified carbohydrate profiles in maturity groups 0 to v. Crop Sci. 2019, 59, 1531–1543. [Google Scholar] [CrossRef]
- Dierking, E.C.; Bilyeu, K.D. Association of a Soybean Raffinose Synthase Gene with Low Raffinose and Stachyose Seed Phenotype. Plant Genome 2008, 1, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Hagely, K.B.; Jo, H.; Kim, J.H.; Hudson, K.A.; Bilyeu, K. Molecular-assisted breeding for improved carbohydrate profiles in soybean seed. Theor. Appl. Genet. 2020, 133, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Hou, A.; Chen, P.; Shi, A.; Zhang, B.; Wang, Y.-J. Sugar Variation in Soybean Seed Assessed with a Rapid Extraction and Quantification Method. Int. J. Agron. 2008, 2009, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Jaureguy, L.M.; Rosso, M.L.; Chen, P.; Rainey, K.M. Selection efficiency of high inorganic phosphorus-low stachyose lines from soybean breeding populations of different genetic backgrounds. J. Crop Improv. 2017, 31, 25–38. [Google Scholar] [CrossRef]
- Kambhampati, S.; Aznar-Moreno, J.A.; Hostetler, C.; Caso, T.; Bailey, S.R.; Hubbard, A.H.; Durrett, T.P.; Allen, D.K. On the inverse correlation of protein and oil: Examining the effects of altered central carbon metabolism on seed composition using soybean fast neutron mutants. Metabolites 2020, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Loman, A.A.; Ju, L.K. Soybean carbohydrate as fermentation feedstock for production of biofuels and value-added chemicals. Process Biochem. 2016, 51, 1046–1057. [Google Scholar] [CrossRef]
- Schirmer-Michel, Â.C.; Flôres, S.H.; Hertz, P.F.; Matos, G.S.; Ayub, M.A.Z. Production of ethanol from soybean hull hydrolysate by osmotolerant Candida guilliermondii NRRL Y-2075. Bioresour. Technol. 2008, 99, 2898–2904. [Google Scholar] [CrossRef]
- Liu, H.M.; Li, H.Y. Application and Conversion of Soybean Hulls. In Soybean-The Basis of Yield, Biomass and Productivity; IntechOpen: London, UK, 2017. [Google Scholar]
- AOAC International. Official Methods of Analysis of AOAC International, 21st ed.; AOAC International: Gaithersburg, MD, USA, 2019. [Google Scholar]
- ICC. ICC Standard No. 123/1. Determination of Starch Content by Hydrochloric Acid Dissolution. Available online: https://www.icc.or.at/publications/icc-standards/standards-overview/123-1-standard-method (accessed on 17 August 2020).
- Xiaoli, X.; Liyi, Y.; Shuang, H.; Wei, L.; Yi, S.; Hao, M.; Jusong, Z.; Xiaoxiong, Z. Determination of oligosaccharide contents in 19 cultivars of chickpea (Cicer arietinum L.) seeds by high performance liquid chromatography. Food Chem. 2008, 111, 215–219. [Google Scholar] [CrossRef]
- Pico, J.; Martínez, M.M.; Martín, M.T.; Gómez, M. Quantification of sugars in wheat flours with an HPAEC-PAD method. Food Chem. 2015, 173, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Gangola, M.P.; Jaiswal, S.; Khedikar, Y.P.; Chibbar, R.N. A reliable and rapid method for soluble sugars and RFO analysis in chickpea using HPAEC–PAD and its comparison with HPLC–RI. FOOD Chem. 2014, 154, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Leach, K.A.; Braun, D.M. Soluble Sugar and Starch Extraction and Quantification from Maize (Zea mays) Leaves. Curr. Protoc. Plant Biol. 2016, 1, 139–161. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Zeeman, S.C. Quantification of starch in plant tissues. Nat. Protoc. 2006, 1, 1342–1345. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Kumar, R.; Wyman, C.E. Fast hemicellulose quantification via a simple one-step acid hydrolysis. Biotechnol. Bioeng. 2014, 111, 1088–1096. [Google Scholar] [CrossRef]
- Foster, C.E.; Martin, T.M.; Pauly, M. Comprehensive compositional analysis of plant cell walls (Lignocellulosic biomass) part II: Carbohydrates. J. Vis. Exp. 2010. [Google Scholar] [CrossRef]
- Mitchell, J.; Johnston, I.G.; Bassel, G.W. Variability in seeds: Biological, ecological, and agricultural implications. J. Exp. Bot. 2017, 68, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Škrbić, B.; Đurišić-Mladenović, N. Distribution of heavy elements in urban and rural surface soils: The Novi Sad city and the surrounding settlements, Serbia. Environ. Monitor. Assess. 2013, 185, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Škrbić, B.; Đurišić-Mladenović, N.; Cvejanov, J. Principal Component Analysis of Trace Elements in Serbian Wheat. J. Agricul. Food Chem. 2005, 53, 2171–2175. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, X.; Wang, Y.J.; Chen, P.; Hou, A. Changes in chemical composition during soybean seed development. Food Chem. 2011, 124. [Google Scholar] [CrossRef]
- Cocuron, J.C.; Anderson, B.; Boyd, A.; Alonso, A.P. Targeted metabolomics of physaria fendleri, an industrial crop producing hydroxy fatty acids. Plant Cell Physiol. 2014, 55, 620–633. [Google Scholar] [CrossRef] [Green Version]
- Pazur, J.H.; Ando, T. The action of an amyloglucosidase of Aspergillus niger on starch and malto-oligosaccharides. J. Biol. Chem. 1959, 234, 1966–1970. [Google Scholar]
- Arakawa, T.; Timasheff, S.N. Stabilization of Protein Structure by Sugars. Biochemistry 1982, 21, 6536–6544. [Google Scholar] [CrossRef]
- Stevenson, D.G.; Doorenbos, R.K.; Jane, J.L.; Inglett, G.E. Structures and functional properties of starch from seeds of three soybean (Glycine max (L.) Merr.) varieties. Starch/Staerke 2006, 58, 509–519. [Google Scholar] [CrossRef]
- Thomas, J.; Boote, K.; Allen, M.; Gallo-Meagher, M.; Davis, J. Elevated Temperature and Carbon Dioxide Effects on Soybean Seed Composition and Transcript Abundance. Crop Sci. 2003, 43, 1548–1557. [Google Scholar] [CrossRef]
- Yu, X.; Yuan, F.; Fu, X.; Zhu, D. Profiling and relationship of water-soluble sugar and protein compositions in soybean seeds. Food Chem. 2016, 196, 776–782. [Google Scholar] [CrossRef]
- Kawamura, S.; Nagao, K.; Kasai, T. Determination of free monosaccharides and detection of sugar alcohols in mature soybean seeds. J. Nutr. Sci. Vitaminol. (Tokyo) 1977, 23, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Martos, I.; Rupérez, P. Soybean oligosaccharides. Potential as new ingredients in functional food. Nutr. Hosp. 2006, 21, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.D.; Borges, L.L.; Good God, P.I.V.; Piovesan, N.D.; Teixeira, A.I.; Cruz, C.D.; De Barros, E.G. Quantification of anti-nutritional factors and their correlations with protein and oil in soybeans. An. Acad. Bras. Cienc. 2018, 90, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravindran, G. Non-Starch polysaccharides of seeds of soybean [Glycine Max. (L)]. J. Natl. Sci. Found. Sri Lanka 1988, 16, 223–228. [Google Scholar] [CrossRef]
- Huisman, M.M.H.; Schols, H.A.; Voragen, A.G.J. Cell wall polysaccharides from soybean (Glycine max.) meal. Isolation and characterisation. Carbohydr. Polym. 1998. [Google Scholar] [CrossRef]
- Brumm, T.J. Quality and Grading Factors of IP Soybeans. Available online: file:///C:/Users/mdpi/AppData/Local/Temp/Chapter_Two_Quality_and_Grading_Factors_of_IP_Soyb.pdf (accessed on 1 June 2020).
- Bellaloui, N. Soybean Seed Phenol, Lignin, and Isoflavones and Sugars Composition Altered by Foliar Boron Application in Soybean under Water Stress. Food Nutr. Sci. 2012, 03, 579–590. [Google Scholar] [CrossRef] [Green Version]
- Grieshop, C.M.; Fahey, G.C. Comparison of quality characteristics of soybeans from Brazil, China, and the United States. J. Agric. Food Chem. 2001, 49, 2669–2673. [Google Scholar] [CrossRef]
- Saravitz, D.M.; Pharr, D.M.; Carter, T.E. Galactinol Synthase Activity and Soluble Sugars in Developing Seeds of Four Soybean Genotypes. Plant Physiol. 1987, 83, 185–189. [Google Scholar] [CrossRef] [Green Version]
- Venton, B.J. Calibration Curves|Protocol. Available online: https://0-www-jove-com.brum.beds.ac.uk/v/10188/calibration-curves (accessed on 21 July 2020).
- Zeleny, L. Ways To Test Seeds For Moisture. Yearb. Agric. 1961, 443–447. [Google Scholar]
- Ng, H.M.; Sin, L.T.; Tee, T.T.; Bee, S.T.; Hui, D.; Low, C.Y.; Rahmat, A.R. Extraction of cellulose nanocrystals from plant sources for application as reinforcing agent in polymers. Compos. Part B Eng. 2015, 75, 176–200. [Google Scholar] [CrossRef]
- Updegraff, D.M. Semimicro determination of cellulose inbiological materials. Anal. Biochem. 1969, 32, 420–424. [Google Scholar] [CrossRef]
- Cocuron, J.C.; Tsogtbaatar, E.; Alonso, A.P. High-throughput quantification of the levels and labeling abundance of free amino acids by liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2017, 1490, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Cocuron, J.-C.; Casas, M.I.; Yang, F.; Grotewold, E.; Alonso, A.P. Beyond the wall: High-throughput quantification of plant soluble and cell-wall bound phenolics by liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2019, 1589, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkaoui, S.; Daali, Y.; Christen, P.; Veuthey, J.L. Development and validation of liquid chromatography and capillary electrophoresis methods for acarbose determination in pharmaceutical tablets. J. Pharm. Biomed. Anal. 1998, 18, 729–735. [Google Scholar] [CrossRef]
- Werz, D.B.; Seeberger, P.H. Carbohydrates as the next frontier in pharmaceutical research. Chem. A Eur. J. 2005, 11, 3194–3206. [Google Scholar] [CrossRef]
- Ahmed, A.B.A.; Adel, M.; Karimi, P.; Peidayesh, M. Pharmaceutical, cosmeceutical, and traditional applications of marine carbohydrates. Adv. Food Nutr. Res. 2014, 73, 197–220. [Google Scholar] [CrossRef]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sugars and Sugar Alcohols | Transition (m/z) | RT | DP | CE | CXP | Linearity Range (nM) | R2 | LOD | LOQ |
|---|---|---|---|---|---|---|---|---|---|
| (min) | (Volts) | (Volts) | (Volts) | (nM) | (nM) | ||||
| Erythritol | 121.1/89.0 | 2.8 | –20 | –13 | –11 | 200–100,000 | 0.9927 | 0.3 | 0.9 |
| Threitol | 121.1/89.0 | 2.8 | −20 | −13 | −11 | 200–100,000 | 0.9903 | 0.3 | 1.0 |
| Arabitol | 150.9/89.0 | 3.8 | −40 | −16 | −37 | 200–100,000 | 0.9956 | 6.0 | 20.0 |
| Ribitol | 150.9/89.0 | 3.8 | −40 | −16 | −37 | 200–100,000 | 0.9923 | 6.1 | 20.3 |
| Xylitol | 150.9/89.0 | 3.8 | −40 | −16 | −37 | 200–100,000 | 0.9993 | 11.2 | 37.4 |
| Pinitol | 193.0/161.0 | 4.4 | −35 | −14 | −9 | 20–100,000 | 0.9942 | 0.7 | 2.4 |
| Fructose | 179.0/89.0 | 4.9 | −40 | −10 | −9 | 200–100,000 | 0.9985 | 14.9 | 49.6 |
| Sorbitol | 181.0/89.0 | 6.1 | −50 | −20 | −9 | 200–100,000 | 0.9992 | 3.6 | 11.9 |
| Mannitol | 181.0/89.0 | 6.8 | −50 | −20 | −9 | 200–100,000 | 0.9815 | 2.6 | 8.6 |
| Galactitol | 181.0/89.0 | 6.8 | −50 | −20 | −9 | 200–200,000 | 0.9965 | 5.6 | 18.7 |
| Glucose | 179.0/89.0 | 7.7 | −40 | −10 | −9 | 2000–200,000 | 0.9902 | 28.0 | 93.5 |
| Chiro-Inositol | 179.0/161.0 | 10.8 | −40 | −16 | −27 | 20–100,000 | 0.9911 | 4.1 | 13.6 |
| Inositol | 178.9/87.0 | 11.1 | −65 | −22 | −9 | 20–10,000 | 0.9940 | 0.5 | 1.7 |
| Sucrose | 341.1/89.0 | 11.9 | −200 | −23 | −21 | 20–100,000 | 0.9974 | 4.5 | 15.0 |
| Maltose | 341.1/161.0 | 13.3 | −50 | −10 | −7 | 200–100,000 | 0.9982 | 2.7 | 8.8 |
| Galactinol | 341.0/179.0 | 17.2 | −120 | −22 | −1 | 20–200,000 | 0.9999 | 4.6 | 15.3 |
| Raffinose | 503.2/178.9 | 17.4 | −140 | −32 | −11 | 200–100,000 | 0.9976 | 1.0 | 3.2 |
| Stachyose | 665.1/383.1 | 21.2 | −220 | −48 | −25 | 200–200,000 | 0.9996 | 0.5 | 1.6 |
| Verbascose | 828.1/545.1 | 23.9 | −280 | −52 | −25 | 2000–500,000 | 0.9786 | 21.1 | 70.2 |
| Accuracy (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Intra-Day Assay (n = 4) | Inter-Day Assay (n = 12) | ||||||||
| Carbo-hydrates | RE (%) | ME (%) | 0.25 µM * | 0.50 µM * | 1.00 µM * | 0.25 µM * | 0.50 µM * | 1.00 µM * | |
| Soluble Sugars | Fructose | 99.39 | 100.46 | −0.99 | −0.96 | −0.94 | −0.99 | −0.97 | −0.95 |
| Sucrose | 95.26 | 109.57 | −1.65 | 1.01 | 1.64 | −0.58 | 1.90 | 3.01 | |
| Raffinose | 89.06 | 84.84 | 0.27 | 0.76 | 1.60 | −0.32 | 0.81 | 2.08 | |
| Stachyose | 114.54 | 101.25 | −0.97 | −0.57 | −0.18 | −0.94 | −0.48 | −0.11 | |
| Verbascose | 86.06 | 86.18 | −0.99 | −0.98 | −0.96 | −0.99 | −0.98 | −0.94 | |
| Glucose | 118.12 | 80.41 | −0.99 | −0.99 | −0.96 | −1.00 | −0.99 | −0.95 | |
| Maltose | 112.80 | 109.89 | −0.99 | −0.95 | −0.95 | −0.99 | −0.98 | −0.97 | |
| Sugar Alcohols | Tetraols | 88.90 | 73.78 | −0.99 | −0.99 | −0.96 | −0.99 | −0.97 | −0.93 |
| Pentitols | 115.12 | 76.44 | −0.99 | −0.96 | −0.90 | −0.98 | −0.95 | −0.89 | |
| Sorbitol | 82.80 | 112.50 | −0.87 | −0.71 | −0.50 | −0.85 | −0.73 | −0.54 | |
| Inositol | 114.53 | 106.55 | −0.95 | −0.59 | −0.49 | −0.99 | −0.79 | −0.69 | |
| Chiro-Inositol | 104.99 | 95.55 | −0.97 | −0.30 | 0.21 | −0.93 | −0.55 | −0.26 | |
| Pinitol | 107.46 | 101.65 | −0.42 | 0.53 | 1.57 | −0.36 | 1.22 | 2.62 | |
| Galactinol | 115.34 | 111.14 | −0.07 | 0.34 | 1.19 | −0.67 | 0.12 | 0.73 | |
| Hexitols | 93.73 | 113.30 | −0.97 | −0.89 | −0.82 | −0.95 | −0.90 | −0.84 | |
| Carbohydrates | RT (min) | R2 | LOD (ng µL−1) | LOQ (ng µL−1) |
|---|---|---|---|---|
| Rhamnose | 10.20 | 0.9982 | 0.54 | 1.79 |
| Fucose | 10.49 | 0.9978 | 0.39 | 1.29 |
| Arabinose | 11.75 | 0.9949 | 0.30 | 1.10 |
| Xylose | 12.80 | 0.9977 | 0.43 | 1.43 |
| Mannose | 14.48 | 0.9981 | 0.77 | 2.56 |
| Galactose | 15.09 | 0.9990 | 0.90 | 3.00 |
| Glucose | 15.79 | 0.9963 | 0.92 | 3.06 |
| Accuracy (%) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Intra-Day Assay (n = 4) | Inter-Day Assay (n = 12) | |||||||
| Carbo-hydrates | RE (%) | ME (%) | 25 µg µL−1 * | 50 µg µL−1 * | 100 µg µL−1 * | 25 µg µL−1 * | 50 µg µL−1 * | 100 µg µL−1 * |
| Rhamnose | 108.84 | 90.72 | 2.13 | 7.52 | 14.12 | 2.87 | 7.62 | 14.88 |
| Fucose | 107.04 | 92.57 | 2.25 | 6.86 | 12.71 | 2.64 | 6.76 | 13.30 |
| Arabinose | 108.98 | 103.13 | 2.89 | 5.58 | 10.82 | 2.62 | 5.32 | 11.84 |
| Xylose | 104.33 | 100.05 | 2.21 | 6.65 | 12.09 | 2.79 | 6.02 | 12.53 |
| Mannose | 108.02 | 103.07 | 2.13 | 6.03 | 13.86 | 1.89 | 5.08 | 13.17 |
| Galactose | 108.06 | 108.64 | 1.06 | 3.87 | 11.94 | 1.53 | 4.24 | 12.52 |
| Glucose | 104.200 | 92.09 | 1.77 | 5.51 | 12.54 | 2.01 | 5.50 | 12.45 |
| Theoretical ng µL−1 Glucose | Measured ng µL−1 Glucose | Standard Deviation | RME (%) | LOD ng µL−1 | LOQ ng µL−1 | Equation R2 | |
|---|---|---|---|---|---|---|---|
| Day 1 n = 4 | 6.67 | 7.15 | 0.036 | 7.23 | 2.62 | 8.72 | y = 0.032x + 0.0260 R2 = 0.9969 |
| 13.33 | 13.11 | 0.055 | −1.70 | ||||
| 20.00 | 19.73 | 0.060 | −1.35 | ||||
| 26.67 | 25.96 | 0.051 | −2.67 | ||||
| 33.33 | 34.06 | 0.072 | 2.17 | ||||
| Day 2 n = 4 | 6.67 | 7.41 | 0.032 | 11.09 | 1.03 | 3.45 | y = 0.0312x + 0.0326 R2 = 0.9966 |
| 13.33 | 12.58 | 0.029 | −5.67 | ||||
| 20.00 | 19.49 | 0.025 | −2.55 | ||||
| 26.67 | 27.00 | 0.065 | 1.23 | ||||
| 33.33 | 33.53 | 0.078 | 0.59 | ||||
| Day 3 n = 4 | 6.67 | 7.47 | 0.014 | 12.11 | 3.00 | 9.99 | y = 0.0311x + 0.0339 R2 = 0.9920 |
| 13.33 | 13.22 | 0.043 | −0.83 | ||||
| 20.00 | 18.43 | 0.014 | −7.87 | ||||
| 26.67 | 26.92 | 0.015 | 0.94 | ||||
| 33.33 | 33.96 | 0.072 | 1.88 | ||||
| Average n = 12 | 6.67 | 7.34 | 0.014 | 10.12 | 2.22 | 7.40 | y = 0.0314x + 0.0225 R2 = 0.9967 |
| 13.33 | 12.97 | 0.043 | −2.72 | ||||
| 20.00 | 19.22 | 0.014 | −3.90 | ||||
| 26.67 | 26.62 | 0.015 | −0.19 | ||||
| 33.33 | 33.85 | 0.072 | 1.55 |
| North Carolina | Arkansas | ||
|---|---|---|---|
| Feature | p-value | Feature | p-value |
| Oil | 3.59E−07 | Sucrose | 0.0002 |
| Xylose | 0.0029 | Oil | 0.0009 |
| Protein | 0.0035 | Maltose | 0.0049 |
| Raffinose | 0.0085 | Raffinose | 0.0073 |
| THC | 0.0166 | Galactinol | 0.0098 |
| Pinitol | 0.0358 | Pentitols | 0.0104 |
| Glucose.1 | 0.0371 | Inositol | 0.0110 |
| Galactose | 0.0388 | Protein | 0.0152 |
| TSA | 0.0489 | TSS | 0.0201 |
| Fucose | 0.0489 | Galactose | 0.0232 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moretti, A.; Arias, C.L.; Mozzoni, L.A.; Chen, P.; McNeece, B.T.; Mian, M.A.R.; McHale, L.K.; Alonso, A.P. Workflow for the Quantification of Soluble and Insoluble Carbohydrates in Soybean Seed. Molecules 2020, 25, 3806. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25173806
Moretti A, Arias CL, Mozzoni LA, Chen P, McNeece BT, Mian MAR, McHale LK, Alonso AP. Workflow for the Quantification of Soluble and Insoluble Carbohydrates in Soybean Seed. Molecules. 2020; 25(17):3806. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25173806
Chicago/Turabian StyleMoretti, Ademar, Cintia L. Arias, Leandro A. Mozzoni, Pengyin Chen, Brant T. McNeece, M. A. Rouf Mian, Leah K. McHale, and Ana P. Alonso. 2020. "Workflow for the Quantification of Soluble and Insoluble Carbohydrates in Soybean Seed" Molecules 25, no. 17: 3806. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25173806