Asymmetric Synthesis of Tertiary α -Hydroxyketones by Enantioselective Decarboxylative Chlorination and Subsequent Nucleophilic Substitution


Abstract
:1. Introduction
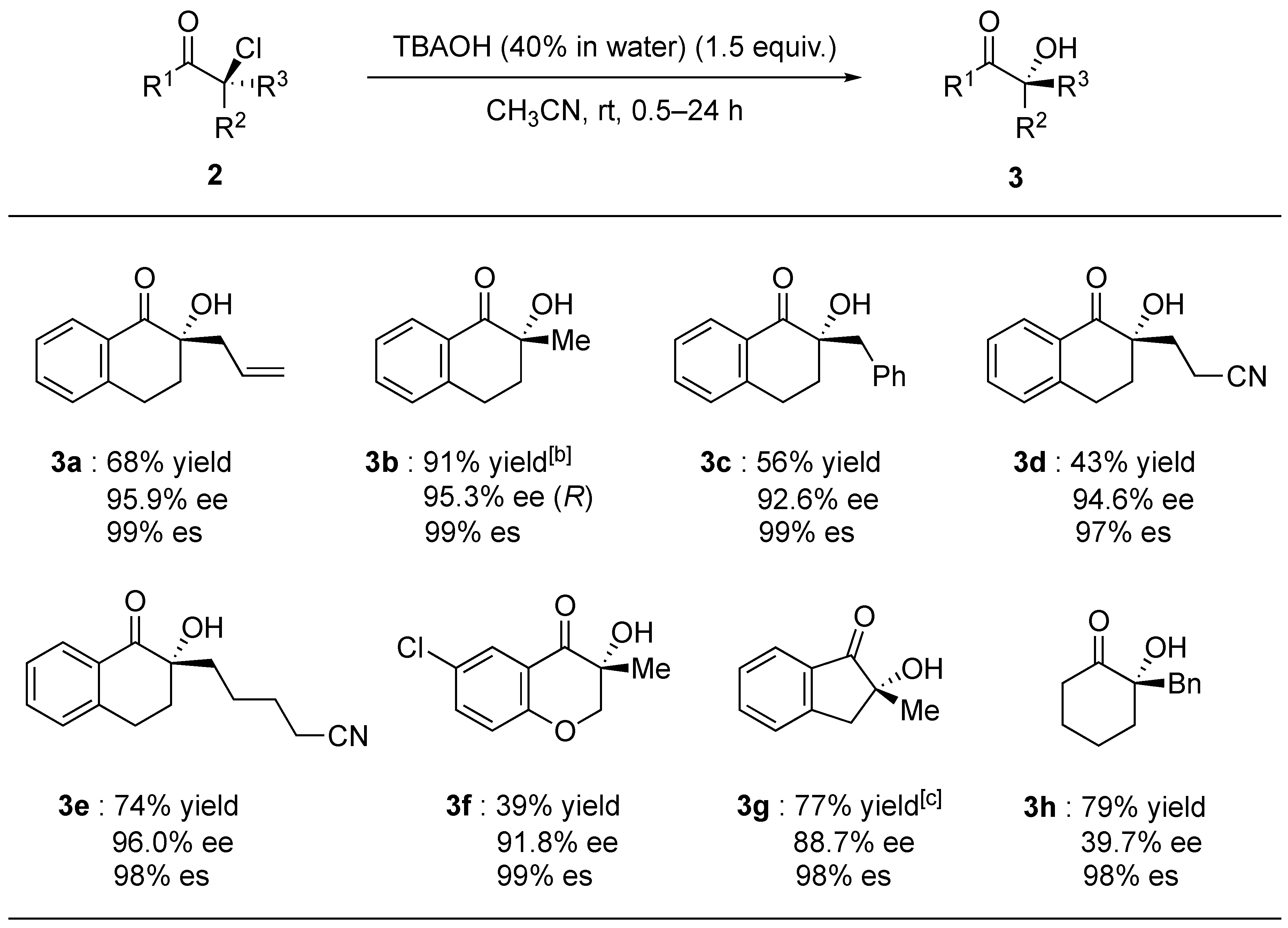
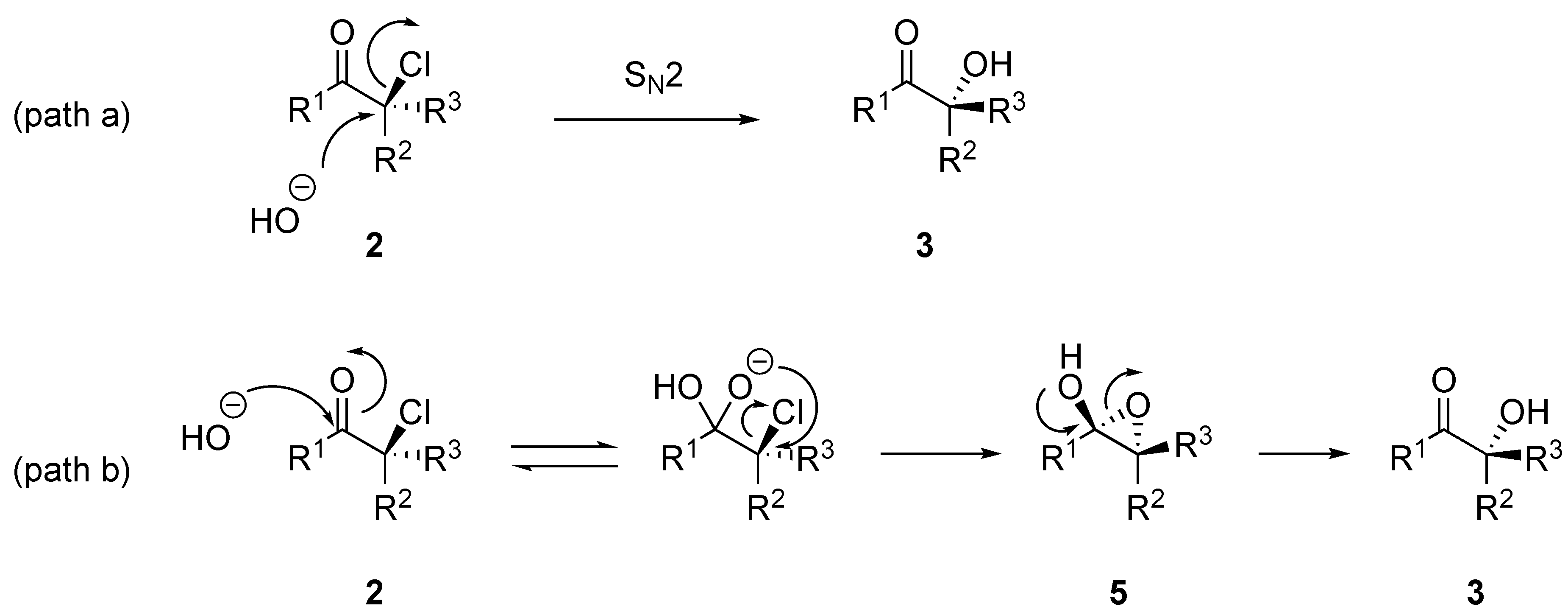
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Materials
3.3. Synthesis of β-ketocarboxylic Acid 1e
3.3.1. Tert-Butyl 2-(2-cyanobutyl)-1-oxo-1,2,3,4-tetrahydronaphthalene-2-carboxylate
3.3.2. 2-(4-Cyanobutyl)-1-oxo-1,2,3,4-tetrahydronaphthalene-2-carboxylic acid (1e)
3.4. Synthesis of α-chloroketone 2e
5-(2-Chloro-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)pentanenitrile (2e)
3.5. Synthesis of α-hydroxyketones 3
3.5.1. 2-Allyl-2-hydroxy-3,4-dihydronaphthalen-1(2H)-one (3a)
3.5.2. (R)-2-Hydroxy-2-methyl-3,4-dihydronaphthalen-1(2H)-one (3b)
3.5.3. 2-Benzyl-2-hydroxy-3,4-dihydronaphthalen-1(2H)-one (3c)
3.5.4. 3-(2-Hydroxy-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)propanenitrile (3d)
3.5.5. 5-(2-Hydroxy-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)pentanenitrile (3e)
3.5.6. 6-Chloro-3-hydroxy-3-methylchroman-4-one (3f)
3.5.7. 2-Hydroxy-2-methyl-2,3-dihydro-1H-inden-1-one (3g)
3.5.8. 2-Benyl-2-hydroxycyclohexan-1-one (3h)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Liu, Y.-L.; Lin, X.-T. Recent advances in catalytic asymmetric synthesis of tertiary alcohols via nucleophilic addition to ketones. Adv. Synth. Catal. 2019, 361, 876–918. [Google Scholar] [CrossRef]
- Olack, G.; Morrison, H. Formation and characterization of lumitetracycline-type photoproducts from members of the tetracycline family. J. Org. Chem. 1991, 56, 4969–4971. [Google Scholar] [CrossRef]
- Lucas-Lopez, C.; Patterson, S.; Blum, T.; Straight, A.F.; Toth, J.; Slawin, A.M.Z.; Mitchison, T.J.; Sellers, J.R.; Westwood, N.J. Absolute stereochemical assignment and fluorescence tuning of the small molecule tool, (–)-blebbistatin. Eur. J. Org. Chem. 2005, 1736–1740. [Google Scholar] [CrossRef]
- Matsuda, H.; Yoshida, K.; Miyagawa, K.; Asao, Y.; Takayama, S.; Nakashima, S.; Xu, F.; Yoshikawa, M. Rotenoids and flavonoids with anti-invasion of HT1080, anti-proliferation of U937, and differentiation-inducing activity in HL-60 from Erycibe expansa. Bioorg. Med. Chem. 2007, 15, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.G.; Kenworthy, M.N.; Kitson, R.R.A.; Scott, M.S.; Taylor, R.J.K. The telescoped intramolecular Michael/olefination (TIMO) approach to α-alkylidene-γ-butyrolactones: Synthesis of (+)-paeonilactone B. Angew. Chem. Int. Ed. 2008, 47, 1935–1937. [Google Scholar] [CrossRef]
- Kusumi, S.; Nakayama, H.; Kobayashi, T.; Kuriki, H.; Matsumoto, Y.; Takahashi, D.; Toshima, K. Total synthesis of aquayamycin. Chem. Eur. J. 2016, 22, 18733–18736. [Google Scholar] [CrossRef]
- Masui, M.; Ando, A.; Shioiri, T. New methods and reagents in organic synthesis. 75. asymmetric synthesis of α-hydroxy ketones using chiral phase transfer catalysts. Tetrahedron Lett. 1988, 29, 2835–2838. [Google Scholar] [CrossRef]
- Morikawa, K.; Park, J.; Andersson, P.G.; Hashiyama, T.; Sharpless, K.B. Catalytic asymmetric dihydroxylation of tetrasubstituted olefins. J. Am. Chem. Soc. 1993, 115, 8463–8464. [Google Scholar] [CrossRef]
- Momiyama, N.; Yamamoto, H. Catalytic enantioselective synthesis of α-aminooxy and α-hydroxy ketone using nitrosobenzene. J. Am. Chem. Soc. 2003, 125, 6038–6039. [Google Scholar] [CrossRef]
- Kawasaki, M.; Li, P.; Yamamoto, H. Enantioselective O-nitroso aldol reaction of silyl enol ethers. Angew. Chem. Int. Ed. 2008, 47, 3795–3797. [Google Scholar] [CrossRef]
- Yanagisawa, A.; Takeshita, S.; Izumi, Y.; Yoshida, K. Enantioselective nitroso aldol reaction catalyzed by quinoxP*·silver(I) complex and tin methoxide. J. Am. Chem. Soc. 2010, 132, 5328–5329. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Odagi, M.; Furukori, K.; Nagasawa, K. Asymmetric α-hydroxylation of a lactone with vinylogous pyridone by using a guanidine-urea bifunctional organocatalyst: Catalytic enantioselective synthesis of a key intermediate for (20S)-camptothecin analogues. Chem. Eur. J. 2014, 20, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.-B.D.; Wang, M.; Zhao, Y. Phase-transfer-catalyzed enantioselective α-hydroxylation of acyclic and cyclic ketones with oxygen. ACS Catal. 2015, 5, 3609–3612. [Google Scholar] [CrossRef]
- Shevchenko, G.A.; Pupo, G.; List, B. Direct asymmetric α-hydroxylation of cyclic α-branched ketones through enol catalysis. Synlett 2019, 30, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Shevchenko, G.A.; Oppelaar, B.; List, B. An unexpected α-oxidation of cyclic ketones with 1,4-benzoquinone by enol catalysis. Angew. Chem. Int. Ed. 2018, 57, 10756–10759. [Google Scholar] [CrossRef]
- Shibatomi, K.; Kitahara, K.; Sasaki, N.; Kawasaki, Y.; Fujisawa, I.; Iwasa, S. Enantioselective decarboxylative chlorination of β-ketocarboxylic acids. Nat. Commun. 2017, 8, 15600. [Google Scholar] [CrossRef] [Green Version]
- Masaki, Y.; Arasaki, H.; Iwata, M. Stereospecific construction of chiral quaternary carbon compounds from chiral secondary alcohol derivatives. Chem. Lett. 2003, 32, 4–5. [Google Scholar] [CrossRef]
- Mascal, M.; Hafezi, N.; Toney, M.D. 1,4,7-Trimethyloxatriquinane: SN2 reaction at tertiary carbon. J. Am. Chem. Soc. 2010, 132, 10662–10664. [Google Scholar] [CrossRef]
- Shibatomi, K.; Soga, Y.; Narayama, A.; Fujisawa, I.; Iwasa, S. Highly enantioselective chlorination of β-keto esters and subsequent SN2 displacement of tertiary chlorides: A flexible method for the construction of quaternary stereogenic centers. J. Am. Chem. Soc. 2012, 134, 9836–9839. [Google Scholar] [CrossRef]
- Pronin, S.V.; Reiher, C.A.; Shenvi, R.A. Stereoinversion of tertiary alcohols to tertiary-alkyl isonitriles and amines. Nature 2013, 501, 195–199. [Google Scholar] [CrossRef]
- Liu, R.Y.; Wasa, M.; Jacobsen, E.N. Enantioselective synthesis of tertiary α-chloro esters by non-covalent catalysis. Tetrahedron Lett. 2015, 56, 3428–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibatomi, K.; Kotozaki, M.; Sasaki, N.; Fujisawa, I.; Iwasa, S. Williamson ether synthesis with phenols at a tertiary stereogenic carbon: Formal enantioselective phenoxylation of β-Keto esters. Chem. Eur. J. 2015, 21, 14095–14098. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Ha, M.W.; Hong, S.; Park, C.; Kim, B.; Yang, J.; Park, H. Enantioselective synthesis of chiral α-azido and α-aryloxy quaternary stereogenic centers via the phase-transfer-catalyzed α-alkylation of α-bromomalonates, followed by SN2 Substitution. J. Org. Chem. 2017, 82, 4936–4943. [Google Scholar] [CrossRef] [PubMed]
- Shibatomi, K.; Kitahara, K.; Okimi, T.; Abe, Y.; Iwasa, S. Enantioselective fluorination of α-branched aldehydes and subsequent conversion to α-hydroxyacetals via stereospecific C–F bond cleavage. Chem. Sci. 2016, 7, 1388–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, A.G.; Townsend, S.D.; Danishefsky, S.J. Halocycloalkenones as Diels−Alder dienophiles. Applications to generating useful structural patterns. J. Org. Chem. 2013, 78, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Davis, F.A.; Weismiller, M.C. Enantioselective synthesis of tertiary α-hydroxy carbonyl compounds using ((8,8-dichlorocamphoryl)sulfonyl)oxaziridine. J. Org. Chem. 1990, 55, 3715–3717. [Google Scholar] [CrossRef]
- Sato, K.; Sekiguchi, T.; Aoki, S. Novel methods for synthesizing 2-chloroaldehyde from carbonyl compounds and converting it into 2-hydroxyaldehyde derivatives. Tetrahedron Lett. 2001, 42, 3625–3628. [Google Scholar] [CrossRef]
- Arai, T.; Sato, T.; Kanoh, H.; Kaneko, K.; Oguma, K.; Yanagisawa, A. Organic-inorganic hybrid polymer-encapsulated magnetic nanobead catalysts. Chem. Eur. J. 2008, 14, 882–885. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Nucleophile | Solvent | Time (h) | Conv. (%) [b] of rac-2a | Yield (%) [c] of rac-3a | Yield (%) [c] of 4a |
|---|---|---|---|---|---|---|
| 1 | NaOH aq. (10 M) | CH3CN | 5 | 19 | 1 | 1 |
| 2 | KOH aq. (10 M) | CH3CN | 5 | 10 | 4 | 3 |
| 3 | TBAF·nH2O (1.0 M in THF) | CH3CN | 5 | 61 | 1 | 38 |
| 4 | TBAOH (40% in water) | CH3CN | 5 | 99 | 68 | 14 |
| 5 | TBAOH (40% in water) | THF | 4 | 96 | 59 | 33 |
| 6 | TBAOH (40% in water) | DMPU | 45 min | 100 | 48 | 39 |
| 7 | TBAOH (40% in water) | DMF | 2 | 100 | 52 | 27 |
| 8 | TBAOH (40% in water) | toluene | 4 | 33 | 22 | 5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kam, M.K.; Sugiyama, A.; Kawanishi, R.; Shibatomi, K. Asymmetric Synthesis of Tertiary α -Hydroxyketones by Enantioselective Decarboxylative Chlorination and Subsequent Nucleophilic Substitution. Molecules 2020, 25, 3902. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25173902
Kam MK, Sugiyama A, Kawanishi R, Shibatomi K. Asymmetric Synthesis of Tertiary α -Hydroxyketones by Enantioselective Decarboxylative Chlorination and Subsequent Nucleophilic Substitution. Molecules. 2020; 25(17):3902. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25173902
Chicago/Turabian StyleKam, Mei Kee, Akira Sugiyama, Ryouta Kawanishi, and Kazutaka Shibatomi. 2020. "Asymmetric Synthesis of Tertiary α -Hydroxyketones by Enantioselective Decarboxylative Chlorination and Subsequent Nucleophilic Substitution" Molecules 25, no. 17: 3902. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25173902