
Design and Synthesis of a Chiral Halogen-Bond Donor with a Sp3-Hybridized Carbon–Iodine Moiety in a Chiral Fluorobissulfonyl Scaffold


Abstract
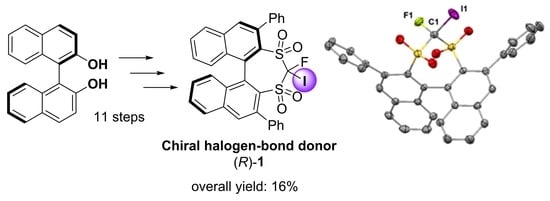
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
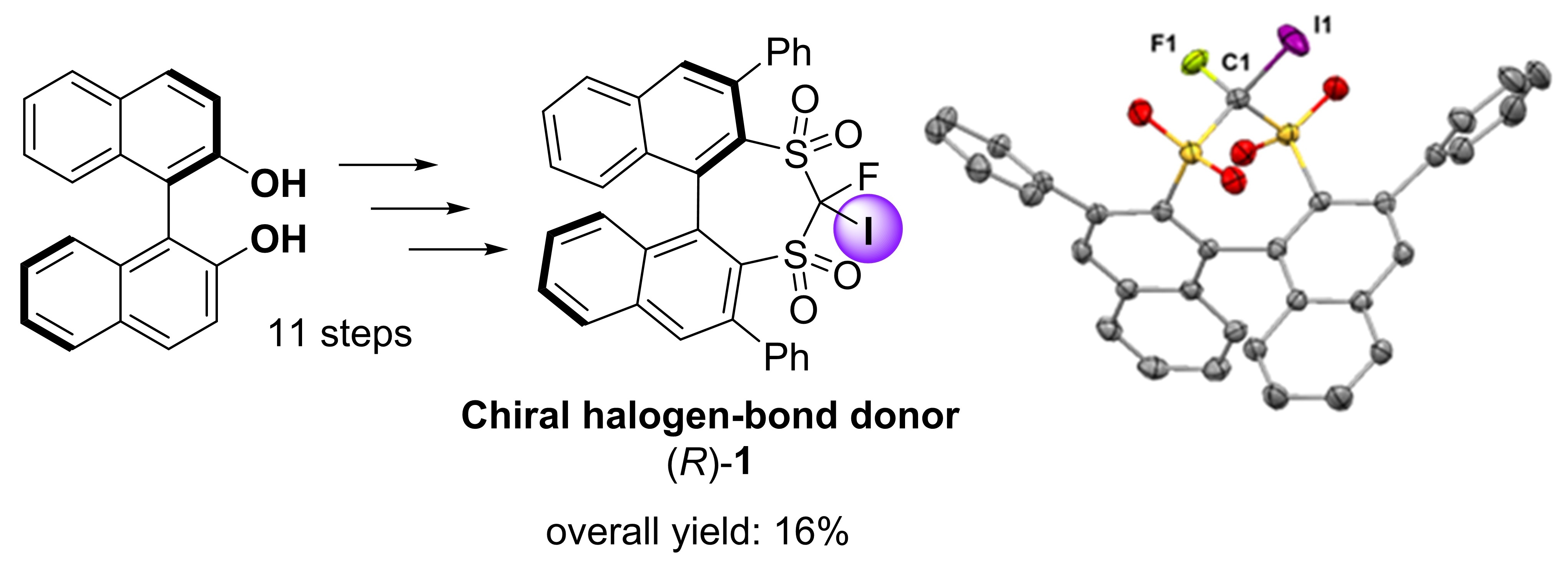
1. Introduction
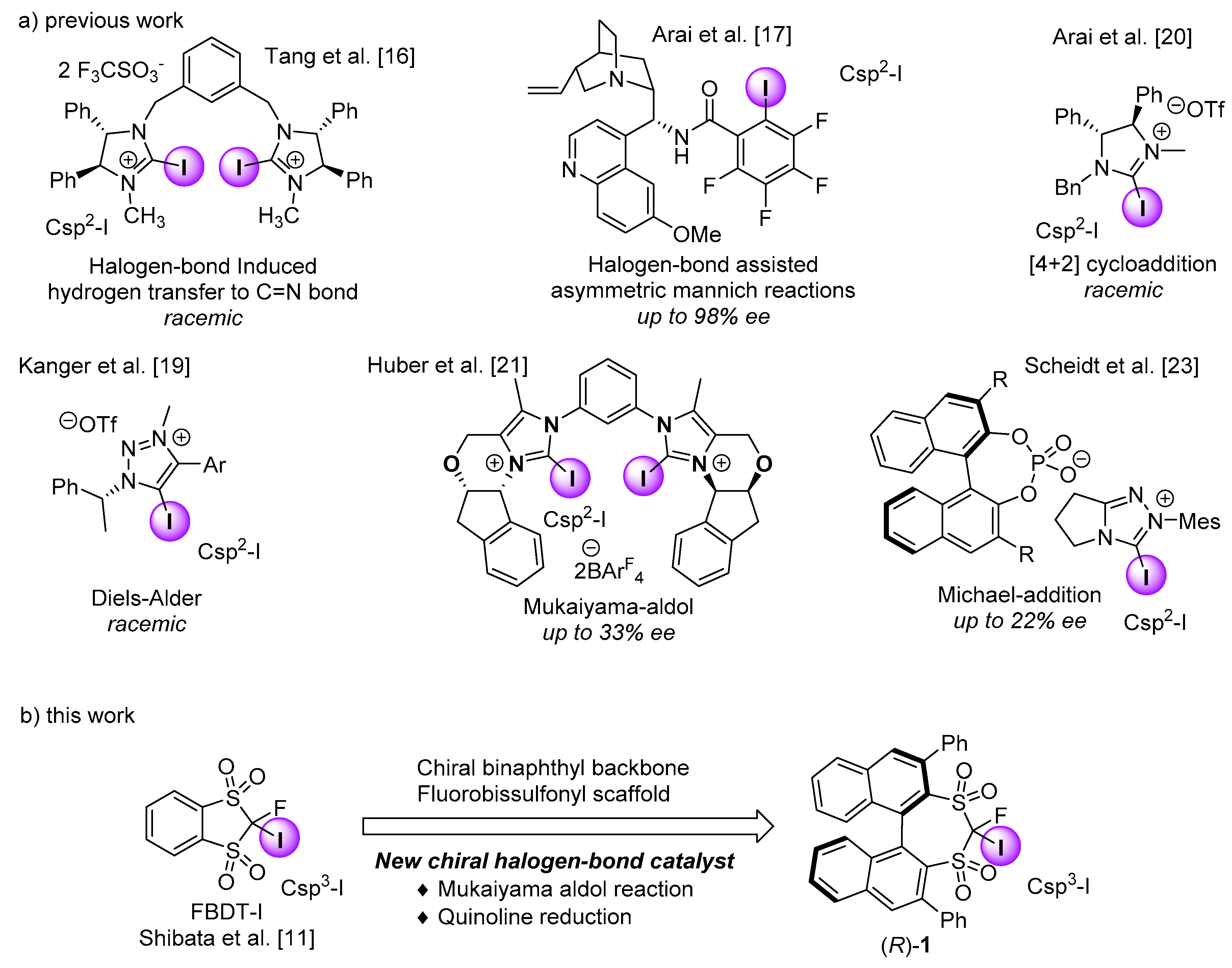
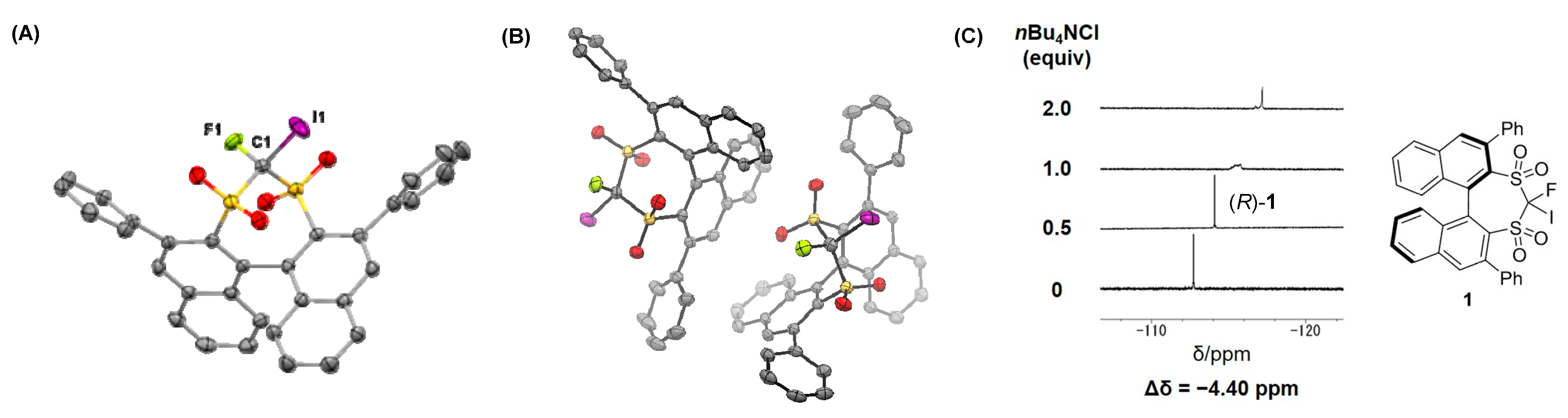
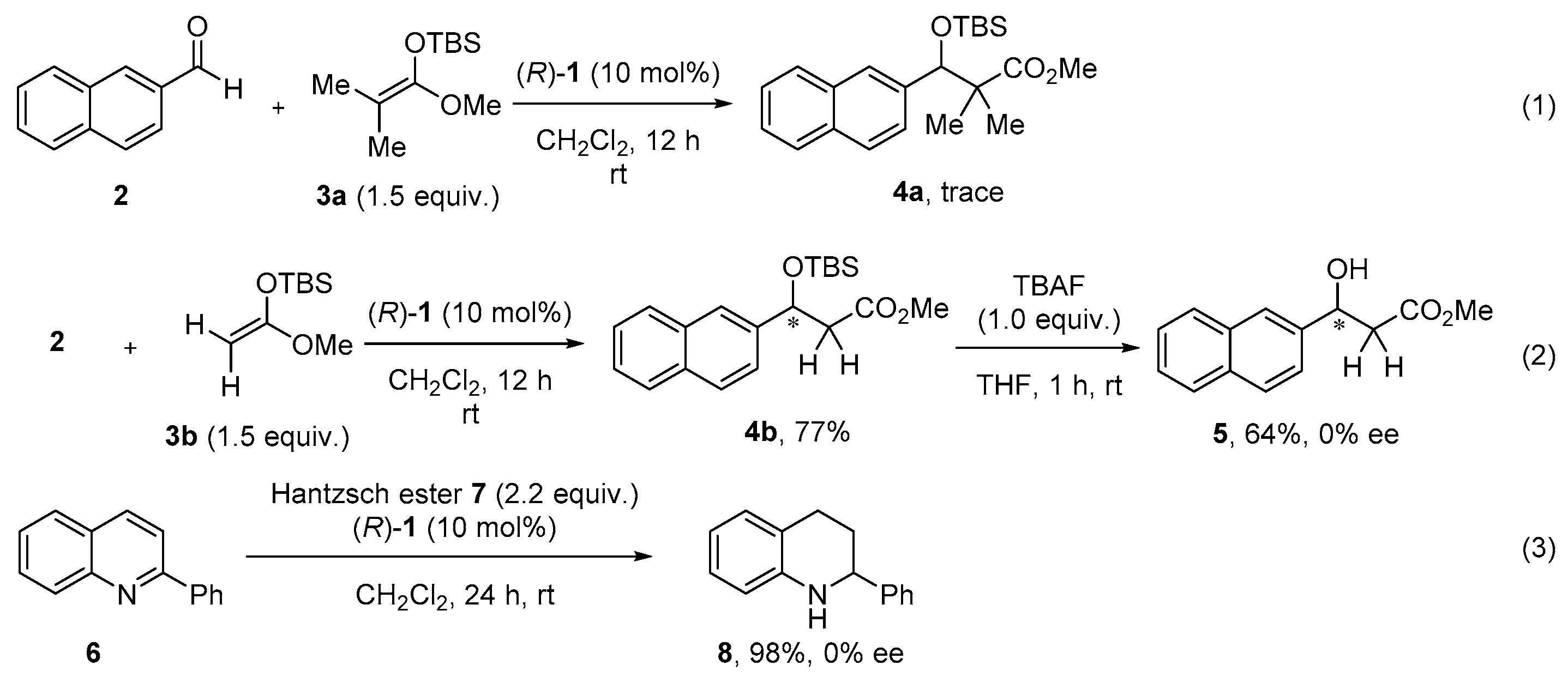
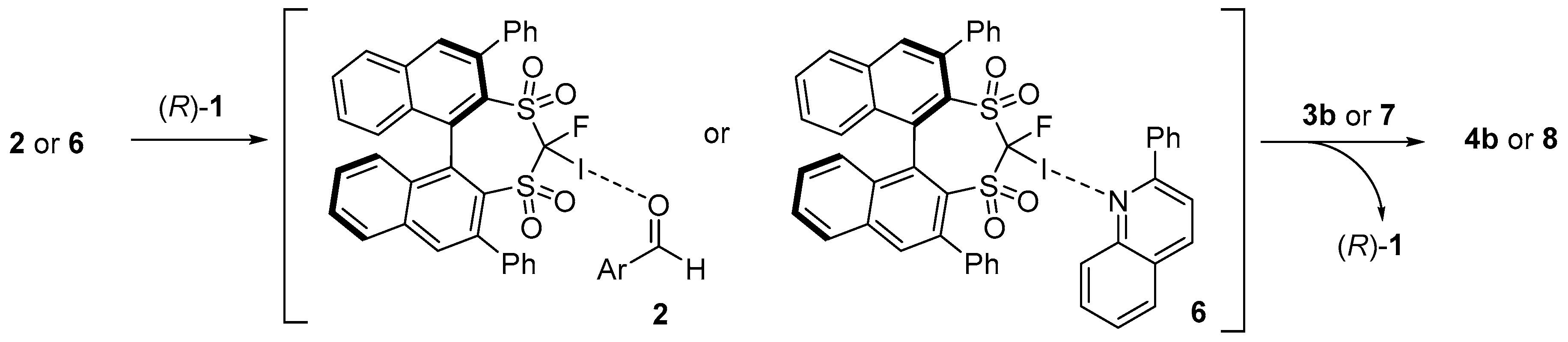
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis of the Chiral Halogen-Bond Donor (R)-1
3.3. Experimental Procedure of 19F NMR Titration of (R)-1 with nBu4NCl in CDCl3
3.4. General Procedure of Mukaiyama Aldol with (R)-1
3.5. General Procedure of Reduction of Quinoline with (R)-1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagorny, P.; Sun, Z. New approaches to organocatalysis based on C–H and C–X bonding for electrophilic substrate activation. Beilstein J. Org. Chem. 2016, 12, 2834–2848. [Google Scholar] [CrossRef] [Green Version]
- Bulfield, D.; Huber, S.M. Halogen Bonding in Organic Synthesis and Organocatalysis. Chem. Eur. J. 2016, 22, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bolm, C.; Bruckmann, A.; Pena, M.A. Organocatalysis through Halogen-Bond Activation. Synlett 2008, 6, 900–902. [Google Scholar] [CrossRef]
- Kniep, F.; Jungbauer, S.H.; Zhang, Q.; Walter, S.M.; Schindler, S.; Schnapperelle, I.; Herdtweck, E.; Huber, S.M. Organocatalysis by Neutral Multidentate Halogen-Bond Donors. Angew. Chem. Int. Ed. 2013, 52, 7028–7032. [Google Scholar] [CrossRef] [PubMed]
- Heinen, F.; Engelage, E.; Dreger, A.; Weiss, R.; Huber, S.M. Iodine(III) Derivatives as Halogen Bonding Organocatalysts. Angew. Chem. Int. Ed. 2018, 57, 3830–3833. [Google Scholar] [CrossRef]
- Takeda, Y.; Hisakuni, D.; Lin, C.-H.; Minakata, S. 2-Halogenoimidazolium Salt Catalyzed Aza-Diels–Alder Reaction through Halogen-Bond Formation. Org. Lett. 2015, 17, 318–321. [Google Scholar] [CrossRef]
- Liu, X.; Toy, P. Halogen Bond-Catalyzed Povarov Reactions. Adv. Synth. Catal. 2020, 362, 3437–3441. [Google Scholar] [CrossRef]
- Gliese, J.-P.; Jungbauer, S.H.; Huber, S.M. A halogen-bonding-catalyzed Michael addition reaction. Chem. Commun. 2017, 53, 12052–12055. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K.; Uno, H.; Tokunaga, E.; Shibata, N. Fluorobissulfonylmethyl Iodides: An Efficient Scaffold for Halogen Bonding Catalysts with an sp3-Hybridized Carbon–Iodine Moiety. ACS Catal. 2018, 8, 6601–6605. [Google Scholar] [CrossRef]
- Sakakura, A.; Ukai, A.; Ishihara, K. Enantioselective halocyclization of polyprenoids induced by nucleophilic phosphoramidites. Nature 2007, 445, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, V.N.G.; Charette, A.B. Design and Synthesis of Chiral Heteroleptic Rhodium(II) Carboxylate Catalysts: Experimental Investigation of Halogen Bond Rigidification Effects in Asymmetric Cyclopropanation. ACS Catal. 2012, 2, 1221–1225. [Google Scholar] [CrossRef]
- Zong, L.; Ban, X.; Kee, C.W.; Tan, C. Catalytic Enantioselective Alkylation of Sulfenate Anions to Chiral Heterocyclic Sulfoxides Using Halogenated Pentanidium Salts. Angew. Chem. Int. Ed. 2014, 126, 12043–12047. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Marques, I.; Ferreira, L.; Félix, V.; Beer, P.D. Enhancing the enantioselective recognition and sensing of chiral anions by halogen bonding. Chem. Commun. 2016, 52, 5527–5530. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Ge, Y.; Tan, C. Halogen-Bonding-Induced Hydrogen Transfer to C═N Bond with Hantzsch Ester. Org. Lett. 2014, 16, 3244–3247. [Google Scholar] [CrossRef]
- Kuwano, S.; Suzuki, T.; Hosaka, Y.; Arai, T. A chiral organic base catalyst with halogen-bonding-donor functionality: Asymmetric Mannich reactions of malononitrile with N-Boc aldimines and ketimines. Chem. Commun. 2018, 54, 3847–3850. [Google Scholar] [CrossRef]
- Kuwano, S.; Nishida, Y.; Suzuki, T.; Arai, T. Catalytic Asymmetric Mannich-Type Reaction of Malononitrile with N-Boc α-Ketiminoesters Using Chiral Organic Base Catalyst with Halogen Bond Donor Functionality. Adv. Synth. Catal. 2020, 362, 1674–1678. [Google Scholar] [CrossRef]
- Kaasik, M.; Metsala, A.; Kaabel, S.; Kriis, K.; Järving, I.; Kanger, T. Halo-1,2,3-triazolium Salts as Halogen Bond Donors for the Activation of Imines in Dihydropyridinone Synthesis. J. Org. Chem. 2019, 84, 4294–4303. [Google Scholar] [CrossRef]
- Kuwano, S.; Suzuki, T.; Yamanaka, M.; Tsutsumi, R.; Arai, T. Catalysis Based on C−I⋅⋅⋅π Halogen Bonds: Electrophilic Activation of 2-Alkenylindoles by Cationic Halogen-Bond Donors for [4+2] Cycloadditions. Angew. Chem. Int. Ed. 2019, 58, 10220–10224. [Google Scholar] [CrossRef]
- Sutar, R.L.; Engelage, E.; Stoll, R.; Huber, S.M. Bidentate Chiral Bis(imidazolium)-Based Halogen-Bond Donors: Synthesis and Applications in Enantioselective Recognition and Catalysis. Angew. Chem. Int. Ed. 2020, 59, 6806–6810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Yang, H.; Heusler, A.; Chua, Z.; Wong, M.W.; Tan, C. Halogen-Bonding-Induced Conjugate Addition of Thiophenes to Enones and Enals. Chem. Asian J. 2019, 14, 2656–2661. [Google Scholar] [CrossRef] [PubMed]
- Squitieri, R.A.; Fitzpatrick, K.P.; Jaworski, A.A.; Scheidt, K.A. Synthesis and Evaluation of Azolium-Based Halogen-Bond Donors. Chem. Eur. J. 2019, 25, 10069–10073. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Antonchick, A.P.; Theissmann, T. A Highly Enantioselective Brønsted Acid Catalyzed Cascade Reaction: Organocatalytic Transfer Hydrogenation of Quinolines and their Application in the Synthesis of Alkaloids. Angew. Chem. Int. Ed. 2006, 45, 3683–3686. [Google Scholar] [CrossRef]
- Wakchaure, V.N.; Kaib, P.S.J.; Leutzsch, M.; List, B. Disulfonimide-Catalyzed Asymmetric Reduction ofN-Alkyl Imines. Angew. Chem. Int. Ed. 2015, 54, 11852–11856. [Google Scholar] [CrossRef]
- Wu, T.R.; Shen, L.; Chong, J.M. Asymmetric Allylboration of Aldehydes and Ketones Using 3,3‘-Disubstitutedbinaphthol-Modified Boronates. Org. Lett. 2004, 6, 2701–2704. [Google Scholar] [CrossRef]
- Gatzenmeier, T.; Van Gemmeren, M.; Xie, Y.; Höfler, D.; Leutzsch, M.; List, B. Asymmetric Lewis acid organocatalysis of the Diels-Alder reaction by a silylated C-H acid. Science 2016, 351, 949–952. [Google Scholar] [CrossRef]
- Wenzel, A.G.; Jacobsen, E.N. Asymmetric Catalytic Mannich Reactions Catalyzed by Urea Derivatives: Enantioselective Synthesis ofβ-Aryl-β-Amino Acids. J. Am. Chem. Soc. 2002, 124, 12964–12965. [Google Scholar] [CrossRef]
- Ratjen, L.; Van Gemmeren, M.; Pesciaioli, F.; List, B. Towards High-Performance Lewis Acid Organocatalysis. Angew. Chem. Int. Ed. 2014, 53, 8765–8769. [Google Scholar] [CrossRef]
- Denmark, S.E.; Beutner, G.L.; Wynn, T.; Eastgate, M.D. Lewis Base Activation of Lewis Acids: Catalytic, Enantioselective Addition of Silyl Ketene Acetals to Aldehydes. J. Am. Chem. Soc. 2005, 127, 3774–3789. [Google Scholar] [CrossRef]
- Zhuo, F.-F.; Xie, W.-W.; Yang, Y.-X.; Zhang, L.; Wang, P.; Yuan, R.; Da, C.-S. TMEDA-Assisted Effective Direct Ortho Arylation of Electron-Deficient N-Heteroarenes with Aromatic Grignard Reagents. J. Org. Chem. 2013, 78, 3243–3249. [Google Scholar] [CrossRef] [PubMed]
- Lachkar, D.; Vilona, D.; Dumont, É.; Lelli, M.; Lacôte, E. Grafting of Secondary Diolamides onto [P2W15V3O62]9− Generates Hybrid Heteropoly Acids. Angew. Chem. Int. Ed. 2016, 55, 5961–5965. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uno, H.; Matsuzaki, K.; Shiro, M.; Shibata, N. Design and Synthesis of a Chiral Halogen-Bond Donor with a Sp3-Hybridized Carbon–Iodine Moiety in a Chiral Fluorobissulfonyl Scaffold. Molecules 2020, 25, 4539. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25194539
Uno H, Matsuzaki K, Shiro M, Shibata N. Design and Synthesis of a Chiral Halogen-Bond Donor with a Sp3-Hybridized Carbon–Iodine Moiety in a Chiral Fluorobissulfonyl Scaffold. Molecules. 2020; 25(19):4539. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25194539
Chicago/Turabian StyleUno, Hiroto, Kohei Matsuzaki, Motoo Shiro, and Norio Shibata. 2020. "Design and Synthesis of a Chiral Halogen-Bond Donor with a Sp3-Hybridized Carbon–Iodine Moiety in a Chiral Fluorobissulfonyl Scaffold" Molecules 25, no. 19: 4539. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25194539