Exploring the Multi-Target Performance of Mitochondriotropic Antioxidants against the Pivotal Alzheimer’s Disease Pathophysiological Hallmarks

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
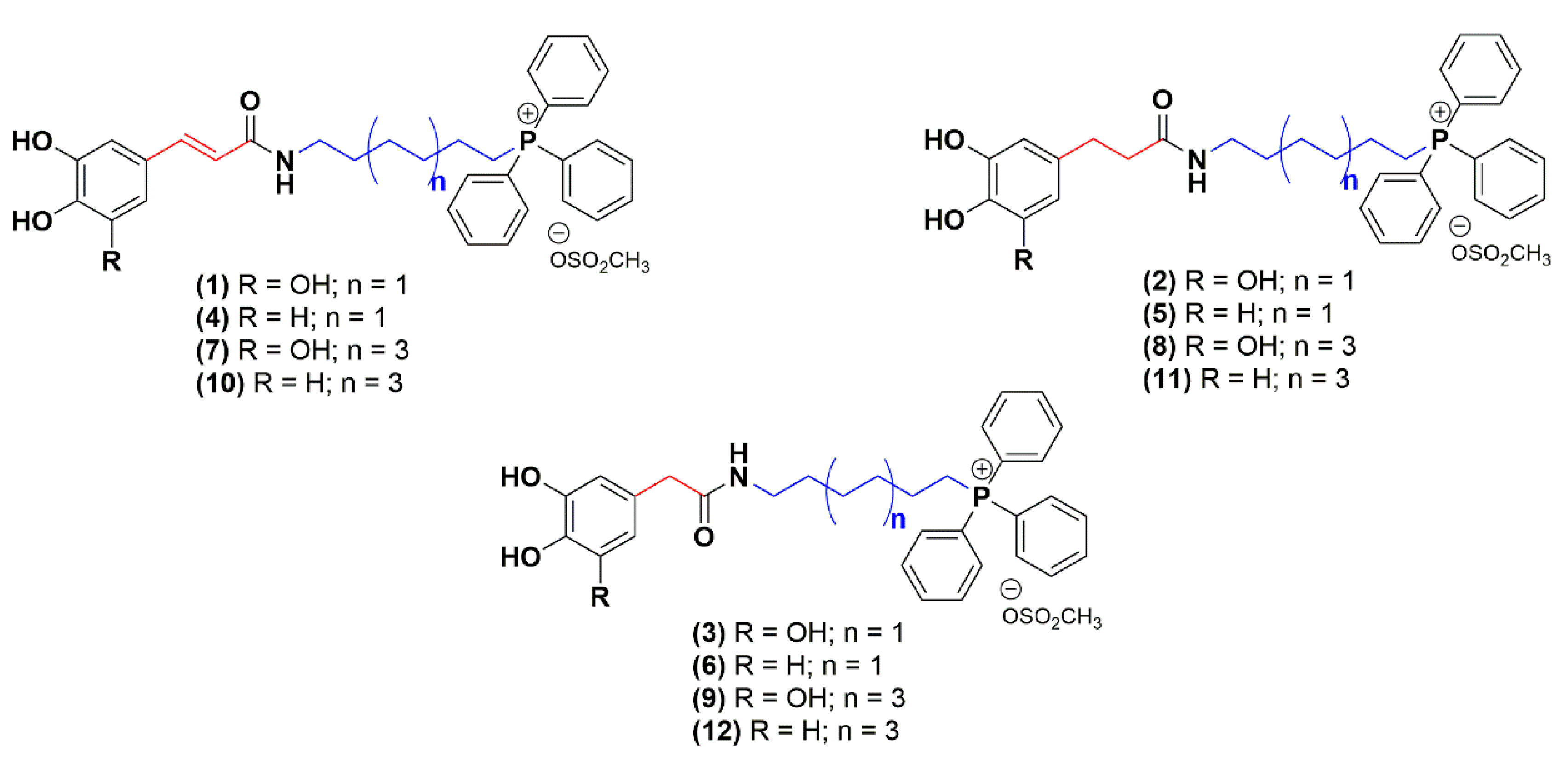
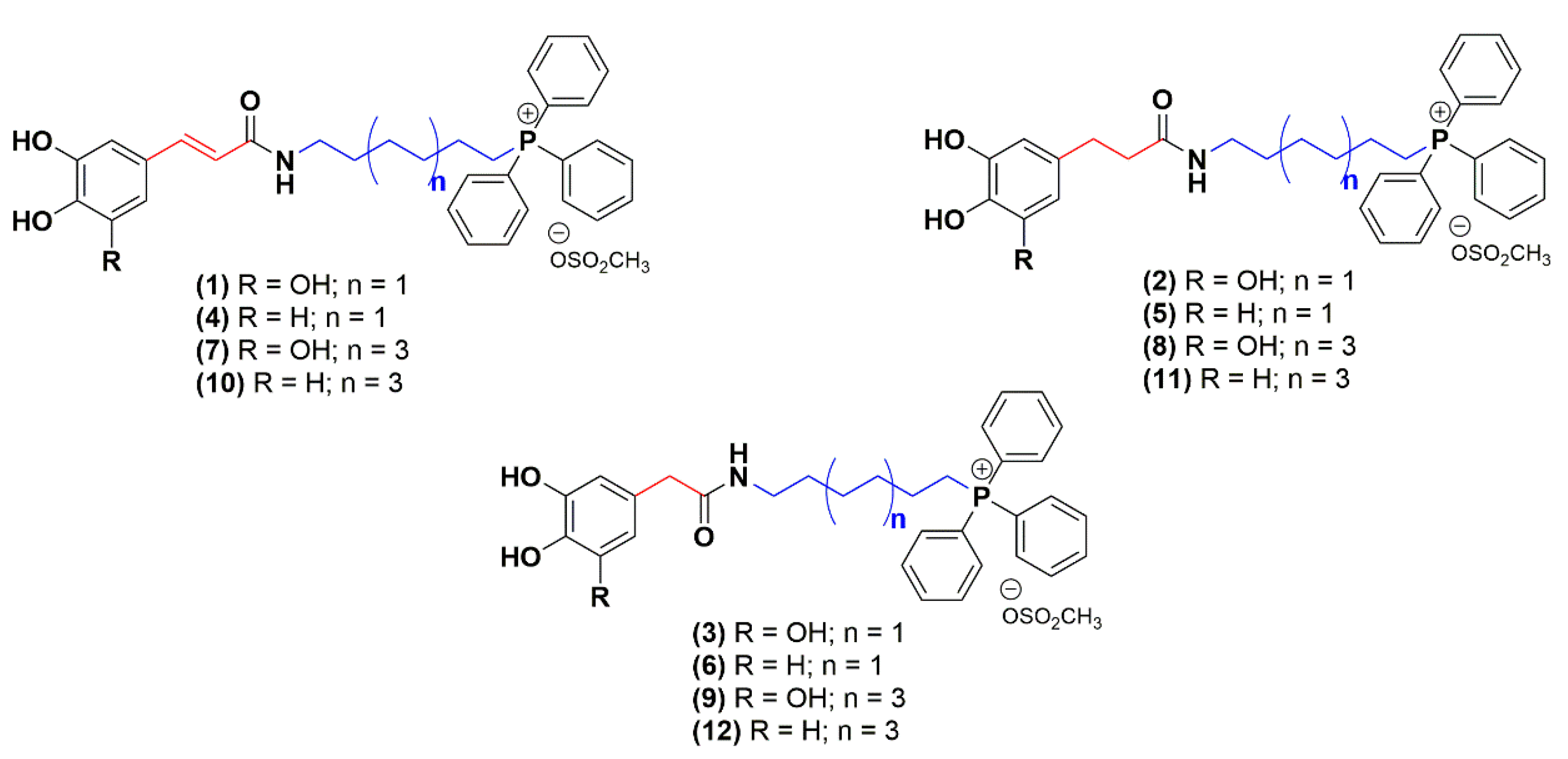
2.1. Chemistry
2.2. Evaluation of Cholinesterase Inhibitory Activity
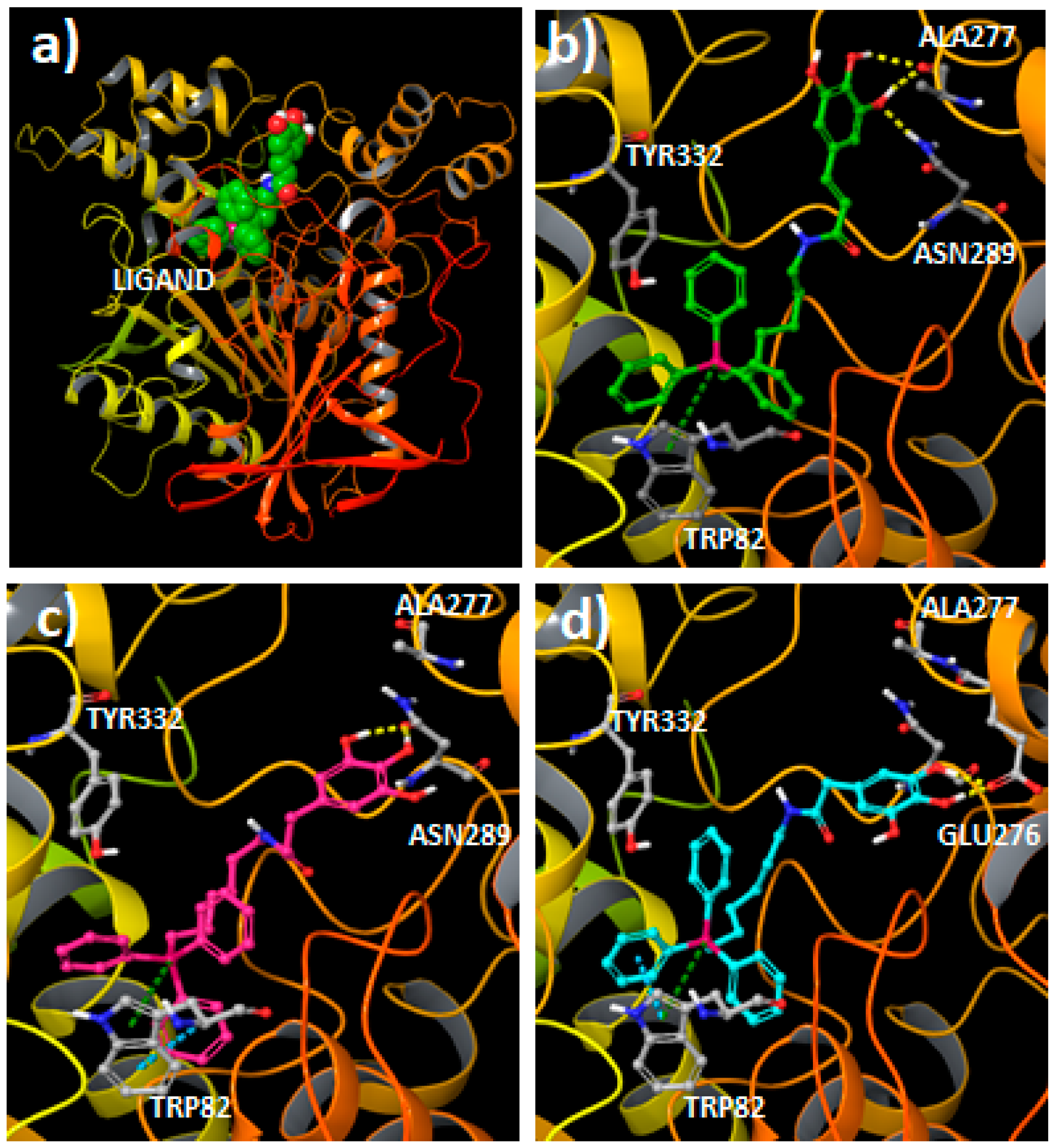
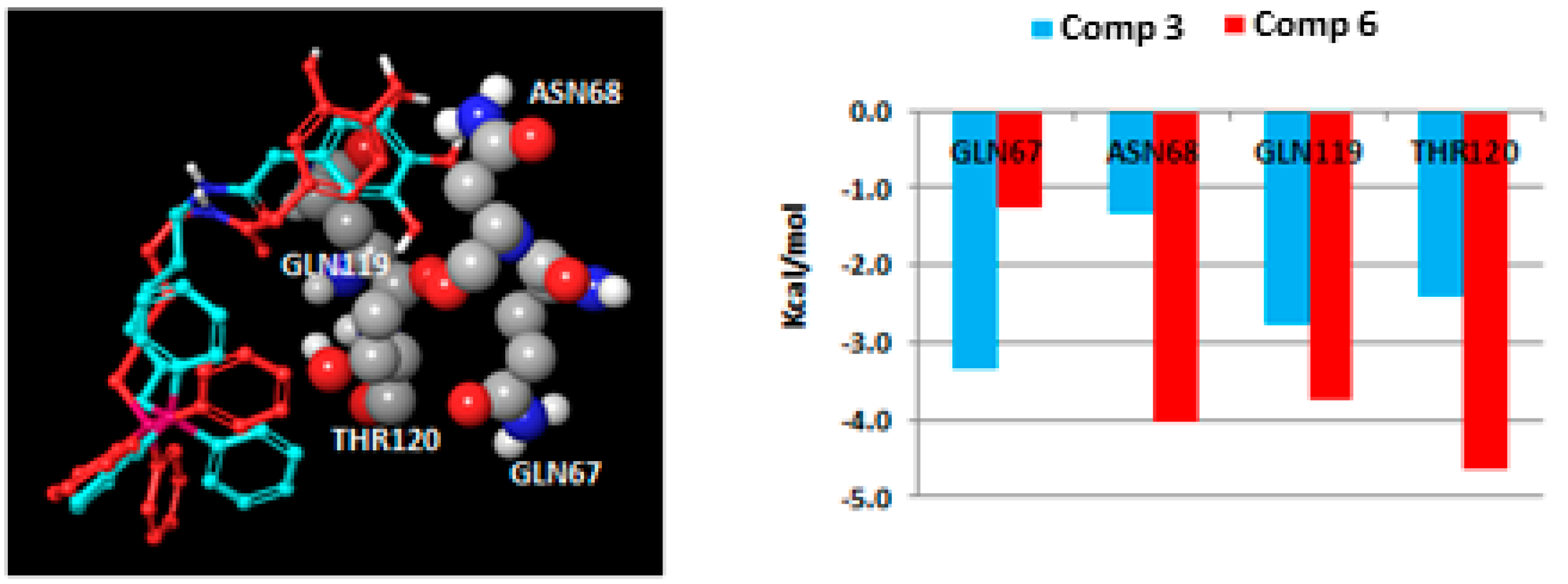
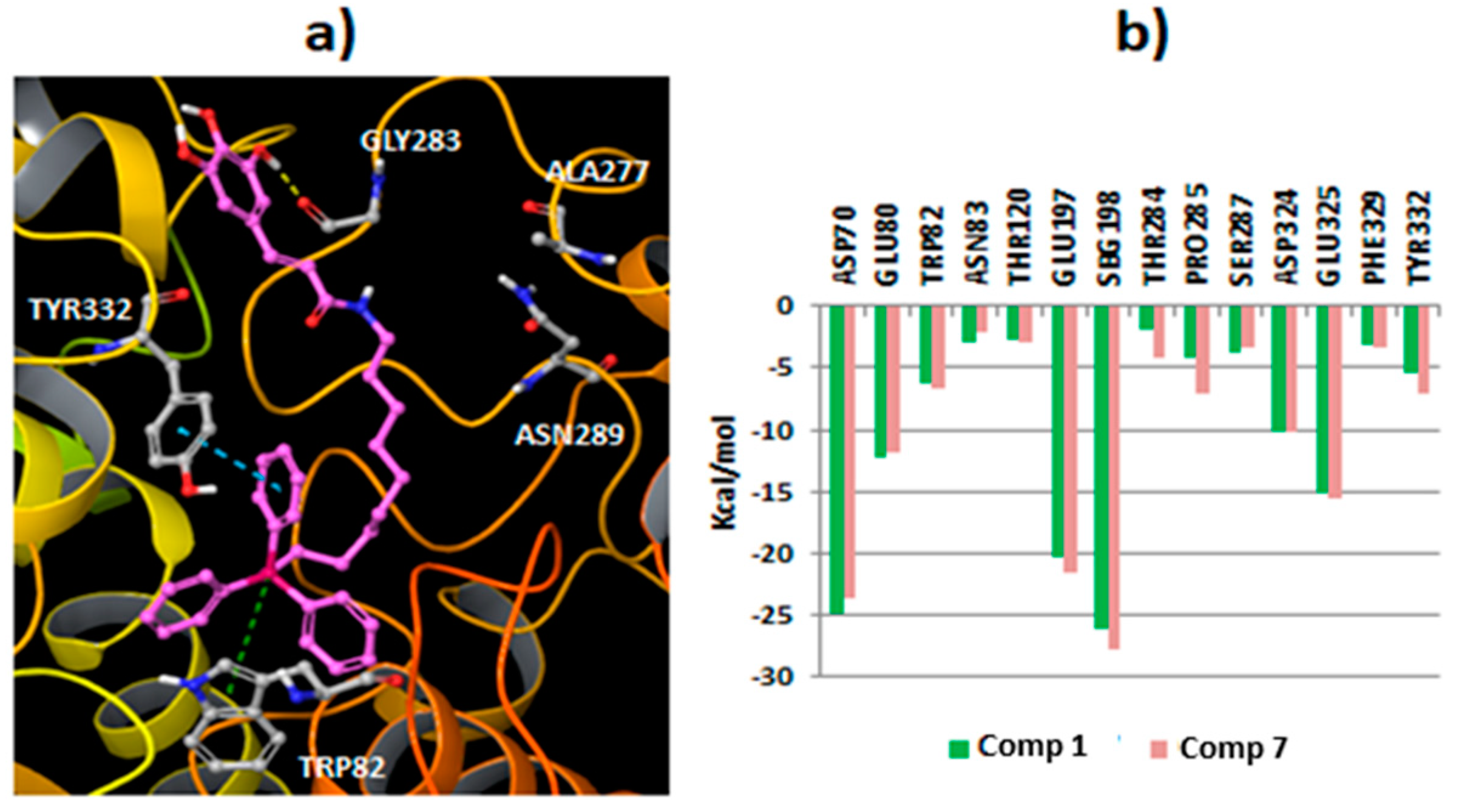
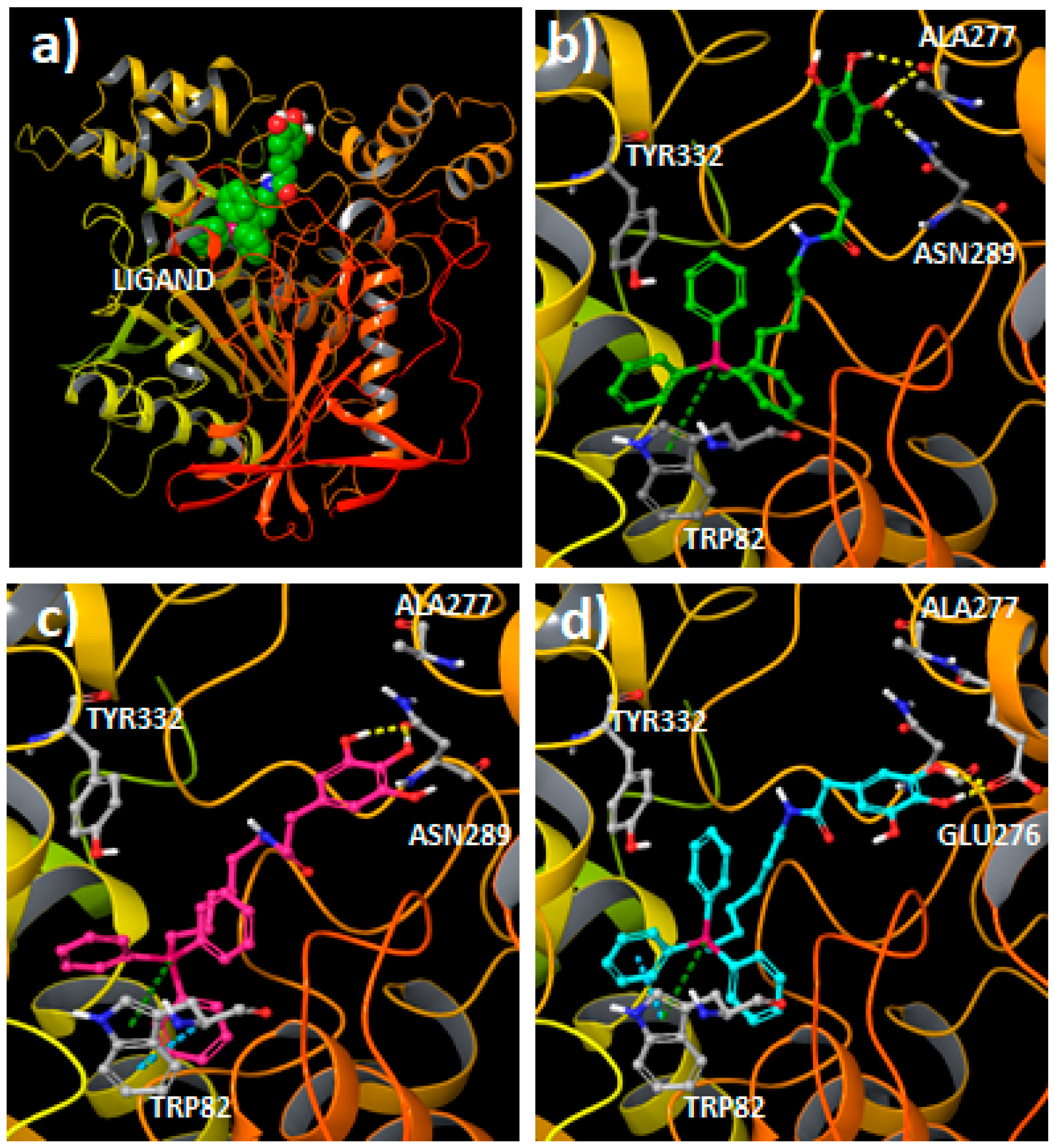
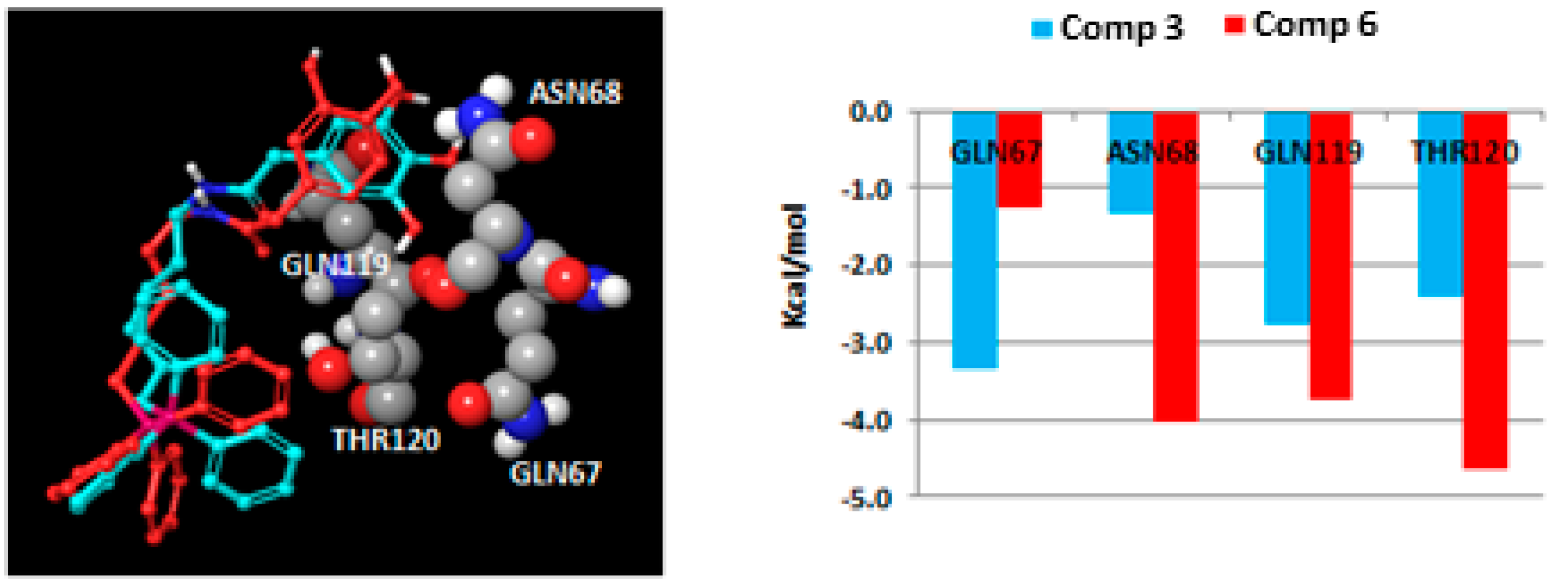
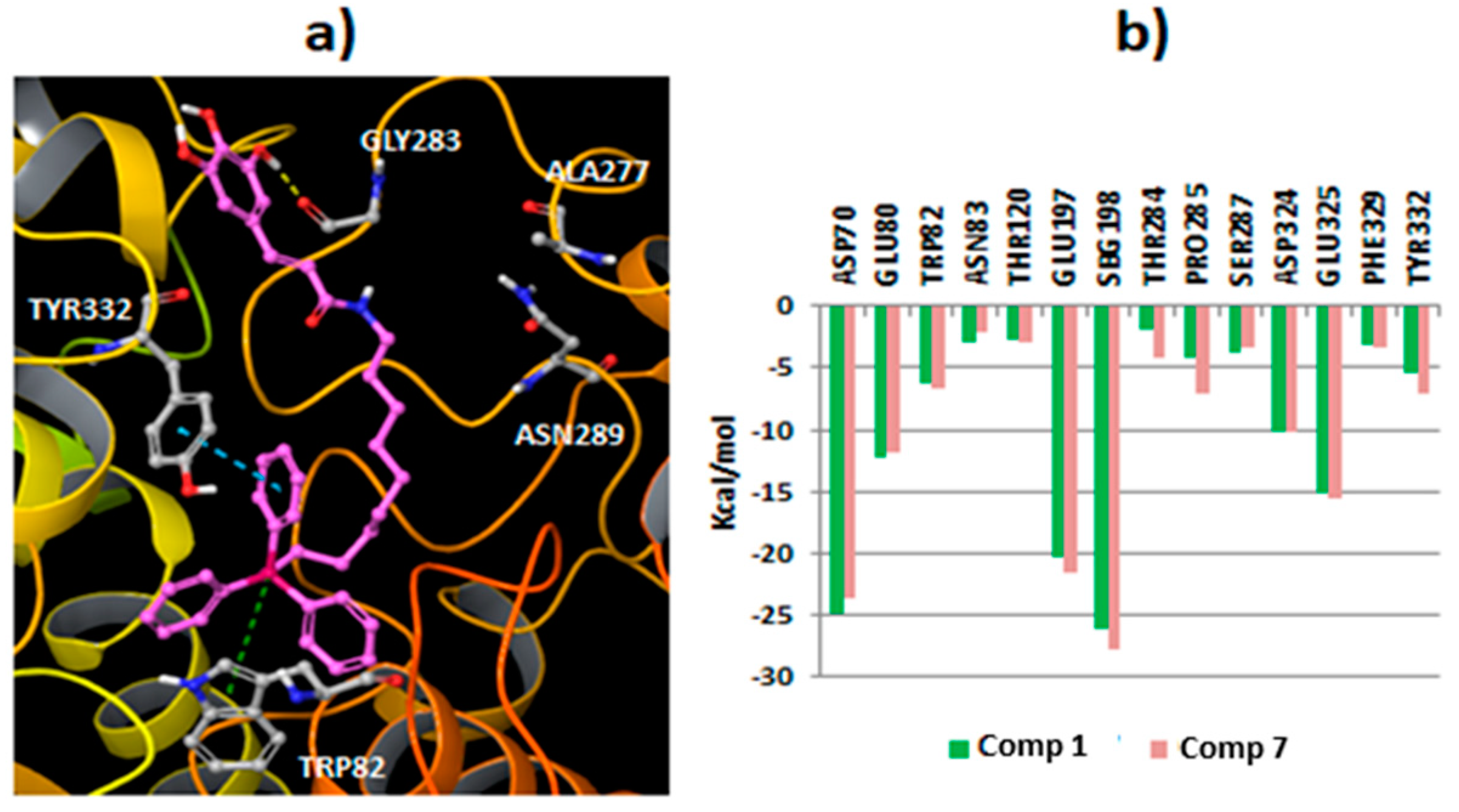
2.3. Molecular Docking Studies
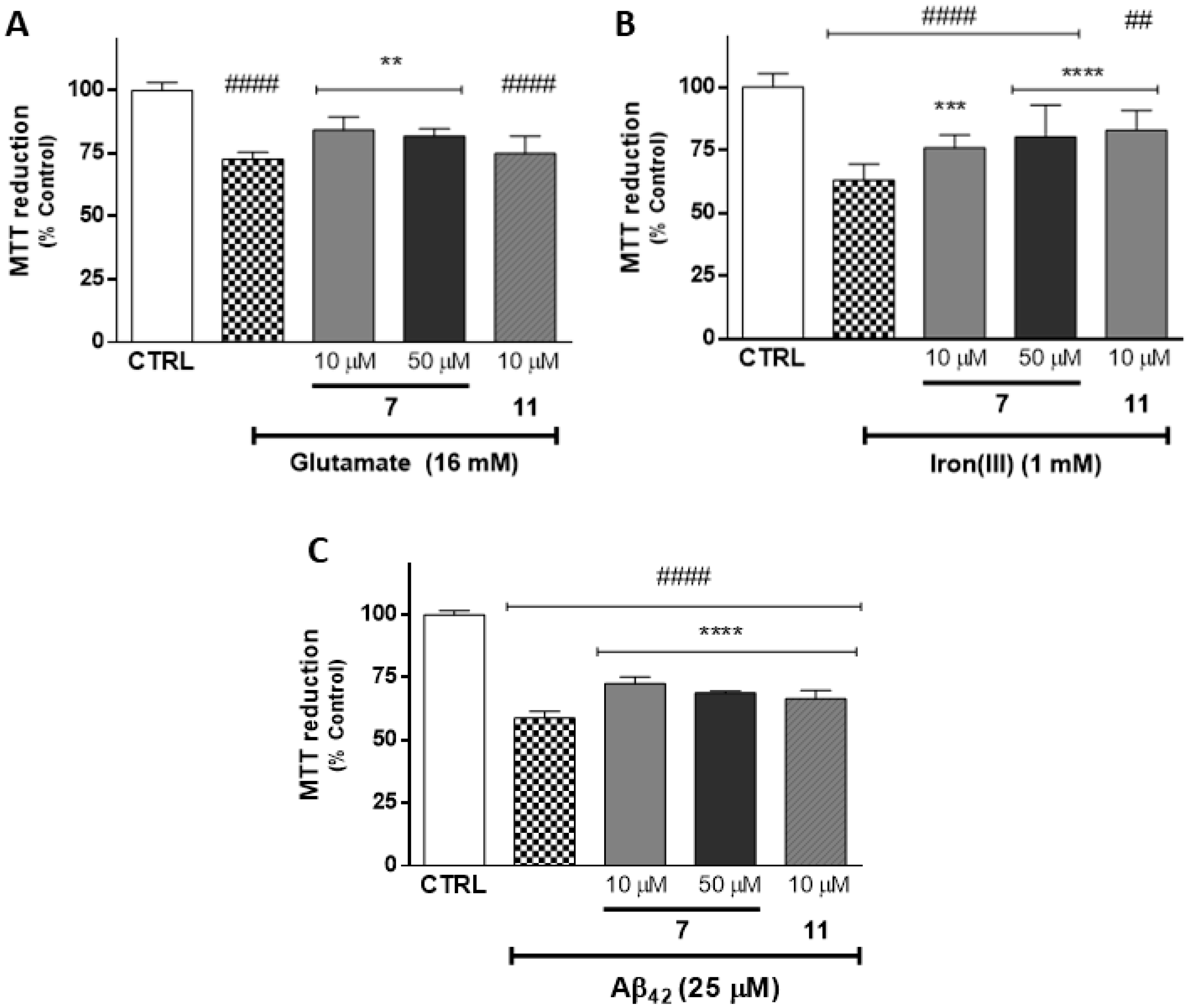
2.4. Evaluation of Neuroprotective Outline
3. Materials and Methods
3.1. Reagents and Apparatus
3.2. Chemistry
3.3. Enzymatic Assays
3.4. Molecular Docking Simulations
3.5. Cellular Culture Conditions
3.6. Evaluation of Neuroprotective Outline in SH-SY5Y Cells
3.7. Statistical Analysis in Cellular Model
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Y.; Wang, H.; Chen, H. AChE Inhibition-based Multi-target-directed Ligands, a Novel Pharmacological Approach for the Symptomatic and Disease-modifying Therapy of Alzheimer’s Disease. Curr. Neuropharmacol. 2016, 14, 364–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markmee, S.; Ruchirawat, S.; Prachyawarakorn, V.; Ingkaninan, K.; Khorana, N. Isoquinoline derivatives as potential acetylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 2170–2172. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; Zhang, T.; Zhang, L.; Mei, X.; Dong, M.; Zhang, K.; Ning, J. Design, synthesis and bioactivity of novel phthalimide derivatives as acetylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2380–2382. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Cholinesterase inhibitors: New roles and therapeutic alternatives. Pharmacol. Res. 2004, 50, 433–440. [Google Scholar] [CrossRef]
- Traub, M.; Freedman, S.B. The Implication of Current Therapeutic Approaches for the Cholinergic Hypothesis of Dementia. Dement. Geriatr. Cogn. 1992, 3, 189–192. [Google Scholar] [CrossRef]
- Pan, L.; Tan, J.H.; Hou, J.Q.; Huang, S.L.; Gu, L.Q.; Huang, Z.S. Design, synthesis and evaluation of isaindigotone derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 3790–3793. [Google Scholar] [CrossRef]
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychop. 2006, 9, 101–124. [Google Scholar] [CrossRef]
- Wright, C.I.; Geula, C.; Mesulam, M.M. Neuroglial cholinesterases in the normal brain and in Alzheimer’s disease: Relationship to plaques, tangles, and patterns of selective vulnerability. Ann. Neurol. 1993, 34, 373–384. [Google Scholar] [CrossRef]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131. [Google Scholar] [CrossRef]
- Silva, T.; Reis, J.; Teixeira, J.; Borges, F. Alzheimer’s disease, enzyme targets and drug discovery struggles: From natural products to drug prototypes. Ageing Res. Rev. 2014, 15, 116–145. [Google Scholar] [CrossRef]
- Li, B.; Stribley, J.A.; Ticu, A.; Xie, W.; Schopfer, L.M.; Hammond, P.; Brimijoin, S.; Hinrichs, S.H.; Lockridge, O. Abundant Tissue Butyrylcholinesterase and Its Possible Function in the Acetylcholinesterase Knockout Mouse. J. Neurochem. 2000, 75, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Utsuki, T.; Yu, Q.S.; Zhu, X.; Holloway, H.W.; Perry, T.; Lee, B.; Ingram, D.K.; Lahiri, D.K. A New Therapeutic Target in Alzheimer’s Disease Treatment: Attention to Butyrylcholinesterase. Curr. Med. Res. Opin. 2001, 17, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; Wieckowska, A.W.; Panek, D.; Malawska, B. Recent Development of Multifunctional Agents as Potential Drug Candidates for the Treatment of Alzheimer’s Disease. Curr. Med. Chem. 2015, 22, 373–404. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Perry, R.H.; Blessed, G.; Tomlinson, B.E. Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol. Appl. Neurobiol. 1978, 4, 273–277. [Google Scholar] [CrossRef]
- Mesulam, M.; Geula, C. Butyrylcholinesterase reactivity differentiates the amyloid plaques of aging from those of dementia. Ann. Neurol. 1994, 36, 722–727. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef] [Green Version]
- Mesulam, M.M.; Guillozet, A.; Shaw, P.; Levey, A.; Duysen, E.G.; Lockridge, O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002, 110, 627–639. [Google Scholar] [CrossRef]
- Reid, G.A.; Chilukuri, N.; Darvesh, S. Butyrylcholinesterase and the cholinergic system. Neuroscience 2013, 234, 53–68. [Google Scholar] [CrossRef] [Green Version]
- Diamant, S.; Podoly, E.; Friedler, A.; Ligumsky, H.; Livnah, O.; Soreq, H. Butyrylcholinesterase attenuates amyloid fibril formation in vitro. Proc. Natl. Acad. Sci. USA 2006, 103, 8628–8633. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M.; Leoutsakos, J.M.; Corcoran, C.D.; Green, R.C.; Norton, M.C.; Welsh-Bohmer, K.A.; Tschanz, J.T.; Lyketsos, C.G. Effects of FDA approved medications for Alzheimer’s disease on clinical progression. Alzheimers Dement. 2012, 8, 180–187. [Google Scholar] [CrossRef] [Green Version]
- García-Ayllón, M.; Small, D.H.; Avila, J.; Sáez-Valero, J. Revisiting the Role of Acetylcholinesterase in Alzheimer’s Disease: Cross-Talk with P-tau and β-Amyloid. Front. Mol. Neurosci. 2011, 4, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacobini, E. Long-term stabilizing effect of cholinesterase inhibitors in the therapy of Alzheimer’ disease. J. Neural. Transm. Suppl. 2002, 62, 181–187. [Google Scholar] [CrossRef]
- Kaduszkiewicz, H.; Zimmermann, T.; Beck-Bornholdt, H.P.; Van den Bussche, H. Cholinesterase inhibitors for patients with Alzheimer’s disease: Systematic review of randomised clinical trials. Br. Med. J. 2005, 331, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folch, J.; Petrov, D.; Ettcheto, M.; Abad, S.; Sánchez-López, E.; García, M.L.; Olloquequi, J.; Beas-Zarate, C.; Auladell, C.; Camins, A. Current Research Therapeutic Strategies for Alzheimer’s Disease Treatment. Neural Plast. 2016, 2016, 8501693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate-induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Amyloid β-Protein and the Genetics of Alzheimer’s Disease. J. Biol. Chem. 1996, 271, 18295–18298. [Google Scholar] [CrossRef] [Green Version]
- Wardman, P.; Candeias, L.P. Fenton Chemistry: An Introduction. Radiat. Res. 1996, 145, 523–531. [Google Scholar] [CrossRef]
- Sun, Z.W.; Zhang, L.; Zhu, S.J.; Chen, W.C.; Mei, B. Excitotoxicity effects of glutamate on human neuroblastoma SH-SY5Y cells via oxidative damage. Neurosci. Bull. 2010, 26, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Benfeito, S.; Oliveira, C.; Fernandes, C.; Cagide, F.; Teixeira, J.; Amorim, R.; Garrido, J.; Martins, C.; Sarmento, B.; Silva, R.; et al. Fine-tuning the neuroprotective and blood-brain barrier permeability profile of multi-target agents designed to prevent progressive mitochondrial dysfunction. Eur. J. Med. Chem. 2019, 167, 525–545. [Google Scholar] [CrossRef]
- Teixeira, J.; Cagide, F.; Benfeito, S.; Soares, P.; Garrido, J.; Baldeiras, I.; Ribeiro, J.A.; Pereira, C.M.; Silva, A.F.; Andrade, P.B.; et al. Development of a Mitochondriotropic Antioxidant Based on Caffeic Acid: Proof of Concept on Cellular and Mitochondrial Oxidative Stress Models. J. Med. Chem. 2017, 60, 7084–7098. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Wandhammer, M.; de Koning, M.; van Grol, M.; Loiodice, M.; Saurel, L.; Noort, D.; Goeldner, M.; Nachon, F. A step toward the reactivation of aged cholinesterases-Crystal structure of ligands binding to aged human butyrylcholinesterase. Chem. Biol. Interact. 2013, 203, 19–23. [Google Scholar] [CrossRef]
- Schrödinger. Schrödinger Schrödinger Suite 2017-2. Available online: http://www.schrodinger.com/ (accessed on 30 August 2017).
- Bacalhau, P.; San Juan, A.A.; Goth, A.; Caldeira, A.T.; Martins, R.; Burke, A.J. Insights into (S)-rivastigmine inhibition of butyrylcholinesterase (BuChE): Molecular docking and saturation transfer difference NMR (STD-NMR). Bioorg. Chem. 2016, 67, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presgraves, S.P.; Ahmed, T.; Borwege, S.; Joyce, J.N. Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotox. Res. 2003, 5, 579–598. [Google Scholar] [CrossRef] [PubMed]
- Krishtal, J.; Bragina, O.; Metsla, K.; Palumaa, P.; Tõugu, V. In situ fibrillizing amyloid-beta 1-42 induces neurite degeneration and apoptosis of differentiated SH-SY5Y cells. PLoS ONE 2017, 12, e0186636. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.; Pinto, M.; Martins, C.; Gomes, M.J.; Sarmento, B.; Oliveira, P.J.; Remião, F.; Borges, F. Development of a PEGylated-Based Platform for Efficient Delivery of Dietary Antioxidants Across the Blood–Brain Barrier. Bioconj. Chem. 2018, 29, 1677–1689. [Google Scholar] [CrossRef]
- Oberley, T.D. Oxidative damage and cancer. Am. J. Pathol. 2002, 160, 403–408. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, S.; Lee, Y.; Lee, Y.; Kim, J. Rac1-NADPH oxidase-regulated generation of reactive oxygen species mediates glutamate-induced apoptosis in SH-SY5Y human neuroblastoma cells. Free Radic. Res. 2005, 39, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Das, P. Emergence of Alternative Structures in Amyloid Beta 1-42 Monomeric Landscape by N-terminal Hexapeptide Amyloid Inhibitors. Sci. Rep. 2017, 7, 9941. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.; Martins, C.; Fonseca, A.; Nunes, R.; Matos, M.J.; Silva, R.; Garrido, J.; Sarmento, B.; Remiao, F.; Otero-Espinar, F.J.; et al. PEGylated PLGA Nanoparticles as a Smart Carrier to Increase the Cellular Uptake of a Coumarin-Based Monoamine Oxidase B Inhibitor. ACS Appl. Mater. Interfaces 2018, 10, 39557–39569. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–12 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | IC50 (µM)—Mean (CI 95%) 1 | |||||
|---|---|---|---|---|---|---|
| Compound | R | A | n | eeAChE 2 | eqBChE 3 | SI 4 |
| 1 | OH | -HC = CH- | 1 | 2.94 (2.58−3.35) | 0.41 (0.36−0.47) | 7.2 |
| 2 | -H2C–CH2- | 6.76 (6.02−7.59) | 3.16 (2.80−3.56) | 2.1 | ||
| 3 | -CH2- | 8.98 (7.98−10.01) | 4.52 (3.52−5.82) | 2.0 | ||
| 4 | H | -HC = CH- | 1 | 6.32 (6.02−6.63) | 0.12 (0.11−0.14) | 52.7 |
| 5 | -H2C–CH2- | 1.08 (1.00−1.18) | 2.52 (1.79−3.53) | 0.4 | ||
| 6 | -CH2- | 3.88 (3.55−4.23) | 0.93 (0.76−1.14) | 4.2 | ||
| 7 | OH | -HC = CH- | 3 | 3.39 (3.03−3.79) | 0.11 (0.09−0.14) | 30.8 |
| 8 | -H2C–CH2- | 5.75 (5.47−6.04) | 0.80 (0.72−0.90) | 7.2 | ||
| 9 | -CH2- | 3.35 (2.78−4.03) | 0.53 (0.43−0.64) | 5.7 | ||
| 10 | H | -HC = CH- | 3 | 3.59 (3.32−3.89) | 0.15 (0.12−0.18) | 23.9 |
| 11 | -H2C–CH2- | 1.59 (1.47−1.73) | 0.32 (0.28−0.40) | 4.9 | ||
| 12 | -CH2- | 2.44 (2.13−2.79) | 0.24 (0.22−0.27) | 10.2 | ||
| Donepezil | 0.045 (0.039−0.052) | 1.98 (1.78−2.19) | --- | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benfeito, S.; Fernandes, C.; Vilar, S.; Remião, F.; Uriarte, E.; Borges, F. Exploring the Multi-Target Performance of Mitochondriotropic Antioxidants against the Pivotal Alzheimer’s Disease Pathophysiological Hallmarks. Molecules 2020, 25, 276. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25020276
Benfeito S, Fernandes C, Vilar S, Remião F, Uriarte E, Borges F. Exploring the Multi-Target Performance of Mitochondriotropic Antioxidants against the Pivotal Alzheimer’s Disease Pathophysiological Hallmarks. Molecules. 2020; 25(2):276. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25020276
Chicago/Turabian StyleBenfeito, Sofia, Carlos Fernandes, Santiago Vilar, Fernando Remião, Eugenio Uriarte, and Fernanda Borges. 2020. "Exploring the Multi-Target Performance of Mitochondriotropic Antioxidants against the Pivotal Alzheimer’s Disease Pathophysiological Hallmarks" Molecules 25, no. 2: 276. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25020276