In Depth Analysis of Kinase Cross Screening Data to Identify CAMKK2 Inhibitory Scaffolds

 , , , , ,
, , , , ,
Abstract
:1. Introduction
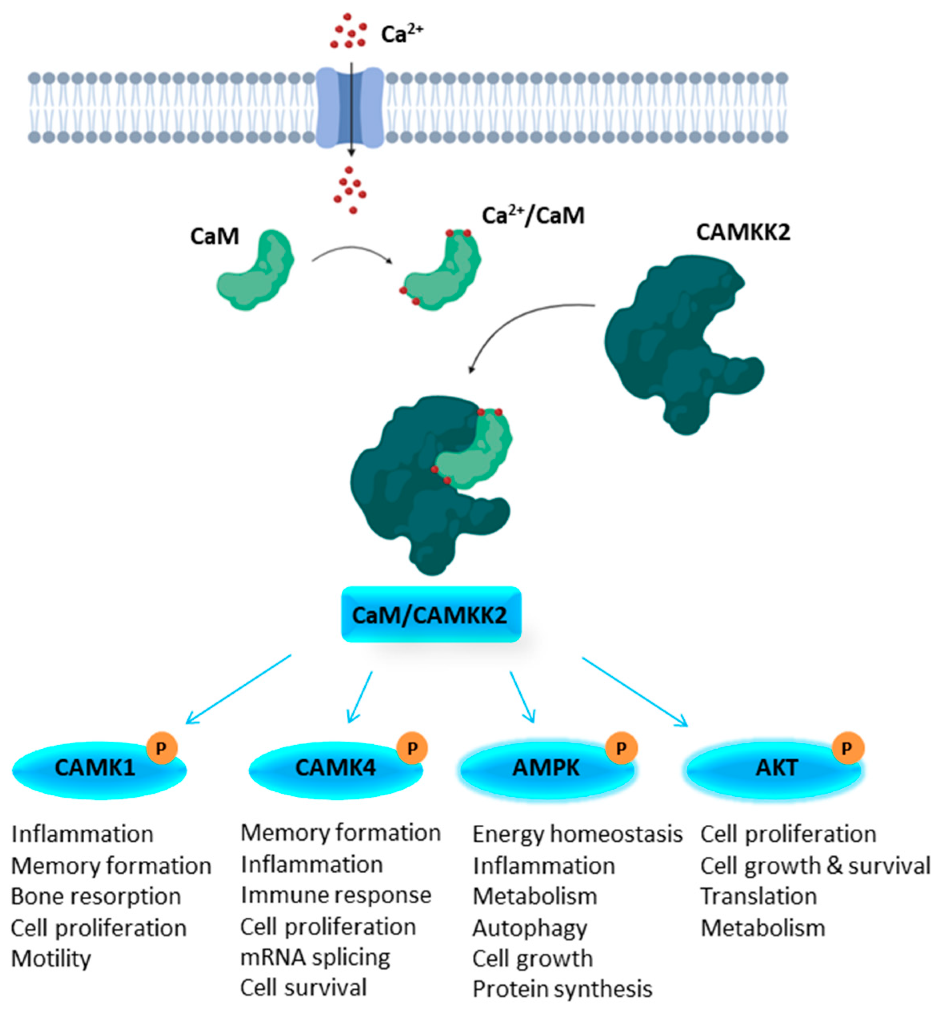
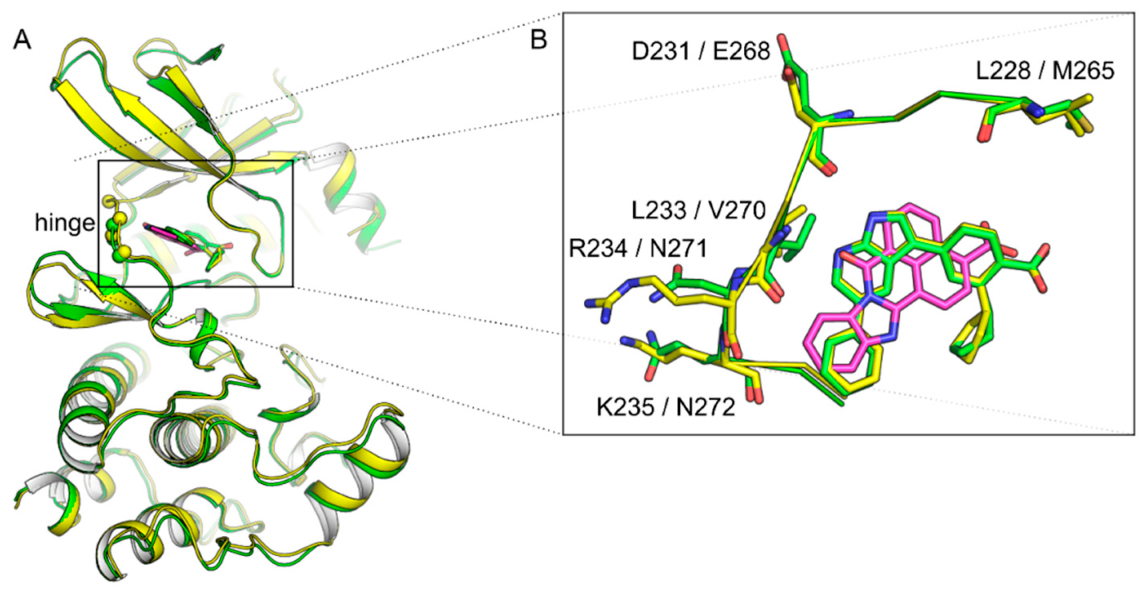
1.1. Structural Features of CAMKK1 & CAMKK2
1.2. CAMKK2 as a Potential Drug Discovery Target
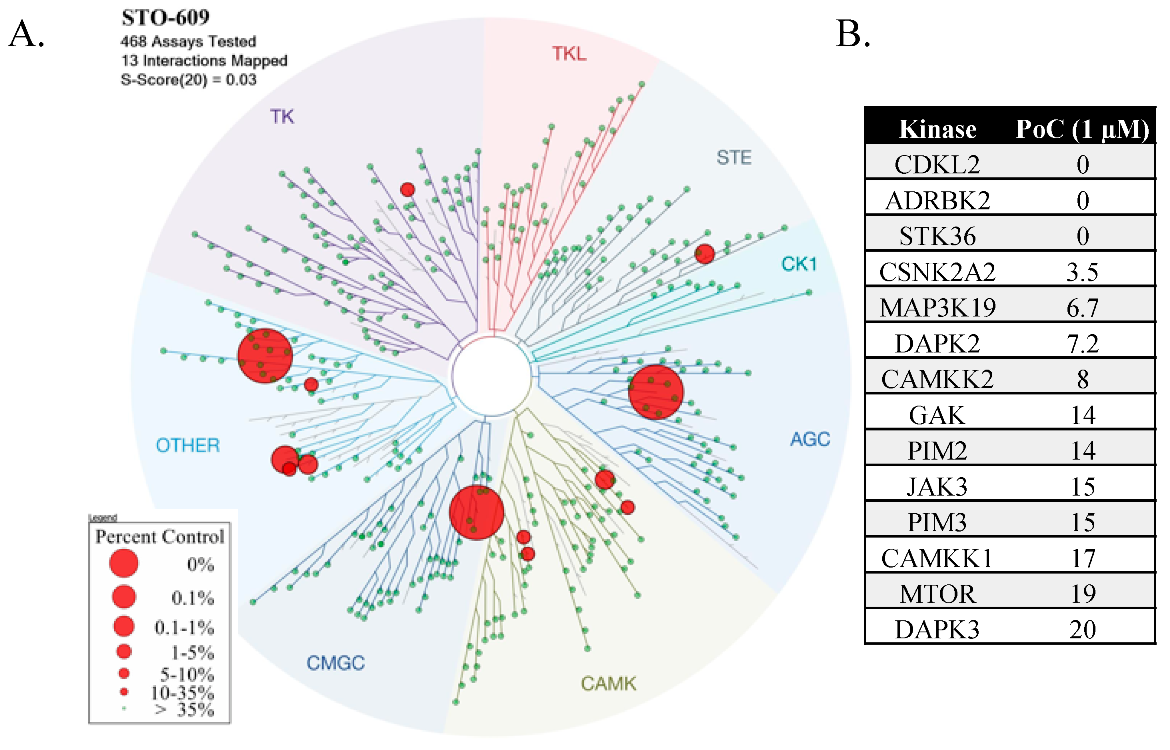
1.3. STO-609
2. Results
2.1. STO-609 Selectivity
2.2. Other CAMKK2 Tool Compounds
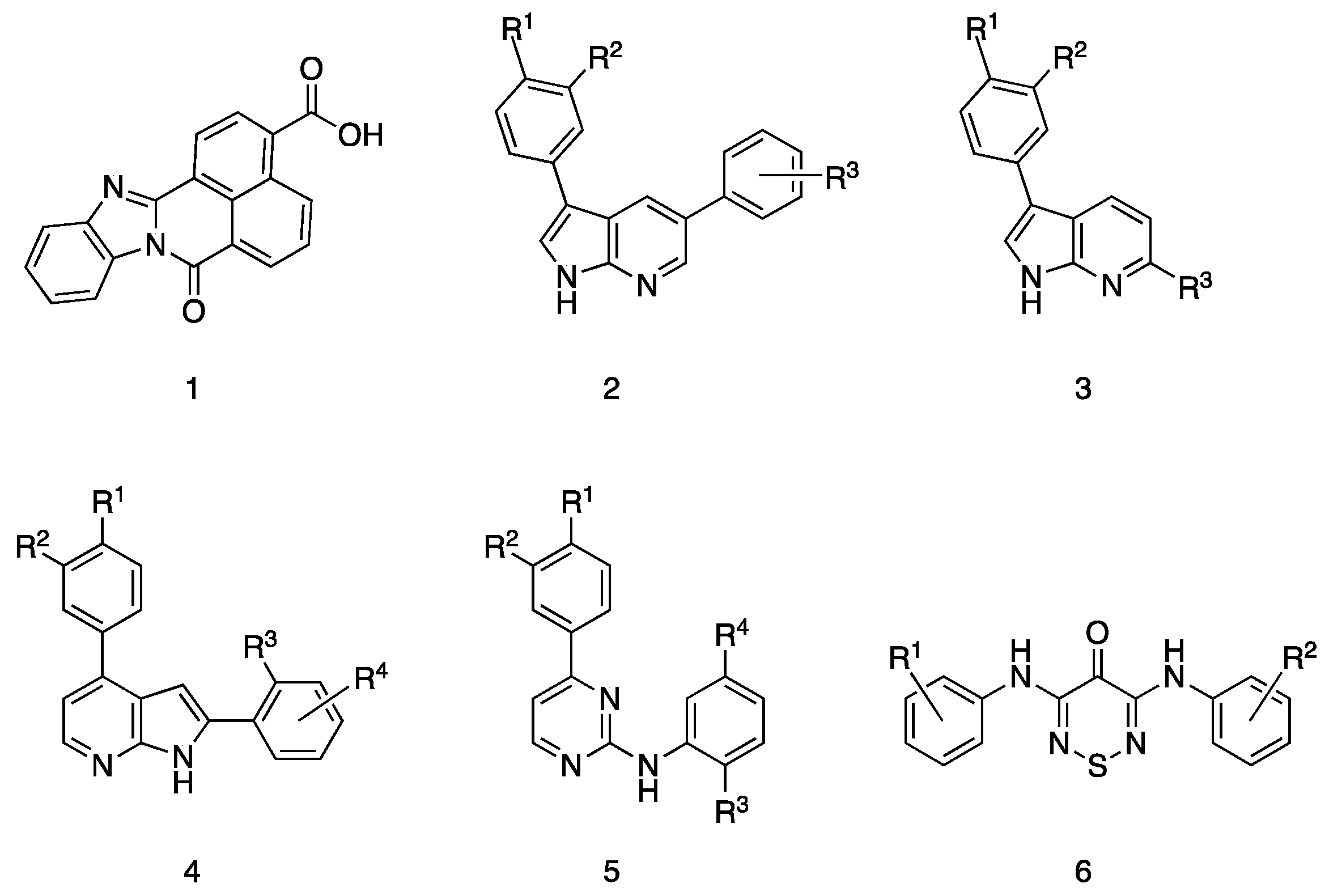
2.3. Literature Survey of CAMKK2 Inhibitor Chemotypes
2.4. Differential Scanning Fluorimetry and Enzymology
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. CAMKK1 and CAMKK2 DSF Assay
5.2. CAMKK2 Enzyme Assay
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Hook, S.S.; Means, A.R. Ca(2+)/CaM-dependent kinases: From activation to function. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 471–505. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.K.; Parameswaran, S. Calmodulin-binding proteins: A journey of 40 years. Cell Calcium 2018, 75, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Valencia, C.A.; Szostak, J.W.; Dong, B.; Liu, R. Scanning the human proteome for calmodulin-binding proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 5969–5974. [Google Scholar] [CrossRef] [Green Version]
- Tidow, H.; Nissen, P. Structural diversity of calmodulin binding to its target sites. FEBS J. 2013, 280, 5551–5565. [Google Scholar] [CrossRef]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Bayer, K.U.; Schulman, H. CaM Kinase: Still Inspiring at 40. Neuron 2019, 103, 380–394. [Google Scholar] [CrossRef]
- Soderling, T.R. The Ca2+-calmodulin-dependent protein kinase cascade. Trends Biochem. Sci. 1999, 24, 232–236. [Google Scholar] [CrossRef]
- De Souza Almeida Matos, A.L.; Oakhill, J.S.; Moreira, J.; Loh, K.; Galic, S.; Scott, J.W. Allosteric regulation of AMP-activated protein kinase by adenylate nucleotides and small-molecule drugs. Biochem. Soc. Trans. 2019, 47, 733–741. [Google Scholar] [CrossRef]
- Racioppi, L.; Means, A.R. Calcium/calmodulin-dependent protein kinase kinase 2: Roles in signaling and pathophysiology. J. Biol. Chem. 2012, 287, 31658–31665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcelo, K.L.; Means, A.R.; York, B. The Ca(2+)/Calmodulin/CaMKK2 Axis: Nature’s Metabolic CaMshaft. Trends Endocrinol. Metab. 2016, 27, 706–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.A.; Means, R.L.; Huang, Q.H.; Kemp, B.E.; Goldstein, E.G.; Selbert, M.A.; Edelman, A.M.; Fremeau, R.T.; Means, A.R. Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase beta. J. Biol. Chem. 1998, 273, 31880–31889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Selbert, M.A.; Goldstein, E.G.; Edelman, A.M.; Carling, D.; Hardie, D.G. 5’-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J. Biol. Chem. 1995, 270, 27186–27191. [Google Scholar] [CrossRef] [Green Version]
- Yano, S.; Tokumitsu, H.; Soderling, T.R. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 1998, 396, 584–587. [Google Scholar] [CrossRef]
- Gocher, A.M.; Azabdaftari, G.; Euscher, L.M.; Dai, S.; Karacosta, L.G.; Franke, T.F.; Edelman, A.M. Akt activation by Ca(2+)/calmodulin-dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J. Biol. Chem. 2017, 292, 14188–14204. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Testa, J.R.; Moore, R.; Larue, L. A portrait of AKT kinases: Human cancer and animal models depict a family with strong individualities. Cancer Biol. Ther. 2004, 3, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Picciotto, M.R.; Zoli, M.; Bertuzzi, G.; Nairn, A.C. Immunochemical localization of calcium/calmodulin-dependent protein kinase I. Synapse 1995, 20, 75–84. [Google Scholar] [CrossRef]
- Kamata, A.; Sakagami, H.; Tokumitsu, H.; Owada, Y.; Fukunaga, K.; Kondo, H. Spatiotemporal expression of four isoforms of Ca2+/calmodulin-dependent protein kinase I in brain and its possible roles in hippocampal dendritic growth. Neurosci. Res. 2007, 57, 86–97. [Google Scholar] [CrossRef]
- Joseph, J.D.; Means, A.R. Identification and characterization of two Ca2+/CaM-dependent protein kinases required for normal nuclear division in Aspergillus nidulans. J. Biol. Chem. 2000, 275, 38230–38238. [Google Scholar] [CrossRef] [Green Version]
- Skelding, K.A.; Rostas, J.A.; Verrills, N.M. Controlling the cell cycle: The role of calcium/calmodulin-stimulated protein kinases I and II. Cell Cycle 2011, 10, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wayman, G.A.; Kaech, S.; Grant, W.F.; Davare, M.; Impey, S.; Tokumitsu, H.; Nozaki, N.; Banker, G.; Soderling, T.R. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J. Neurosci. 2004, 24, 3786–3794. [Google Scholar] [CrossRef]
- Ang, E.S.; Zhang, P.; Steer, J.H.; Tan, J.W.; Yip, K.; Zheng, M.H.; Joyce, D.A.; Xu, J. Calcium/calmodulin-dependent kinase activity is required for efficient induction of osteoclast differentiation and bone resorption by receptor activator of nuclear factor kappa B ligand (RANKL). J. Cell Physiol. 2007, 212, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Condon, J.C.; Pezzi, V.; Drummond, B.M.; Yin, S.; Rainey, W.E. Calmodulin-dependent kinase I regulates adrenal cell expression of aldosterone synthase. Endocrinology 2002, 143, 3651–3657. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, J.M.; Guire, E.S.; Saneyoshi, T.; Soderling, T.R. Calmodulin-dependent kinase kinase/calmodulin kinase I activity gates extracellular-regulated kinase-dependent long-term potentiation. J. Neurosci. 2005, 25, 1281–1290. [Google Scholar] [CrossRef]
- Takemoto-Kimura, S.; Ageta-Ishihara, N.; Nonaka, M.; Adachi-Morishima, A.; Mano, T.; Okamura, M.; Fujii, H.; Fuse, T.; Hoshino, M.; Suzuki, S.; et al. Regulation of dendritogenesis via a lipid-raft-associated Ca2+/calmodulin-dependent protein kinase CLICK-III/CaMKIgamma. Neuron 2007, 54, 755–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmstede, C.A.; Jensen, K.F.; Sahyoun, N.E. Ca2+/calmodulin-dependent protein kinase enriched in cerebellar granule cells. Identification of a novel neuronal calmodulin-dependent protein kinase. J. Biol. Chem. 1989, 264, 5866–5875. [Google Scholar]
- Kitsos, C.M.; Sankar, U.; Illario, M.; Colomer-Font, J.M.; Duncan, A.W.; Ribar, T.J.; Reya, T.; Means, A.R. Calmodulin-dependent protein kinase IV regulates hematopoietic stem cell maintenance. J. Biol. Chem. 2005, 280, 33101–33108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.Y.; Means, A.R. Ca(2+)/calmodulin-dependent protein kinase IV is expressed in spermatids and targeted to chromatin and the nuclear matrix. J. Biol. Chem. 2000, 275, 7994–7999. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Y.; Gonzalez-Robayna, I.J.; Richards, J.S.; Means, A.R. Female fertility is reduced in mice lacking Ca2+/calmodulin-dependent protein kinase IV. Endocrinology 2000, 141, 4777–4783. [Google Scholar] [CrossRef]
- Kimura, Y.; Corcoran, E.E.; Eto, K.; Gengyo-Ando, K.; Muramatsu, M.A.; Kobayashi, R.; Freedman, J.H.; Mitani, S.; Hagiwara, M.; Means, A.R.; et al. CaMK cascade activates CRE-mediated transcription in neurons of Caenorhabditis elegans. EMBO Rep. 2002, 3, 962–966. [Google Scholar] [CrossRef] [Green Version]
- Bleier, J.; Toliver, A. Exploring the Role of CaMKIV in Homeostatic Plasticity. J. Neurosci. 2017, 37, 11520–11522. [Google Scholar] [CrossRef] [PubMed]
- Takemura, M.; Mishima, T.; Wang, Y.; Kasahara, J.; Fukunaga, K.; Ohashi, K.; Mizuno, K. Ca2+/calmodulin-dependent protein kinase IV-mediated LIM kinase activation is critical for calcium signal-induced neurite outgrowth. J. Biol. Chem. 2009, 284, 28554–28562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, F.; Qiu, C.S.; Liauw, J.; Robinson, D.A.; Ho, N.; Chatila, T.; Zhuo, M. Calcium calmodulin-dependent protein kinase IV is required for fear memory. Nat. Neurosci. 2002, 5, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Racioppi, L.; Means, A.R. Calcium/calmodulin-dependent kinase IV in immune and inflammatory responses: Novel routes for an ancient traveller. Trends Immunol. 2008, 29, 600–607. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Gradinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond. Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y.; Tokumitsu, H.; Inuzuka, H.; Murata-Hori, M.; Hosoya, H.; Kobayashi, R. Identification and characterization of novel components of a Ca2+/calmodulin-dependent protein kinase cascade in HeLa cells. FEBS Lett. 2003, 550, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Hsu, L.S.; Chen, G.D.; Lee, L.S.; Chi, C.W.; Cheng, J.F.; Chen, J.Y. Human Ca2+/calmodulin-dependent protein kinase kinase beta gene encodes multiple isoforms that display distinct kinase activity. J. Biol. Chem. 2001, 276, 31113–31123. [Google Scholar] [CrossRef] [Green Version]
- Santiago, A.D.S.; Counago, R.M.; Ramos, P.Z.; Godoi, P.H.C.; Massirer, K.B.; Gileadi, O.; Elkins, J.M. Structural Analysis of Inhibitor Binding to CAMKK1 Identifies Features Necessary for Design of Specific Inhibitors. Sci. Rep. 2018, 8, 14800. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Iwabu, M.; Ishikawa, Y.; Kobayashi, R. Differential regulatory mechanism of Ca2+/calmodulin-dependent protein kinase kinase isoforms. Biochemistry 2001, 40, 13925–13932. [Google Scholar] [CrossRef] [PubMed]
- Edelman, A.M.; Mitchelhill, K.I.; Selbert, M.A.; Anderson, K.A.; Hook, S.S.; Stapleton, D.; Goldstein, E.G.; Means, A.R.; Kemp, B.E. Multiple Ca(2+)-calmodulin-dependent protein kinase kinases from rat brain. Purification, regulation by Ca(2+)-calmodulin, and partial amino acid sequence. J. Biol. Chem. 1996, 271, 10806–10810. [Google Scholar] [CrossRef] [Green Version]
- Green, M.F.; Scott, J.W.; Steel, R.; Oakhill, J.S.; Kemp, B.E.; Means, A.R. Ca2+/Calmodulin-dependent protein kinase kinase beta is regulated by multisite phosphorylation. J. Biol. Chem. 2011, 286, 28066–28079. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.W.; Park, E.; Rodriguiz, R.M.; Oakhill, J.S.; Issa, S.M.; O’Brien, M.T.; Dite, T.A.; Langendorf, C.G.; Wetsel, W.C.; Means, A.R.; et al. Autophosphorylation of CaMKK2 generates autonomous activity that is disrupted by a T85S mutation linked to anxiety and bipolar disorder. Sci. Rep. 2015, 5, 14436. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, M.T.; Oakhill, J.S.; Ling, N.X.; Langendorf, C.G.; Hoque, A.; Dite, T.A.; Means, A.R.; Kemp, B.E.; Scott, J.W. Impact of Genetic Variation on Human CaMKK2 Regulation by Ca(2+)-Calmodulin and Multisite Phosphorylation. Sci. Rep. 2017, 7, 43264. [Google Scholar] [CrossRef]
- Wayman, G.A.; Tokumitsu, H.; Soderling, T.R. Inhibitory cross-talk by cAMP kinase on the calmodulin-dependent protein kinase cascade. J. Biol. Chem. 1997, 272, 16073–16076. [Google Scholar] [CrossRef] [Green Version]
- Price, D.J.; Drewry, D.H.; Schaller, L.T.; Thompson, B.D.; Reid, P.R.; Maloney, P.R.; Liang, X.; Banker, P.; Buckholz, R.G.; Selley, P.K.; et al. An orally available, brain-penetrant CAMKK2 inhibitor reduces food intake in rodent model. Bioorg. Med. Chem. Lett. 2018, 28, 1958–1963. [Google Scholar] [CrossRef] [PubMed]
- Kukimoto-Niino, M.; Yoshikawa, S.; Takagi, T.; Ohsawa, N.; Tomabechi, Y.; Terada, T.; Shirouzu, M.; Suzuki, A.; Lee, S.; Yamauchi, T.; et al. Crystal structure of the Ca(2)(+)/calmodulin-dependent protein kinase kinase in complex with the inhibitor STO-609. J. Biol. Chem. 2011, 286, 22570–22579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asquith, C.R.M.; Godoi, P.H.; Counago, R.M.; Laitinen, T.; Scott, J.W.; Langendorf, C.G.; Oakhill, J.S.; Drewry, D.H.; Zuercher, W.J.; Koutentis, P.A.; et al. 1,2,6-Thiadiazinones as Novel Narrow Spectrum Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CaMKK2) Inhibitors. Molecules 2018, 23, 1221. [Google Scholar] [CrossRef] [Green Version]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [Green Version]
- Karacosta, L.G.; Foster, B.A.; Azabdaftari, G.; Feliciano, D.M.; Edelman, A.M. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J. Biol. Chem. 2012, 287, 24832–24843. [Google Scholar] [CrossRef] [Green Version]
- Shima, T.; Mizokami, A.; Miyagi, T.; Kawai, K.; Izumi, K.; Kumaki, M.; Ofude, M.; Zhang, J.; Keller, E.T.; Namiki, M. Down-regulation of calcium/calmodulin-dependent protein kinase kinase 2 by androgen deprivation induces castration-resistant prostate cancer. Prostate 2012, 72, 1789–1801. [Google Scholar] [CrossRef] [Green Version]
- Subbannayya, Y.; Syed, N.; Barbhuiya, M.A.; Raja, R.; Marimuthu, A.; Sahasrabuddhe, N.; Pinto, S.M.; Manda, S.S.; Renuse, S.; Manju, H.C.; et al. Calcium calmodulin dependent kinase kinase 2-a novel therapeutic target for gastric adenocarcinoma. Cancer Biol. Ther. 2015, 16, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.; Marcelo, K.L.; Rajapakshe, K.; Coarfa, C.; Dean, A.; Wilganowski, N.; Robinson, H.; Sevick, E.; Bissig, K.D.; Goldie, L.C.; et al. The camKK2/camKIV relay is an essential regulator of hepatic cancer. Hepatology 2015, 62, 505–520. [Google Scholar] [CrossRef]
- Liu, D.M.; Wang, H.J.; Han, B.; Meng, X.Q.; Chen, M.H.; Yang, D.B.; Sun, Y.; Li, Y.L.; Jiang, C.L. CAMKK2, Regulated by Promoter Methylation, is a Prognostic Marker in Diffuse Gliomas. CNS Neurosci. Ther. 2016, 22, 518–524. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Mora, O.G.; LaHair, M.M.; McCubrey, J.A.; Franklin, R.A. Calcium/calmodulin-dependent kinase I and calcium/calmodulin-dependent kinase kinase participate in the control of cell cycle progression in MCF-7 human breast cancer cells. Cancer Res. 2005, 65, 5408–5416. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, J.M.; Abell, E.; Wagner, A.; Davare, M.A. ERK activation and cell growth require CaM kinases in MCF-7 breast cancer cells. Mol. Cell. Biochem. 2010, 335, 155–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davare, M.A.; Saneyoshi, T.; Soderling, T.R. Calmodulin-kinases regulate basal and estrogen stimulated medulloblastoma migration via Rac1. J. Neurooncol. 2011, 104, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Frigo, D.E.; Howe, M.K.; Wittmann, B.M.; Brunner, A.M.; Cushman, I.; Wang, Q.; Brown, M.; Means, A.R.; McDonnell, D.P. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011, 71, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; He, H.C.; Han, Z.D.; Wan, Y.P.; Luo, H.W.; Huang, Y.Q.; Cai, C.; Liang, Y.X.; Dai, Q.S.; Jiang, F.N.; et al. MicroRNA-224 and its target CAMKK2 synergistically influence tumor progression and patient prognosis in prostate cancer. Tumour Biol. 2015, 36, 1983–1991. [Google Scholar] [CrossRef]
- Ma, Z.; Wen, D.; Wang, X.; Yang, L.; Liu, T.; Liu, J.; Zhu, J.; Fang, X. Growth inhibition of human gastric adenocarcinoma cells in vitro by STO-609 is independent of calcium/calmodulin-dependent protein kinase kinase-beta and adenosine monophosphate-activated protein kinase. Am. J. Transl. Res. 2016, 8, 1164–1171. [Google Scholar]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Lonergan, P.E.; Tindall, D.J. Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 2011, 10, 20. [Google Scholar]
- Yasui, K.; Hashimoto, E.; Komorizono, Y.; Koike, K.; Arii, S.; Imai, Y.; Shima, T.; Kanbara, Y.; Saibara, T.; Mori, T.; et al. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clin. Gastroenterol. Hepatol. 2011, 9, 428–433, quiz e50. [Google Scholar] [CrossRef]
- Takuma, Y.; Nouso, K. Nonalcoholic steatohepatitis-associated hepatocellular carcinoma: Our case series and literature review. World J. Gastroenterol. 2010, 16, 1436–1441. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.A.; Ribar, T.J.; Lin, F.; Noeldner, P.K.; Green, M.F.; Muehlbauer, M.J.; Witters, L.A.; Kemp, B.E.; Means, A.R. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008, 7, 377–388. [Google Scholar] [CrossRef] [Green Version]
- York, B.; Li, F.; Lin, F.; Marcelo, K.L.; Mao, J.; Dean, A.; Gonzales, N.; Gooden, D.; Maity, S.; Coarfa, C.; et al. Pharmacological inhibition of CaMKK2 with the selective antagonist STO-609 regresses NAFLD. Sci. Rep. 2017, 7, 11793. [Google Scholar] [CrossRef] [PubMed]
- Racioppi, L.; Nelson, E.R.; Huang, W.; Mukherjee, D.; Lawrence, S.A.; Lento, W.; Masci, A.M.; Jiao, Y.; Park, S.; York, B.; et al. CaMKK2 in myeloid cells is a key regulator of the immune-suppressive microenvironment in breast cancer. Nat. Commun. 2019, 10, 2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cary, R.L.; Waddell, S.; Racioppi, L.; Long, F.; Novack, D.V.; Voor, M.J.; Sankar, U. Inhibition of Ca(2)(+)/calmodulin-dependent protein kinase kinase 2 stimulates osteoblast formation and inhibits osteoclast differentiation. J. Bone Miner. Res. 2013, 28, 1599–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, Z.J.; Cary, R.L.; Yang, C.; Novack, D.V.; Voor, M.J.; Sankar, U. Inhibition of CaMKK2 reverses age-associated decline in bone mass. Bone 2015, 75, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.; Ribar, T.J.; Means, A.R. The Ca2+/calmodulin-dependent protein kinase kinase, CaMKK2, inhibits preadipocyte differentiation. Endocrinology 2011, 152, 3668–3679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, X.; Cui, C.; Wang, C.; Wu, G.; Chen, H.; Lu, Z.; Chen, X.; Wang, L.; Huang, J.; Geng, H.; et al. CAMKs support development of acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, T.J.; Macdonald, S.J. The impact of aromatic ring count on compound developability-are too many aromatic rings a liability in drug design? Drug Discov. Today 2009, 14, 1011–1020. [Google Scholar] [CrossRef]
- Ritchie, T.J.; Macdonald, S.J.; Young, R.J.; Pickett, S.D. The impact of aromatic ring count on compound developability: Further insights by examining carbo- and hetero-aromatic and -aliphatic ring types. Drug Discov. Today 2011, 16, 164–171. [Google Scholar] [CrossRef]
- Timm, M.; Saaby, L.; Moesby, L.; Hansen, E.W. Considerations regarding use of solvents in in vitro cell based assays. Cytotechnology 2013, 65, 887–894. [Google Scholar] [CrossRef] [Green Version]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Morishita, D.; Katayama, R.; Sekimizu, K.; Tsuruo, T.; Fujita, N. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res. 2008, 68, 5076–5085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, C.; Nakamoto, Y.; Lu, P.; Tsuneyama, K.; Popivanova, B.K.; Kaneko, S.; Mukaida, N. Aberrant expression of serine/threonine kinase Pim-3 in hepatocellular carcinoma development and its role in the proliferation of human hepatoma cell lines. Int. J. Cancer 2005, 114, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.Y.; Popivanova, B.K.; Nagai, Y.; Ishikura, H.; Fujii, C.; Mukaida, N. Pim-3, a proto-oncogene with serine/threonine kinase activity, is aberrantly expressed in human pancreatic cancer and phosphorylates bad to block bad-mediated apoptosis in human pancreatic cancer cell lines. Cancer Res. 2006, 66, 6741–6747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popivanova, B.K.; Li, Y.Y.; Zheng, H.; Omura, K.; Fujii, C.; Tsuneyama, K.; Mukaida, N. Proto-oncogene, Pim-3 with serine/threonine kinase activity, is aberrantly expressed in human colon cancer cells and can prevent Bad-mediated apoptosis. Cancer Sci. 2007, 98, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.C.; Tsuneyama, K.; Takahashi, H.; Miwa, S.; Sugiyama, T.; Popivanova, B.K.; Fujii, C.; Nomoto, K.; Mukaida, N.; Takano, Y. Aberrant Pim-3 expression is involved in gastric adenoma-adenocarcinoma sequence and cancer progression. J. Cancer Res. Clin. Oncol. 2008, 134, 481–488. [Google Scholar] [CrossRef]
- Monteiro, P.; Gilot, D.; Langouet, S.; Fardel, O. Activation of the aryl hydrocarbon receptor by the calcium/calmodulin-dependent protein kinase kinase inhibitor 7-oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic acid (STO-609). Drug Metab. Dispos. 2008, 36, 2556–2563. [Google Scholar] [CrossRef]
- Landesman-Bollag, E.; Romieu-Mourez, R.; Song, D.H.; Sonenshein, G.E.; Cardiff, R.D.; Seldin, D.C. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 2001, 20, 3247–3257. [Google Scholar] [CrossRef] [Green Version]
- Daya-Makin, M.; Sanghera, J.S.; Mogentale, T.L.; Lipp, M.; Parchomchuk, J.; Hogg, J.C.; Pelech, S.L. Activation of a Tumor-associated Protein Kinase (p40TAK) and Casein Kinase 2 in Human Squamous Cell Carcinomas and Adenocarcinomas of the Lung. Cancer Res. 1994, 54, 2262–2268. [Google Scholar]
- Yenice, S.; Davis, A.T.; Goueli, S.A.; Akdas, A.; Limas, C.; Ahmed, K. Nuclear casein kinase 2 (CK-2) activity in human normal, benign hyperplastic, and cancerous prostate. Prostate 1994, 24, 11–16. [Google Scholar] [CrossRef]
- Stalter, G.; Siemer, S.; Becht, E.; Ziegler, M.; Remberger, K.; Issinger, O.G. Asymmetric expression of protein kinase CK2 subunits in human kidney tumors. Biochem. Biophys. Res. Commun. 1994, 202, 141–147. [Google Scholar] [CrossRef]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. Acta 2008, 1784, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, C.; Amato, R.J.; Chang, J.T.; Du, G.; Li, W. CDKL2 promotes epithelial-mesenchymal transition and breast cancer progression. Oncotarget 2014, 5, 10840–10853. [Google Scholar] [CrossRef] [PubMed]
- Billard, M.J.; Fitzhugh, D.J.; Parker, J.S.; Brozowski, J.M.; McGinnis, M.W.; Timoshchenko, R.G.; Serafin, D.S.; Lininger, R.; Klauber-Demore, N.; Sahagian, G.; et al. G Protein Coupled Receptor Kinase 3 Regulates Breast Cancer Migration, Invasion, and Metastasis. PLoS ONE 2016, 11, e0152856. [Google Scholar] [CrossRef] [PubMed]
- Maloveryan, A.; Finta, C.; Osterlund, T.; Kogerman, P. A possible role of mouse Fused (STK36) in Hedgehog signaling and Gli transcription factor regulation. J. Cell Commun. Signal. 2007, 1, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Arrowsmith, C.H.; Audia, J.E.; Austin, C.; Baell, J.; Bennett, J.; Blagg, J.; Bountra, C.; Brennan, P.E.; Brown, P.J.; Bunnage, M.E.; et al. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541. [Google Scholar] [CrossRef] [Green Version]
- Blagg, J.; Workman, P. Choose and Use Your Chemical Probe Wisely to Explore Cancer Biology. Cancer Cell 2017, 32, 9–25. [Google Scholar] [CrossRef] [Green Version]
- Ghose, A.K.; Herbertz, T.; Pippin, D.A.; Salvino, J.M.; Mallamo, J.P. Knowledge based prediction of ligand binding modes and rational inhibitor design for kinase drug discovery. J. Med. Chem. 2008, 51, 5149–5171. [Google Scholar] [CrossRef] [Green Version]
- Krishna, S.N.; Luan, C.H.; Mishra, R.K.; Xu, L.; Scheidt, K.A.; Anderson, W.F.; Bergan, R.C. A fluorescence-based thermal shift assay identifies inhibitors of mitogen activated protein kinase kinase 4. PLoS ONE 2013, 8, e81504. [Google Scholar] [CrossRef]
- Simeonov, A. Recent developments in the use of differential scanning fluorometry in protein and small molecule discovery and characterization. Expert Opin. Drug Discov. 2013, 8, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Profeta, G.S.; Dos Reis, C.V.; Santiago, A.D.S.; Godoi, P.H.C.; Fala, A.M.; Wells, C.I.; Sartori, R.; Salmazo, A.P.T.; Ramos, P.Z.; Massirer, K.B.; et al. Binding and structural analyses of potent inhibitors of the human Ca(2+)/calmodulin dependent protein kinase kinase 2 (CAMKK2) identified from a collection of commercially-available kinase inhibitors. Sci. Rep. 2019, 9, 16452. [Google Scholar] [CrossRef]
- Zhang, S.; Anjum, R.; Squillace, R.; Nadworny, S.; Zhou, T.; Keats, J.; Ning, Y.; Wardwell, S.D.; Miller, D.; Song, Y.; et al. The Potent ALK Inhibitor Brigatinib (AP26113) Overcomes Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in Preclinical Models. Clin. Cancer Res. 2016, 22, 5527–5538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Gohda, J.; Suzuki, K.; Liu, K.; Xie, X.; Takeuchi, H.; Inoue, J.I.; Kawaguchi, Y.; Ishida, T. BI-2536 and BI-6727, dual Polo-like kinase/bromodomain inhibitors, effectively reactivate latent HIV-1. Sci. Rep. 2018, 8, 3521. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Klug-Mcleod, J.; Rai, B.; Lunney, E.A. Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg. Med. Chem. 2015, 23, 6520–6527. [Google Scholar] [CrossRef]
- Fedorov, O.; Niesen, F.H.; Knapp, S. Kinase Inhibitor Selectivity Profiling Using Differential Scanning Fluorimetry. In Kinase Inhibitors: Methods and Protocols; Kuster, B., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 109–118. [Google Scholar]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
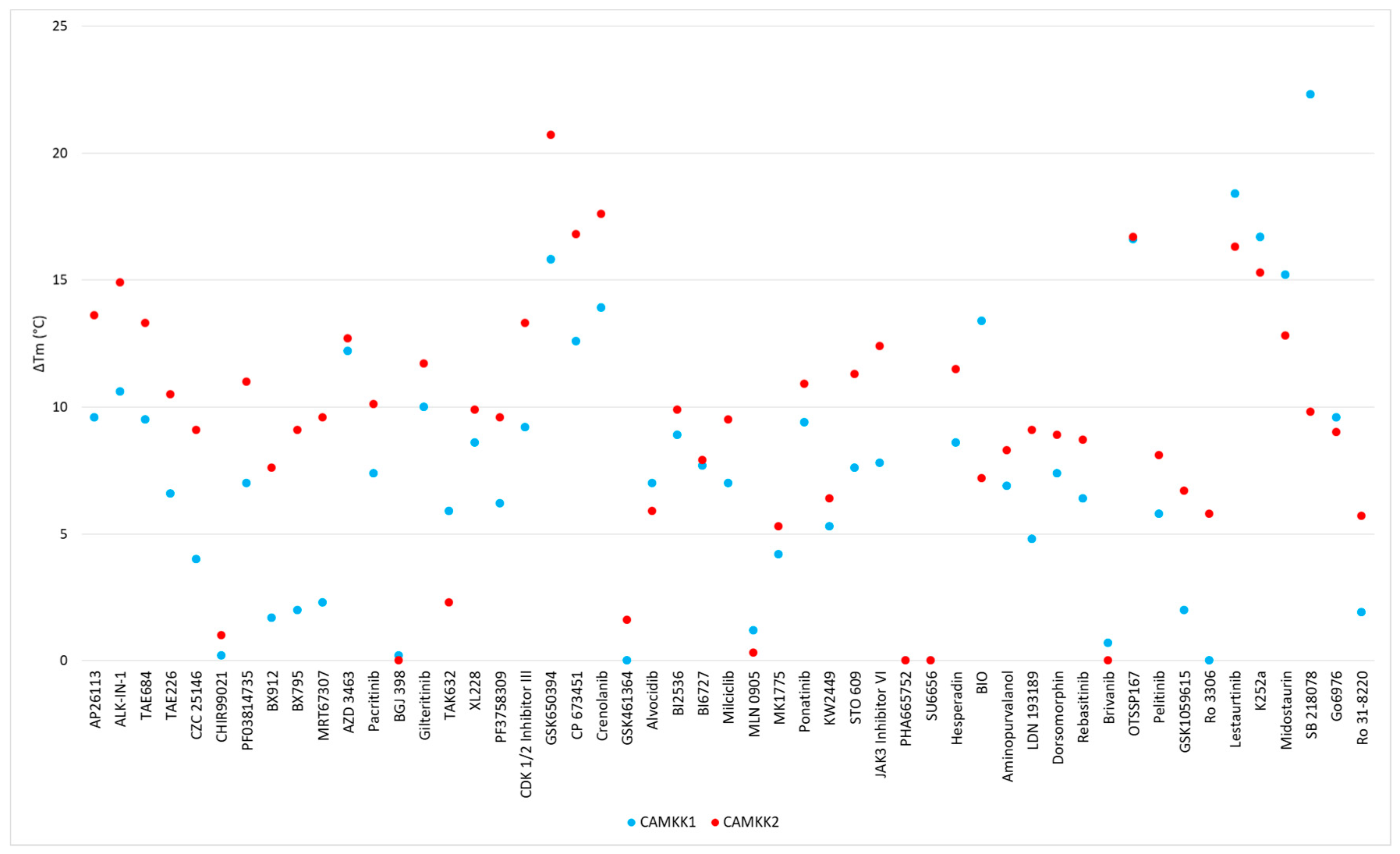
| Scaffold | Inhibitor | CAMKK1 ΔTm (°C) | CAMKK2 ΔTm (°C) | PoC (1.0, 0.1, 0.01 µM) | IC50 (nM) | ||
|---|---|---|---|---|---|---|---|
| 2,4-dianilinopyrimidine | AP26113 | 9.6 | 13.6 | 2 | 20 | 60 | 16 |
| 2,4-dianilinopyrimidine | ALK-IN-1 | 10.6 | 14.9 | 3 | 34 | 62 | 28 |
| 2,4-dianilinopyrimidine | TAE684 | 9.5 | 13.3 | 42 | 87 | 97 | |
| 2,4-dianilinopyrimidine | TAE226 | 6.6 | 10.5 | 90 | 97 | 104 | |
| 2,4-dianilinopyrimidine | CZC 25146 | 4 | 9.1 | 93 | 101 | 96 | |
| 2-aminopyrimidine | CHIR99021 | 0.2 | 1 | 96 | 101 | 98 | |
| 2-anilino-4-alkyl-aminopyrimidine | PF03814735 | 7 | 11 | 20 | 70 | 92 | 227 |
| 2-anilino-4-alkyl-aminopyrimidine- | BX912 | 1.7 | 7.6 | 26 | 90 | 100 | |
| 2-anilino-4-alkyl aminopyrimidine | BX795 | 2 | 9.1 | 33 | 80 | 97 | |
| 2-anilino-4-alkyl-aminopyrimidine | MRT67307 | 2.3 | 9.6 | 35 | 86 | 92 | |
| 2-anilino-4-aryl-pyrimidine | AZD 3463 | 12.2 | 12.7 | 26 | 74 | 103 | 185 |
| 2-anilino-4-aryl-pyrimidine | Pacritinib | 7.4 | 10.1 | 31 | 78 | 96 | |
| 4,6-disubstituted-pyrimidine | BGJ 398 | 0.2 | 0 | 96 | 99 | 100 | |
| Amide hinge binder | Gilteritinib | 10 | 11.7 | 18 | 74 | 84 | 204 |
| Aminobenzothiazole (Type II) | TAK632 | 5.9 | 2.3 | 82 | 87 | 92 | |
| Aminopyrazole | XL228 | 8.6 | 9.9 | 26 | 65 | 94 | |
| Aminopyrazole | PF3758309 | 6.2 | 9.6 | 71 | 78 | 95 | |
| Aminotriazole | CDK 1/2 Inhibitor III | 9.2 | 13.3 | 3 | 17 | 69 | 19 |
| Azaindole | GSK650394 | 15.8 | 20.7 | 3 | 4 | 14 | 3 |
| Benzimidazole | CP 673451 | 12.6 | 16.8 | 17 | 46 | 88 | 73 |
| Benzimidazole | Crenolanib | 13.9 | 17.6 | 47 | 92 | 118 | |
| Benzimidazole | GSK461364 | 0 | 1.6 | 92 | 96 | 95 | |
| Flavone | Alvocidib | 7 | 5.9 | 29 | 62 | 102 | |
| Fused pyrimidine | BI2536 | 8.9 | 9.9 | 54 | 70 | 101 | 96 |
| Fused pyrimidine | BI6727 | 7.7 | 7.9 | 75 | 94 | 104 | |
| Fused pyrimidine | Milciclib | 7 | 9.5 | 76 | 105 | 107 | |
| Fused pyrimidine | MLN 0905 | 1.2 | 0.3 | 86 | 95 | 99 | |
| Fused pyrimidine | MK1775 | 4.2 | 5.3 | 99 | 99 | 101 | |
| Imidazopyridazine (Type II) | Ponatinib | 9.4 | 10.9 | 53 | 92 | 99 | |
| Indazole | KW2449 | 5.3 | 6.4 | 42 | 84 | 96 | |
| Miscellaneous | STO 609 | 7.6 | 11.3 | 21 | 56 | 63 | 58 |
| Oxindole | JAK3 Inhibitor VI | 7.8 | 12.4 | 21 | 81 | 87 | 26 |
| Oxindole | PHA665752 | * | * | 67 | 93 | 109 | |
| Oxindole | SU6656 | * | * | 71 | 86 | 95 | |
| Oxindole | Hesperadin | 8.6 | 11.5 | 84 | 95 | 99 | |
| Oxindole | BIO | 13.4 # | 7.2 # | 89 | 100 | 102 | |
| Purine | Aminopurvalanol | 6.9 | 8.3 | 22 | 80 | 100 | |
| Pyrazolo-pyrimidine | LDN 193189 | 4.8 | 9.1 | 50 | 62 | 68 | 146 |
| Pyrazolo-pyrimidine | Dorsomorphin | 7.4 | 8.9 | 58 | 96 | 100 | |
| Pyridine (Type II) | Rebasitinib | 6.4 | 8.7 | 81 | 93 | 95 | |
| Pyrrolotriazine | Brivanib | 0.7 | 0 | 100 | 100 | 101 | |
| Quinoline | OTSSP167 | 16.6 | 16.7 | 0 | 49 | 84 | 86 |
| Quinoline | Pelitinib | 5.8 | 8.1 | 60 | 84 | 100 | |
| Quinolines | GSK1059615 | 2 | 6.7 | 34 | 81 | 92 | |
| Quinolines | Ro 3306 | 0 | 5.8 | 88 | 94 | 94 | |
| Staurosporine Analogue | Lestaurtinib | 18.4 | 16.3 | 3 | 33 | 64 | 25 |
| Staurosporine Analogue | K252a | 16.7 | 15.3 | 31 | 4 | 99 | |
| Staurosporine Analogue | Midostaurin | 15.2 | 12.8 | 38 | 82 | 97 | |
| Staurosporine Analogue | SB 218078 | 22.3 | 9.8 | 64 | 86 | 98 | |
| Staurosporine Analogue | Go6976 | 9.6 | 9 | 80 | 92 | 97 | |
| Staurosporine Analogue | Ro 31-8220 | 1.9 | 5.7 | 85 | 100 | 100 | |
| Triazolopyridine | Filgotinib | 0 | 0.5 | 72 | 86 | 96 | |
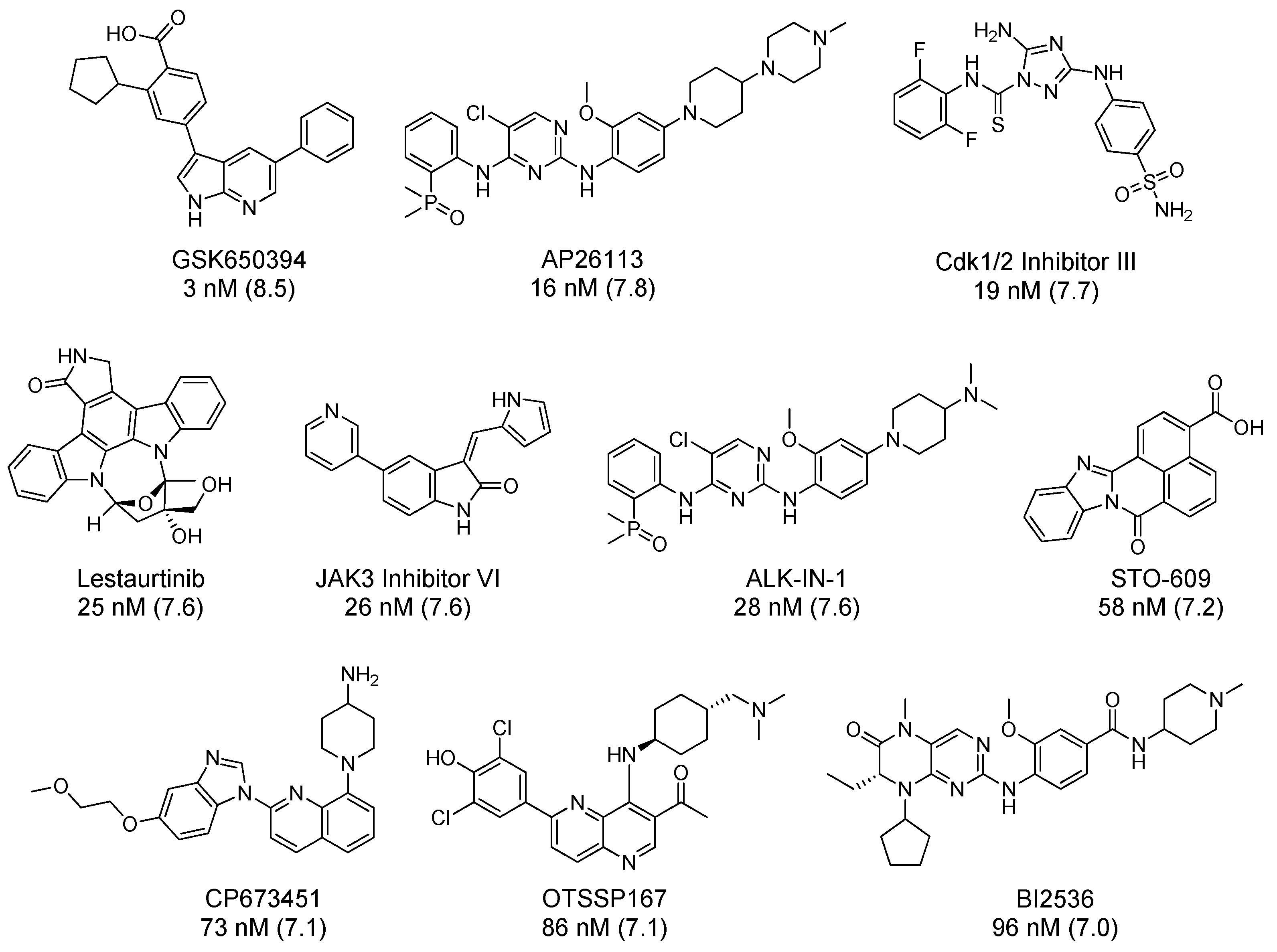
| Inhibitor | IC50 (nM) | Selectivity Metric | Method (#Kinases in Panel) | Source |
|---|---|---|---|---|
| GSK650394 | 3 | 0.083 | S10 @ 1 µM (334) | RBC |
| AP26113 | 16 | 0.125 | IC50 ≤ 50 nM (289) | Literature [101] |
| CDK 1/2 Inhibitor III | 19 | 0.278 | S10 @ 0.5 µM (300) | RBC |
| Lestaurtinib | 25 | 0.356 | Kd ≤ 50 nM (443) | LINCS |
| JAK3 Inhibitor VI | 26 | 0.163 | S10 @ 0.5 µM (300) | RBC |
| ALK-IN-1 | 28 | - | - | |
| STO 609 | 58 | 0.015 | S10 @ 1 µM (409) | KINOMEscan |
| CP 673451 | 73 | - | - | |
| OTSSP167 | 86 | 0.66 | S10 @ 1 µM (141) | MRC |
| BI2536 | 96 | 0.007 | S10 @ 1 µM (131) | MRC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Byrne, S.N.; Scott, J.W.; Pilotte, J.R.; Santiago, A.d.S.; Langendorf, C.G.; Oakhill, J.S.; Eduful, B.J.; Couñago, R.M.; Wells, C.I.; Zuercher, W.J.; et al. In Depth Analysis of Kinase Cross Screening Data to Identify CAMKK2 Inhibitory Scaffolds. Molecules 2020, 25, 325. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25020325
O’Byrne SN, Scott JW, Pilotte JR, Santiago AdS, Langendorf CG, Oakhill JS, Eduful BJ, Couñago RM, Wells CI, Zuercher WJ, et al. In Depth Analysis of Kinase Cross Screening Data to Identify CAMKK2 Inhibitory Scaffolds. Molecules. 2020; 25(2):325. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25020325
Chicago/Turabian StyleO’Byrne, Sean N., John W. Scott, Joseph R. Pilotte, André da S. Santiago, Christopher G. Langendorf, Jonathan S. Oakhill, Benjamin J. Eduful, Rafael M. Couñago, Carrow I. Wells, William J. Zuercher, and et al. 2020. "In Depth Analysis of Kinase Cross Screening Data to Identify CAMKK2 Inhibitory Scaffolds" Molecules 25, no. 2: 325. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25020325