
Preparation of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Drug Delivery and the Effects of Preparation Parameters of Solvent Injection Method



Abstract
:1. Introduction
2. Methods for SLNs and NLCs Preparation
2.1. High-Pressure Homogenization Method
2.2. High-Speed Stirring and Ultra-Sonication Methods
2.3. Microemulsion Method
2.4. Solvent Emulsification-Diffusion Method
2.5. Solvent Emulsification-Evaporation Method
2.6. Double Emulsion Method
2.7. Phase Inversion Temperature (PIT) Method
2.8. Membrane Contactor Method
2.9. Supercritical Fluid-Based Methods
2.10. Coacervation Method
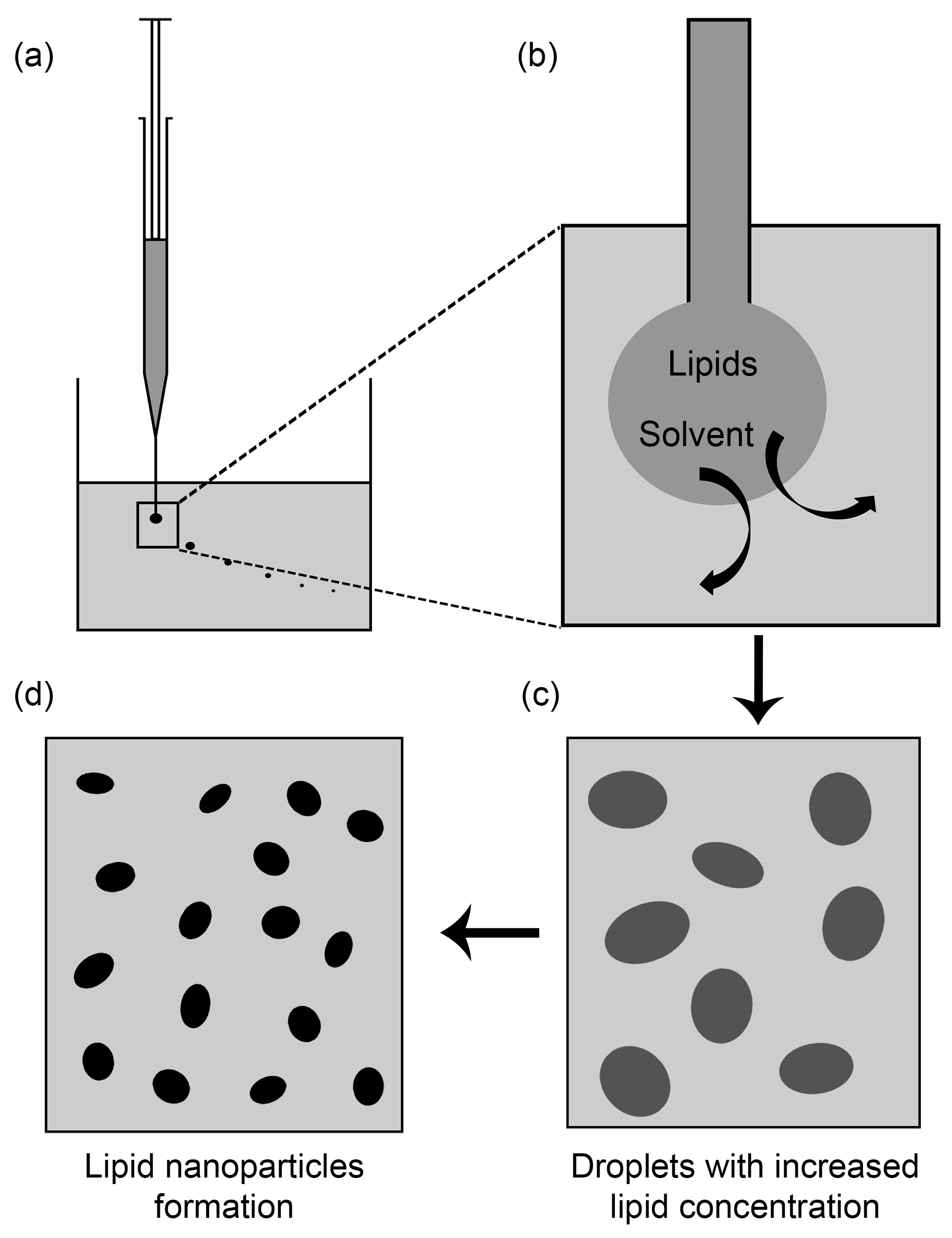
2.11. Solvent Injection Method
3. Preparation of SLNs and NLCs Using Solvent Injection Method in Recent Studies
3.1. Oral Delivery
3.2. Parenteral Delivery
3.3. Topical Delivery
3.4. Nose-to-Brain Delivery
4. Effect of Process Parameters on SLNs and NLCs Produced by Solvent Injection Method
4.1. Aqueous Phase
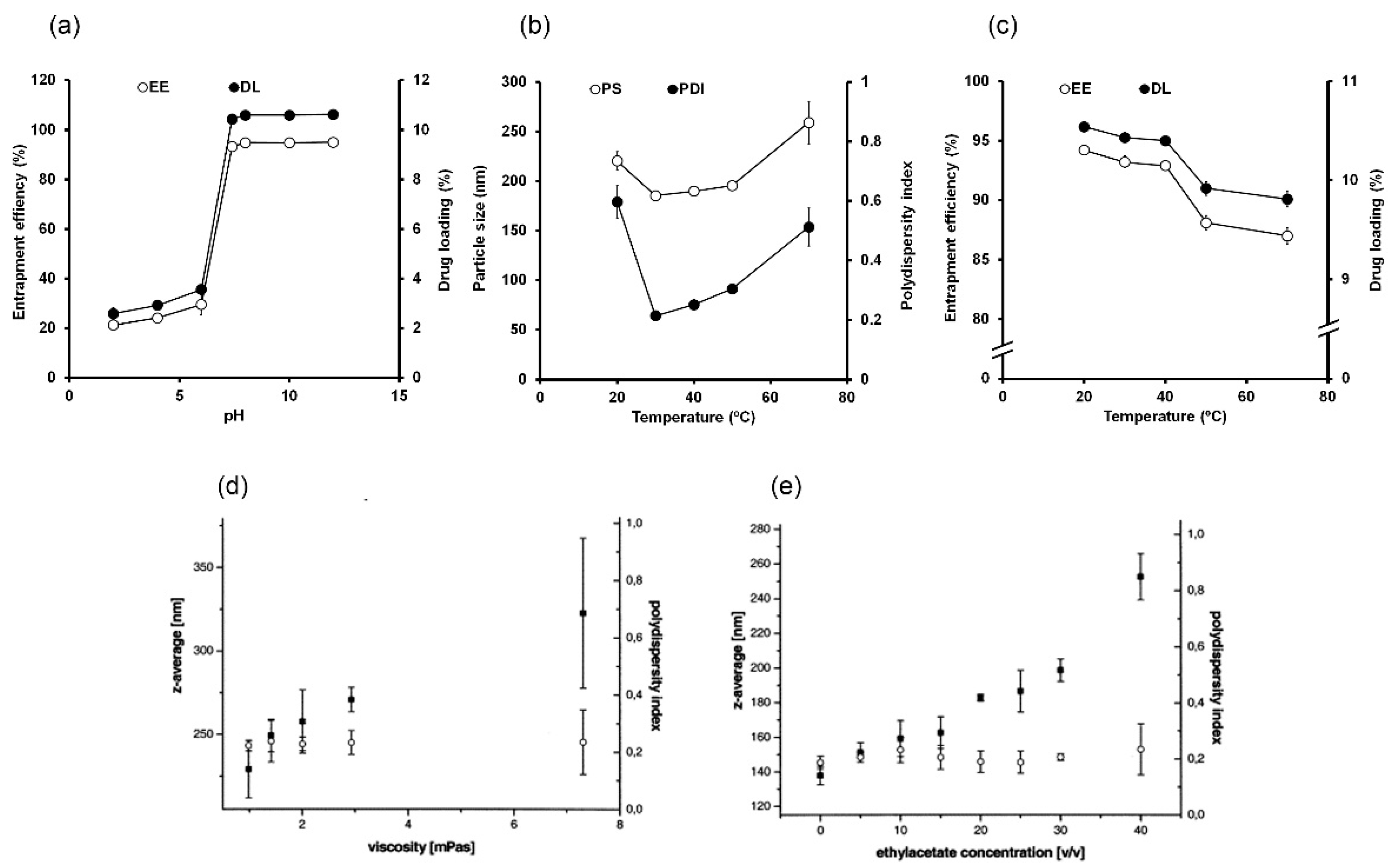
4.1.1. pH
4.1.2. Temperature
4.1.3. Viscosity
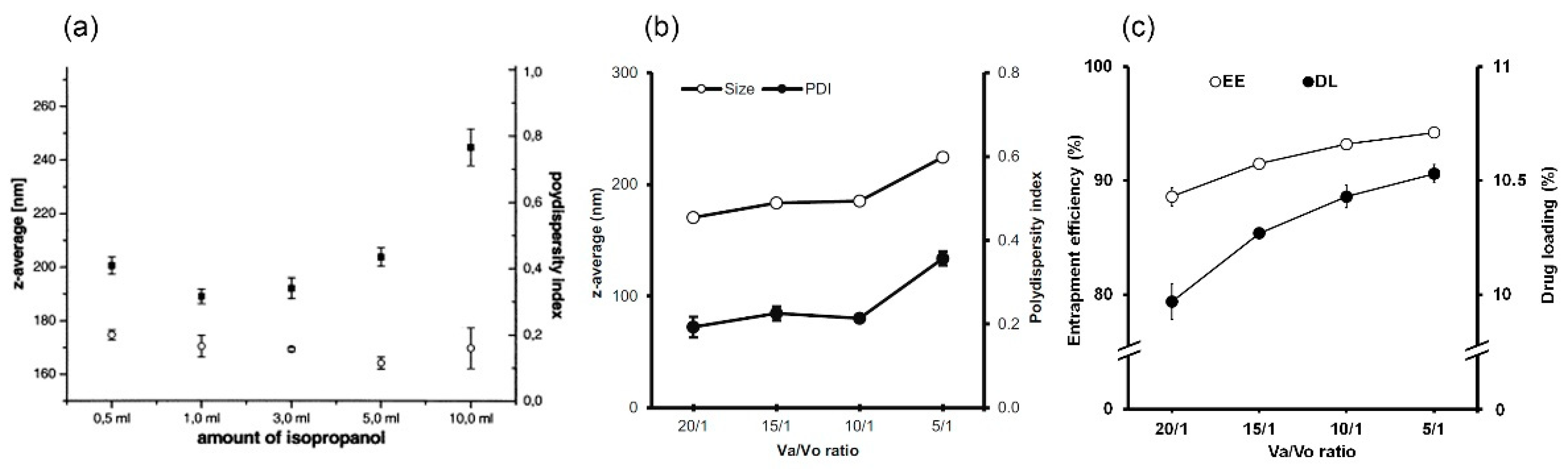
4.2. Organic Phase
4.3. Ratio of Aqueous Phase to Organic Phase
4.4. Dispersion Energy
5. Effect of Formulation Parameters on SLNs and NLCs Produced by Solvent Injection Method
5.1. Total Lipid Concentration
5.2. Liquid Lipid Level
5.3. Drug Hydrophilicity and Drug Amount
5.4. Emulsifier
6. Authors’ Outlook and Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Mehnert, W.; Mäder, K. Solid lipid nanoparticles: Production, characterization and applications. Adv. Drug Deliv. Rev. 2001, 47, 165–196. [Google Scholar] [CrossRef]
- Müller, R.H.; Mäder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery-a review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Wissing, S.A.; Kayser, O.; Müller, R.H. Solid lipid nanoparticles for parenteral drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Marcial, S.P.; Carneiro, G.; Leite, E.A. Lipid-based nanoparticles as drug delivery system for paclitaxel in breast cancer treatment. J. Nanoparticle Res. 2017, 19, 340. [Google Scholar] [CrossRef]
- Doktorovova, S.; Souto, E.B.; Silva, A.M. Nanotoxicology applied to solid lipid nanoparticles and nanostructured lipid carriers-A systematic review of in vitro data. Eur. J. Pharm. Biopharm. 2014, 87, 1–18. [Google Scholar] [CrossRef]
- Pardeike, J.; Hommoss, A.; Müller, R.H. Lipid nanoparticles (SLN, NLC) in cosmetic and pharmaceutical dermal products. Int. J. Pharm. 2009, 366, 170–184. [Google Scholar] [CrossRef]
- Bhaskar, K.; Anbu, J.; Ravichandiran, V.; Venkateswarlu, V.; Rao, Y.M. Lipid nanoparticles for transdermal delivery of flurbiprofen: Formulation, in vitro, ex vivo and in vivo studies. Lipids Health Dis. 2009, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Joshi, M.D.; Müller, R.H. Lipid nanoparticles for parenteral delivery of actives. Eur. J. Pharm. Biopharm. 2009, 71, 161–172. [Google Scholar] [CrossRef]
- Hou, D.; Xie, C.; Huang, K.; Zhu, C. The production and characteristics of solid lipid nanoparticles (SLNs). Biomaterials 2003, 24, 1781–1785. [Google Scholar] [CrossRef]
- Schwarz, C.; Mehnert, W.; Lucks, J.S.; Müller, R.H. Solid lipid nanoparticles (SLN) for controlled drug delivery. I. Production, characterization and sterilization. J. Control. Release 1994, 30, 83–96. [Google Scholar] [CrossRef]
- Shah, K.A.; Date, A.A.; Joshi, M.D.; Patravale, V.B. Solid lipid nanoparticles (SLN) of tretinoin: Potential in topical delivery. Int. J. Pharm. 2007, 345, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Zur Mühlen, A.; Schwarz, C.; Mehnert, W. Solid lipid nanoparticles (SLN) for controlled drug delivery—Drug release and release mechanism. Eur. J. Pharm. Biopharm. 1998, 45, 149–155. [Google Scholar] [CrossRef]
- Lu, B.; Xiong, S.-B.; Yang, H.; Yin, X.-D.; Chao, R.-B. Solid lipid nanoparticles of mitoxantrone for local injection against breast cancer and its lymph node metastases. Eur. J. Pharm. Sci. 2006, 28, 86–95. [Google Scholar] [CrossRef]
- Zielińska, A.; Ferreira, N.R.; Durazzo, A.; Lucarini, M.; Cicero, N.; Mamouni, S.E.; Silva, A.M.; Nowak, I.; Santini, A.; Souto, E.B. Development and Optimization of Alpha-Pinene-Loaded Solid Lipid Nanoparticles (SLN) Using Experimental Factorial Design and Dispersion Analysis. Molecules 2019, 24, 2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qushawy, M.; Prabahar, K.; Abd-Alhaseeb, M.; Swidan, S.; Nasr, A. Preparation and Evaluation of Carbamazepine Solid Lipid Nanoparticle for Alleviating Seizure Activity in Pentylenetetrazole-Kindled Mice. Molecules 2019, 24, 3971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, M.; Patravale, V. Nanostructured lipid carrier (NLC) based gel of celecoxib. Int. J. Pharm. 2008, 346, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Dasgupta, S.; Dey, S.; Ramani, Y.R.; Ray, S.; Mazumder, B. Nanostructured Lipid Carriers (NLC)-Based Gel for the Topical Delivery of Aceclofenac: Preparation, characterization, and in vivo evaluation. Sci. Pharm. 2012, 80, 749–764. [Google Scholar] [CrossRef] [Green Version]
- Taratula, O.; Kuzmov, A.; Shah, M.; Garbuzenko, O.B.; Minko, T. Nanostructured lipid carriers as multifunctional nanomedicine platform for pulmonary co-delivery of anticancer drugs and siRNA. J. Control. Release 2013, 171, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Fathi, H.A.; Allam, A.; Elsabahy, M.; Fetih, G.; El-Badry, M. Nanostructured lipid carriers for improved oral delivery and prolonged antihyperlipidemic effect of simvastatin. Colloids Surf. B Biointerfaces 2018, 162, 236–245. [Google Scholar] [CrossRef]
- Devkar, T.B.; Tekade, A.R.; Khandelwal, K.R. Surface engineered nanostructured lipid carriers for efficient nose to brain delivery of ondansetron HCl using Delonix regia gum as a natural mucoadhesive polymer. Colloids Surf. B Biointerfaces 2014, 122, 143–150. [Google Scholar] [CrossRef]
- Garcês, A.; Amaral, M.H.; Sousa Lobo, J.M.; Silva, A.C. Formulations based on solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) for cutaneous use: A review. Eur. J. Pharm. Sci. 2018, 112, 159–167. [Google Scholar] [CrossRef]
- Müller, R.H.; Radtke, M.; Wissing, S.A. Nanostructured lipid matrices for improved microencapsulation of drugs. Int. J. Pharm. 2002, 242, 121–128. [Google Scholar] [CrossRef]
- Westesen, K.; Bunjes, H.; Koch, M.H.J. Physicochemical characterization of lipid nanoparticles and evaluation of their drug loading capacity and sustained release potential. J. Control. Release 1997, 48, 223–236. [Google Scholar] [CrossRef]
- Luan, J.; Zheng, F.; Yang, X.; Yu, A.; Zhai, G. Nanostructured lipid carriers for oral delivery of baicalin: In vitro and in vivo evaluation. Colloids Surf. A Physicochem. Eng. Asp. 2015, 466, 154–159. [Google Scholar] [CrossRef]
- Araújo, J.; Nikolic, S.; Egea, M.A.; Souto, E.B.; Garcia, M.L. Nanostructured lipid carriers for triamcinolone acetonide delivery to the posterior segment of the eye. Colloids Surf. B Biointerfaces 2011, 88, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Radtke, M.; Wissing, S.A. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv. Drug Deliv. Rev. 2002, 54, S131–S155. [Google Scholar] [CrossRef]
- Weber, S.; Zimmer, A.; Pardeike, J. Solid Lipid Nanoparticles (SLN) and Nanostructured Lipid Carriers (NLC) for pulmonary application: A review of the state of the art. Eur. J. Pharm. Biopharm. 2014, 86, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Ravani, L.; Esposito, E.; Bories, C.; Moal, V.L.-L.; Loiseau, P.M.; Djabourov, M.; Cortesi, R.; Bouchemal, K. Clotrimazole-loaded nanostructured lipid carrier hydrogels: Thermal analysis and in vitro studies. Int. J. Pharm. 2013, 454, 695–702. [Google Scholar] [CrossRef]
- Esposito, E.; Sguizzato, M.; Drechsler, M.; Mariani, P.; Carducci, F.; Nastruzzi, C.; Cortesi, R. Progesterone lipid nanoparticles: Scaling up and in vivo human study. Eur. J. Pharm. Biopharm. 2017, 119, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Bondì, M.L.; Craparo, E.F. Solid lipid nanoparticles for applications in gene therapy: A review of the state of the art. Expert Opin. Drug Deliv. 2010, 7, 7–18. [Google Scholar] [CrossRef]
- Hörmann, K.; Zimmer, A. Drug delivery and drug targeting with parenteral lipid nanoemulsions-A review. J. Control. Release 2016, 223, 85–98. [Google Scholar] [CrossRef]
- Batzri, S.; Korn, E.D. Single bilayer liposomes prepared without sonication. Biochim. et Biophys. Acta (BBA)-Biomembr. 1973, 298, 1015–1019. [Google Scholar] [CrossRef]
- Lujan, H.; Griffin, W.C.; Taube, J.H.; Sayes, C.M. Synthesis and characterization of nanometer-sized liposomes for encapsulation and microRNA transfer to breast cancer cells. Int. J. Nanomed. 2019, 14, 5159–5173. [Google Scholar] [CrossRef] [Green Version]
- Gentine, P.; Bubel, A.; Crucifix, C.; Bourel-Bonnet, L.; Frisch, B. Manufacture of liposomes by isopropanol injection: Characterization of the method. J. Liposome Res. 2012, 22, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Lv, B.; Li, L.; Lee, R.J.; Xie, J.; Qiu, Z.; Teng, L. A Liposomal Formulation for Improving Solubility and Oral Bioavailability of Nifedipine. Molecules 2020, 25, 338. [Google Scholar] [CrossRef] [Green Version]
- Schubert, M.A.; Müller-Goymann, C.C. Solvent injection as a new approach for manufacturing lipid nanoparticles—Evaluation of the method and process parameters. Eur. J. Pharm. Biopharm. 2003, 55, 125–131. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Md, S.; Sahni, J.K.; Baboota, S.; Dang, S.; Ali, J. Nanostructured lipid carriers system: Recent advances in drug delivery. J. Drug Target. 2012, 20, 813–830. [Google Scholar] [CrossRef]
- Jain, A.; Agarwal, A.; Majumder, S.; Lariya, N.; Khaya, A.; Agrawal, H.; Majumdar, S.; Agrawal, G.P. Mannosylated solid lipid nanoparticles as vectors for site-specific delivery of an anti-cancer drug. J. Control. Release 2010, 148, 359–367. [Google Scholar] [CrossRef]
- Luo, C.-F.; Yuan, M.; Chen, M.-S.; Liu, S.-M.; Zhu, L.; Huang, B.-Y.; Liu, X.-W.; Xiong, W. Pharmacokinetics, tissue distribution and relative bioavailability of puerarin solid lipid nanoparticles following oral administration. Int. J. Pharm. 2011, 410, 138–144. [Google Scholar] [CrossRef]
- Arıca Yegin, B.; Benoît, J.-P.; Lamprecht, A. Paclitaxel-Loaded Lipid Nanoparticles Prepared by Solvent Injection or Ultrasound Emulsification. Drug Dev. Ind. Pharm. 2006, 32, 1089–1094. [Google Scholar] [CrossRef]
- Stancampiano, A.; Acquaviva, R.; Campisi, A.; Vanella, L.; Ventura, C.; Puglisi, G.; Pignatello, R. Technological and biological characterization of idebenone-loaded solid lipid nanoparticles prepared by a modified solvent injection technique. J. Biomed. Nanotechnol. 2006, 2, 253–270. [Google Scholar] [CrossRef]
- Tiwari, R.; Pathak, K. Nanostructured lipid carrier versus solid lipid nanoparticles of simvastatin: Comparative analysis of characteristics, pharmacokinetics and tissue uptake. Int. J. Pharm. 2011, 415, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.S.; Patel, H.S.; Belgamwar, V.S.; Agrawal, A.; Tekade, A.R. Solid lipid nanoparticles of ondansetron HCl for intranasal delivery: Development, optimization and evaluation. J. Mater. Sci.: Mater. Med. 2012, 23, 2163–2175. [Google Scholar] [CrossRef] [PubMed]
- Vaghasiya, H.; Kumar, A.; Sawant, K. Development of solid lipid nanoparticles based controlled release system for topical delivery of terbinafine hydrochloride. Eur. J. Pharm. Sci. 2013, 49, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Calva-Estrada, S.d.J.; García, O.; Mendoza, M.R.; Jiménez, M. Characterization of O/W emulsions of carotenes in blackberry juice performed by ultrasound and high-pressure homogenization. J. Disper. Sci. Technol. 2018, 39, 181–189. [Google Scholar] [CrossRef]
- Galvão, K.C.S.; Vicente, A.A.; Sobral, P.J.A. Development, Characterization, and Stability of O/W Pepper Nanoemulsions Produced by High-Pressure Homogenization. Food Bioprocess. Technol. 2018, 11, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Amasya, G.; Aksu, B.; Badilli, U.; Onay-Besikci, A.; Tarimci, N. QbD guided early pharmaceutical development study: Production of lipid nanoparticles by high pressure homogenization for skin cancer treatment. Int. J. Pharm. 2019, 563, 110–121. [Google Scholar] [CrossRef]
- Shegokar, R.; Singh, K.K.; Müller, R.H. Production & stability of stavudine solid lipid nanoparticles—From lab to industrial scale. Int. J. Pharm. 2011, 416, 461–470. [Google Scholar]
- Hu, C.; Qian, A.; Wang, Q.; Xu, F.; He, Y.; Xu, J.; Xia, Y.; Xia, Q. Industrialization of lipid nanoparticles: From laboratory-scale to large-scale production line. Eur. J. Pharm. Biopharm. 2016, 109, 206–213. [Google Scholar] [CrossRef]
- Suter, F.; Schmid, D.; Wandrey, F.; Zülli, F. Heptapeptide-loaded solid lipid nanoparticles for cosmetic anti-aging applications. Eur. J. Pharm. Biopharm. 2016, 108, 304–309. [Google Scholar] [CrossRef]
- Štecová, J.; Mehnert, W.; Blaschke, T.; Kleuser, B.; Sivaramakrishnan, R.; Zouboulis, C.C.; Seltmann, H.; Korting, H.C.; Kramer, K.D.; Schäfer-Korting, M. Cyproterone Acetate Loading to Lipid Nanoparticles for Topical Acne Treatment: Particle Characterisation and Skin Uptake. Pharm. Res. 2007, 24, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kesarla, R.; Chotai, N.; Misra, A.; Omri, A. Systematic Approach for the Formulation and Optimization of Solid Lipid Nanoparticles of Efavirenz by High Pressure Homogenization Using Design of Experiments for Brain Targeting and Enhanced Bioavailability. BioMed Res. Int. 2017, 2017, 5984014. [Google Scholar] [CrossRef]
- Üner, M.; Yener, G.; Ergüven, M. Design of colloidal drug carriers of celecoxib for use in treatment of breast cancer and leukemia. Mater. Sci. Eng. C 2019, 103, 109874. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Schiavone, N.; Guzman-Aranguez, A.; Giansanti, F.; Papucci, L.; Perez de Lara, M.J.; Singh, M.; Kaur, I.P. Atorvastatin-loaded solid lipid nanoparticles as eye drops: Proposed treatment option for age-related macular degeneration (AMD). Drug Deliv. Transl. Res. 2020, 10, 919–944. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chaudhury, A. Recent Advances in Lipid Nanoparticle Formulations with Solid Matrix for Oral Drug Delivery. AAPS PharmSciTech 2011, 12, 62–76. [Google Scholar] [CrossRef] [Green Version]
- Irby, D.; Du, C.; Li, F. Lipid-Drug Conjugate for Enhancing Drug Delivery. Mol. Pharm. 2017, 14, 1325–1338. [Google Scholar] [CrossRef] [Green Version]
- Olbrich, C.; Gessner, A.; Kayser, O.; Müller, R.H. Lipid-Drug-Conjugate (LDC) Nanoparticles as Novel Carrier System for the Hydrophilic Antitrypanosomal Drug Diminazenediaceturate. J. Drug Target. 2002, 10, 387–396. [Google Scholar] [CrossRef]
- Duan, Y.; Dhar, A.; Patel, C.; Khimani, M.; Neogi, S.; Sharma, P.; Siva Kumar, N.; Vekariya, R.L. A brief review on solid lipid nanoparticles: Part and parcel of contemporary drug delivery systems. RSC Adv. 2020, 10, 26777–26791. [Google Scholar] [CrossRef]
- Sharma, G.; Chopra, K.; Puri, S.; Bishnoi, M.; Rishi, P.; Kaur, I.P. Topical delivery of TRPsiRNA-loaded solid lipid nanoparticles confer reduced pain sensation via TRPV1 silencing, in rats. J. Drug Target. 2018, 26, 135–149. [Google Scholar] [CrossRef]
- Zhang, S.-J.; Zhang, Y.-T.; Zhao, J.-H.; Shen, L.-N.; Shi, F.; Feng, N.-P. Preparation and in vitro anti-tumor properties of toad venom extract-loaded solid lipid nanoparticles. Die Pharm. 2013, 68, 653–660. [Google Scholar]
- Duong, V.-A.; Nguyen, T.-T.-L.; Maeng, H.-J.; Chi, S.-C. Nanostructured lipid carriers containing ondansetron hydrochloride by cold high-pressure homogenization method: Preparation, characterization, and pharmacokinetic evaluation. J. Drug Deliv. Sci. Technol. 2019, 53, 101185. [Google Scholar] [CrossRef]
- Kasongo, K.W.; Müller, R.H.; Walker, R.B. The use of hot and cold high pressure homogenization to enhance the loading capacity and encapsulation efficiency of nanostructured lipid carriers for the hydrophilic antiretroviral drug, didanosine for potential administration to paediatric patients. Pharm. Dev. Technol. 2012, 17, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhao, P.; Cui, F.; Li, X. Preparation and characterization of solid lipid nanoparticles loaded with total flavones of Hippophae rhamnoides (TFH). PDA J. Pharm. Sci. Technol. 2007, 61, 110–120. [Google Scholar]
- Duong, V.-A.; Nguyen, T.-T.-L.; Maeng, H.-J.; Chi, S.-C. Data on optimization and drug release kinetics of nanostructured lipid carriers containing ondansetron hydrochloride prepared by cold high-pressure homogenization method. Data Brief. 2019, 26, 104475. [Google Scholar] [CrossRef] [PubMed]
- Neupane, Y.R.; Sabir, M.D.; Ahmad, N.; Ali, M.; Kohli, K. Lipid drug conjugate nanoparticle as a novel lipid nanocarrier for the oral delivery of decitabine: Ex vivo gut permeation studies. Nanotechnology 2013, 24, 415102. [Google Scholar] [CrossRef] [PubMed]
- Souto, E.B.; Doktorovova, S.; Zielinska, A.; Silva, A.M. Key production parameters for the development of solid lipid nanoparticles by high shear homogenization. Pharm. Dev. Technol. 2019, 24, 1181–1185. [Google Scholar] [CrossRef]
- Severino, P.; Santana, M.H.A.; Souto, E.B. Optimizing SLN and NLC by 22 full factorial design: Effect of homogenization technique. Mater. Sci. Eng. C 2012, 32, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Zhu, L.; Dong, Z.; Wang, X.; Wang, Y.; Li, X.; Zhou, W. Preparation, characterization and pharmacokinetics of enrofloxacin-loaded solid lipid nanoparticles: Influences of fatty acids. Colloids Surf. B Biointerfaces 2011, 83, 382–387. [Google Scholar] [CrossRef]
- Tamjidi, F.; Shahedi, M.; Varshosaz, J.; Nasirpour, A. Design and characterization of astaxanthin-loaded nanostructured lipid carriers. Innov. Food Sci. Emerg. Technol. 2014, 26, 366–374. [Google Scholar] [CrossRef]
- Akhoond Zardini, A.; Mohebbi, M.; Farhoosh, R.; Bolurian, S. Production and characterization of nanostructured lipid carriers and solid lipid nanoparticles containing lycopene for food fortification. J. Food Sci. Technol. 2018, 55, 287–298. [Google Scholar] [CrossRef]
- Fazly Bazzaz, B.S.; Khameneh, B.; Namazi, N.; Iranshahi, M.; Davoodi, D.; Golmohammadzadeh, S. Solid lipid nanoparticles carrying Eugenia caryophyllata essential oil: The novel nanoparticulate systems with broad-spectrum antimicrobial activity. Lett. Appl. Microbiol. 2018, 66, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Chavda, K.; Vyas, B.; Patel, S. Formulation development of linagliptin solid lipid nanoparticles for oral bioavailability enhancement: Role of P-gp inhibition. Drug Deliv. Transl. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Li, W.; Wang, X.; Xue, J.; Zhao, L.; Song, Y.; Zhou, T.; Zhang, M. Thiopental sodium loaded solid lipid nano-particles attenuates obesity-induced cardiac dysfunction and cardiac hypertrophy via inactivation of inflammatory pathway. Drug Deliv. 2020, 27, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Jannin, V.; Blas, L.; Chevrier, S.; Miolane, C.; Demarne, F.; Spitzer, D. Evaluation of the digestibility of solid lipid nanoparticles of glyceryl dibehenate produced by two techniques: Ultrasonication and spray-flash evaporation. Eur. J. Pharm. Sci. 2018, 111, 91–95. [Google Scholar] [CrossRef]
- Cortesi, R.; Esposito, E.; Luca, G.; Nastruzzi, C. Production of lipospheres as carriers for bioactive compounds. Biomaterials 2002, 23, 2283–2294. [Google Scholar] [CrossRef]
- Duong, V.-A.; Nguyen, T.-T.-L.; Maeng, H.-J.; Chi, S.-C. Preparation of Ondansetron Hydrochloride-Loaded Nanostructured Lipid Carriers Using Solvent Injection Method for Enhancement of Pharmacokinetic Properties. Pharm. Res. 2019, 36, 138. [Google Scholar] [CrossRef]
- Gasco, M.R. Method for Producing Solid Lipid Microspheres Having a Narrow Size Distribution. U.S. Patents 5,250,236, 5 October 1993. [Google Scholar]
- Shah, R.M.; Eldridge, D.S.; Palombo, E.A.; Harding, I.H. Microwave-assisted microemulsion technique for production of miconazole nitrate- and econazole nitrate-loaded solid lipid nanoparticles. Eur. J. Pharm. Biopharm. 2017, 117, 141–150. [Google Scholar] [CrossRef]
- Mohd, Y.; Praveen Kumar, G.; Dinesh, P.; Preeti, S.; Shanmugam Sadish, K. Solid Lipid Nanoparticles Approach for Lymphatic Targeting Through Intraduodenal Delivery of Quetiapine Fumarate. Curr. Drug Deliv. 2018, 15, 818–828. [Google Scholar] [CrossRef]
- Masiiwa, W.L.; Gadaga, L.L. Intestinal Permeability of Artesunate-Loaded Solid Lipid Nanoparticles Using the Everted Gut Method. J. Drug Deliv. 2018, 2018, 3021738. [Google Scholar] [CrossRef]
- Souza, L.G.; Silva, E.J.; Martins, A.L.L.; Mota, M.F.; Braga, R.C.; Lima, E.M.; Valadares, M.C.; Taveira, S.F.; Marreto, R.N. Development of topotecan loaded lipid nanoparticles for chemical stabilization and prolonged release. Eur. J. Pharm. Biopharm. 2011, 79, 189–196. [Google Scholar] [CrossRef]
- Fathy Abd-Ellatef, G.-E.; Gazzano, E.; Chirio, D.; Ragab Hamed, A.; Belisario, D.C.; Zuddas, C.; Peira, E.; Rolando, B.; Kopecka, J.; Assem Said Marie, M.; et al. Curcumin-Loaded Solid Lipid Nanoparticles Bypass P-Glycoprotein Mediated Doxorubicin Resistance in Triple Negative Breast Cancer Cells. Pharmaceutics 2020, 12, 96. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Wairkar, S.; Bhatt, L.K. Isotretinoin and α-tocopherol acetate-loaded solid lipid nanoparticle topical gel for the treatment of acne. J. Microencapsul. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Igartua, M.; Saulnier, P.; Heurtault, B.; Pech, B.; Proust, J.E.; Pedraz, J.L.; Benoit, J.P. Development and characterization of solid lipid nanoparticles loaded with magnetite. Int. J. Pharm. 2002, 233, 149–157. [Google Scholar] [CrossRef]
- Trotta, M.; Debernardi, F.; Caputo, O. Preparation of solid lipid nanoparticles by a solvent emulsification–diffusion technique. Int. J. Pharm. 2003, 257, 153–160. [Google Scholar] [CrossRef]
- Hu, F.Q.; Yuan, H.; Zhang, H.H.; Fang, M. Preparation of solid lipid nanoparticles with clobetasol propionate by a novel solvent diffusion method in aqueous system and physicochemical characterization. Int. J. Pharm. 2002, 239, 121–128. [Google Scholar] [CrossRef]
- Dhaundiyal, A.; Jena, S.K.; Samal, S.K.; Sonvane, B.; Chand, M.; Sangamwar, A.T. Alpha-lipoic acid–stearylamine conjugate-based solid lipid nanoparticles for tamoxifen delivery: Formulation, optimization, in-vivo pharmacokinetic and hepatotoxicity study. J. Pharm. Pharmacol. 2016, 68, 1535–1550. [Google Scholar] [CrossRef] [PubMed]
- Yassemi, A.; Kashanian, S.; Zhaleh, H. Folic acid receptor-targeted solid lipid nanoparticles to enhance cytotoxicity of letrozole through induction of caspase-3 dependent-apoptosis for breast cancer treatment. Pharm. Dev. Technol. 2020, 25, 397–407. [Google Scholar] [CrossRef]
- Patravale, V.B.; Mirani, A.G. Preparation and Characterization of Solid Lipid Nanoparticles-Based Gel for Topical Delivery. In Pharmaceutical Nanotechnology: Basic Protocols; Weissig, V., Elbayoumi, T., Eds.; Springer New York: New York, NY, USA, 2019; pp. 293–302. [Google Scholar]
- Trotta, M.; Cavalli, R.; Carlotti, M.E.; Battaglia, L.; Debernardi, F. Solid lipid micro-particles carrying insulin formed by solvent-in-water emulsion–diffusion technique. Int. J. Pharm. 2005, 288, 281–288. [Google Scholar] [CrossRef]
- Battaglia, L.; Trotta, M.; Gallarate, M.; Carlotti, M.E.; Zara, G.P.; Bargoni, A. Solid lipid nanoparticles formed by solvent-in-water emulsion–diffusion technique: Development and influence on insulin stability. J. Microencapsul. 2007, 24, 672–684. [Google Scholar] [CrossRef]
- Urbán-Morlán, Z.; Ganem-Rondero, A.; Melgoza-Contreras, L.M.; Escobar-Chávez, J.J.; Nava-Arzaluz, M.G.; Quintanar-Guerrero, D. Preparation and characterization of solid lipid nanoparticles containing cyclosporine by the emulsification-diffusion method. Int. J. Nanomed. 2010, 5, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Sjöström, B.; Kaplun, A.; Talmon, Y.; Cabane, B. Structures of Nanoparticles Prepared from Oil-in-Water Emulsions. Pharm. Res. 1995, 12, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Pooja, D.; Tunki, L.; Kulhari, H.; Reddy, B.B.; Sistla, R. Characterization, biorecognitive activity and stability of WGA grafted lipid nanostructures for the controlled delivery of Rifampicin. Chem. Phys. Lipids 2015, 193, 11–17. [Google Scholar] [CrossRef]
- Pooja, D.; Kulhari, H.; Tunki, L.; Chinde, S.; Kuncha, M.; Grover, P.; Rachamalla, S.S.; Sistla, R. Nanomedicines for targeted delivery of etoposide to non-small cell lung cancer using transferrin functionalized nanoparticles. RSC Adv. 2015, 5, 49122–49131. [Google Scholar] [CrossRef]
- Patel, K.K.; Gade, S.; Anjum, M.M.; Singh, S.K.; Maiti, P.; Agrawal, A.K.; Singh, S. Effect of penetration enhancers and amorphization on transdermal permeation flux of raloxifene-encapsulated solid lipid nanoparticles: An ex vivo study on human skin. Appl. Nanosci. 2019, 9, 1383–1394. [Google Scholar] [CrossRef]
- Singh, B.; Vuddanda, P.R.; Vijayakumar, M.R.; Kumar, V.; Saxena, P.S.; Singh, S. Cefuroxime axetil loaded solid lipid nanoparticles for enhanced activity against S. aureus biofilm. Colloids Surf. B Biointerfaces 2014, 121, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, A.; Costa, B.; Ferreira, M.V.; Miguéis, D.; Louros, J.M.S.; Durazzo, A.; Lucarini, M.; Eder, P.; Chaud, M.V.; Morsink, M.; et al. Nanotoxicology and Nanosafety: Safety-by-Design and Testing at a Glance. Int. J. Environ. Res. Public Health 2020, 17, 4657. [Google Scholar] [CrossRef] [PubMed]
- García-Fuentes, M.; Torres, D.; Alonso, M.J. Design of lipid nanoparticles for the oral delivery of hydrophilic macromolecules. Colloids Surf. B Biointerfaces 2003, 27, 159–168. [Google Scholar] [CrossRef]
- Li, Z.; Yu, L.; Zheng, L.; Geng, F. Studies on crystallinity state of puerarin loaded solid lipid nanoparticles prepared by double emulsion method. J. Therm. Anal. Calorim. 2010, 99, 689–693. [Google Scholar] [CrossRef]
- Singh, S.; Dobhal, A.K.; Jain, A.; Pandit, J.K.; Chakraborty, S. Formulation and Evaluation of Solid Lipid Nanoparticles of a Water Soluble Drug: Zidovudine. Chem. Pharm. Bull. 2010, 58, 650–655. [Google Scholar] [CrossRef] [Green Version]
- Fonte, P.; Nogueira, T.; Gehm, C.; Ferreira, D.; Sarmento, B. Chitosan-coated solid lipid nanoparticles enhance the oral absorption of insulin. Drug Deliv. Transl. Res. 2011, 1, 299–308. [Google Scholar] [CrossRef]
- Trotta, M.; Carlotti, M.E.; Gallarate, M.; Zara, G.P.; Muntoni, E.; Battaglia, L. Insulin-Loaded SLN Prepared with the Emulsion Dilution Technique: In Vivo Tracking of Nanoparticles after Oral Administration to Rats. J. Dispers. Sci. Technol. 2011, 32, 1041–1045. [Google Scholar] [CrossRef]
- Sarmento, B.; Martins, S.; Ferreira, D.; Souto, E.B. Oral insulin delivery by means of solid lipid nanoparticles. Int. J. Nanomed. 2007, 2, 743–749. [Google Scholar]
- Nabi-Meibodi, M.; Vatanara, A.; Najafabadi, A.R.; Rouini, M.R.; Ramezani, V.; Gilani, K.; Etemadzadeh, S.M.H.; Azadmanesh, K. The effective encapsulation of a hydrophobic lipid-insoluble drug in solid lipid nanoparticles using a modified double emulsion solvent evaporation method. Colloids Surf. B Biointerfaces 2013, 112, 408–414. [Google Scholar] [CrossRef]
- Mazur, K.L.; Feuser, P.E.; Valério, A.; Poester Cordeiro, A.; de Oliveira, C.I.; Assolini, J.P.; Pavanelli, W.R.; Sayer, C.; Araújo, P.H.H. Diethyldithiocarbamate loaded in beeswax-copaiba oil nanoparticles obtained by solventless double emulsion technique promote promastigote death in vitro. Colloids Surf. B Biointerfaces 2019, 176, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Becker Peres, L.; Becker Peres, L.; de Araújo, P.H.H.; Sayer, C. Solid lipid nanoparticles for encapsulation of hydrophilic drugs by an organic solvent free double emulsion technique. Colloids Surf. B Biointerfaces 2016, 140, 317–323. [Google Scholar] [CrossRef]
- Gallarate, M.; Trotta, M.; Battaglia, L.; Chirio, D. Preparation of solid lipid nanoparticles from W/O/W emulsions: Preliminary studies on insulin encapsulation. J. Microencapsul. 2009, 26, 394–402. [Google Scholar] [CrossRef]
- Severino, P.; Silveira, E.F.; Loureiro, K.; Chaud, M.V.; Antonini, D.; Lancellotti, M.; Sarmento, V.H.; da Silva, C.F.; Santana, M.H.A.; Souto, E.B. Antimicrobial activity of polymyxin-loaded solid lipid nanoparticles (PLX-SLN): Characterization of physicochemical properties and in vitro efficacy. Eur. J. Pharm. Sci. 2017, 106, 177–184. [Google Scholar] [CrossRef]
- Izquierdo, P.; Esquena, J.; Tadros, T.F.; Dederen, C.; Garcia, M.J.; Azemar, N.; Solans, C. Formation and Stability of Nano-Emulsions Prepared Using the Phase Inversion Temperature Method. Langmuir 2002, 18, 26–30. [Google Scholar] [CrossRef]
- Heurtault, B.; Saulnier, P.; Pech, B.; Proust, J.-E.; Benoit, J.-P. A Novel Phase Inversion-Based Process for the Preparation of Lipid Nanocarriers. Pharmaceutical Research 2002, 19, 875–880. [Google Scholar] [CrossRef]
- Roger, K.; Cabane, B.; Olsson, U. Formation of 10–100 nm Size-Controlled Emulsions through a Sub-PIT Cycle. Langmuir 2010, 26, 3860–3867. [Google Scholar] [CrossRef]
- Montenegro, L.; Sarpietro, M.G.; Ottimo, S.; Puglisi, G.; Castelli, F. Differential scanning calorimetry studies on sunscreen loaded solid lipid nanoparticles prepared by the phase inversion temperature method. Int. J. Pharm. 2011, 415, 301–306. [Google Scholar] [CrossRef]
- Montenegro, L.; Sinico, C.; Castangia, I.; Carbone, C.; Puglisi, G. Idebenone-loaded solid lipid nanoparticles for drug delivery to the skin: In vitro evaluation. Int. J. Pharm. 2012, 434, 169–174. [Google Scholar] [CrossRef]
- Ali, H.; Singh, S.K. Preparation and characterization of solid lipid nanoparticles of furosemide using quality by design. Part. Sci. Technol. 2018, 36, 695–709. [Google Scholar] [CrossRef]
- Gao, S.; McClements, D.J. Formation and stability of solid lipid nanoparticles fabricated using phase inversion temperature method. Colloids Surf. A Physicochem. Eng. Asp. 2016, 499, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Shinde, U.A.; Parmar, S.J.; Easwaran, S. Metronidazole-loaded nanostructured lipid carriers to improve skin deposition and retention in the treatment of rosacea. Drug Dev. Ind. Pharm. 2019, 45, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L.; Carbone, C.; Condorelli, G.; Drago, R.; Puglisi, G. Effect of Oil Phase Lipophilicity on In Vitro Drug Release from O/W Microemulsions with Low Surfactant Content. Drug Dev. Ind. Pharm. 2006, 32, 539–548. [Google Scholar] [CrossRef]
- Montenegro, L.; Campisi, A.; Sarpietro, M.G.; Carbone, C.; Acquaviva, R.; Raciti, G.; Puglisi, G. In vitro evaluation of idebenone-loaded solid lipid nanoparticles for drug delivery to the brain. Drug Dev. Ind. Pharm. 2011, 37, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Gomes, G.V.L.; Sola, M.R.; Rochetti, A.L.; Fukumasu, H.; Vicente, A.A.; Pinho, S.C. β-carotene and α-tocopherol coencapsulated in nanostructured lipid carriers of murumuru (Astrocaryum murumuru) butter produced by phase inversion temperature method: Characterisation, dynamic in vitro digestion and cell viability study. J. Microencapsul. 2019, 36, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, C.; Tomasello, B.; Ruozi, B.; Renis, M.; Puglisi, G. Preparation and optimization of PIT solid lipid nanoparticles via statistical factorial design. Eur. J. Med. Chem. 2012, 49, 110–117. [Google Scholar] [CrossRef]
- Izquierdo, P.; Feng, J.; Esquena, J.; Tadros, T.F.; Dederen, J.C.; Garcia, M.J.; Azemar, N.; Solans, C. The influence of surfactant mixing ratio on nano-emulsion formation by the pit method. J. Colloid Interface Sci. 2005, 285, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Charcosset, C.; El-Harati, A.; Fessi, H. Preparation of solid lipid nanoparticles using a membrane contactor. J. Control. Release 2005, 108, 112–120. [Google Scholar] [CrossRef]
- D’oria, C.; Charcosset, C.; Barresi, A.A.; Fessi, H. Preparation of solid lipid particles by membrane emulsification-Influence of process parameters. Colloids Surf. A Physicochem. Eng. Asp. 2009, 338, 114–118. [Google Scholar] [CrossRef]
- Ahmed El-Harati, A.; Charcosset, C.; Fessi, H. Influence of the Formulation for Solid Lipid Nanoparticles Prepared with a Membrane Contactor. Pharm. Dev. Technol. 2006, 11, 153–157. [Google Scholar] [CrossRef]
- Laouini, A.; Andrieu, V.; Vecellio, L.; Fessi, H.; Charcosset, C. Characterization of different vitamin E carriers intended for pulmonary drug delivery. Int. J. Pharm. 2014, 471, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Khayata, N.; Abdelwahed, W.; Chehna, M.F.; Charcosset, C.; Fessi, H. Preparation of vitamin E loaded nanocapsules by the nanoprecipitation method: From laboratory scale to large scale using a membrane contactor. Int. J. Pharm. 2012, 423, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, P.; Shekunov, B.Y.; Yim, D.; Cipolla, D.; Boyd, B.; Farr, S. Production of solid lipid nanoparticle suspensions using supercritical fluid extraction of emulsions (SFEE) for pulmonary delivery using the AERx system. Adv. Drug Deliv. Rev. 2007, 59, 444–453. [Google Scholar] [CrossRef]
- Andrade, L.N.; Oliveira, D.M.L.; Chaud, M.V.; Alves, T.F.R.; Nery, M.; da Silva, C.F.; Gonsalves, J.K.C.; Nunes, R.S.; Corrêa, C.B.; Amaral, R.G.; et al. Praziquantel-Solid Lipid Nanoparticles Produced by Supercritical Carbon Dioxide Extraction: Physicochemical Characterization, Release Profile, and Cytotoxicity. Molecules 2019, 24, 3881. [Google Scholar] [CrossRef] [Green Version]
- Campardelli, R.; Cherain, M.; Perfetti, C.; Iorio, C.; Scognamiglio, M.; Reverchon, E.; Della Porta, G. Lipid nanoparticles production by supercritical fluid assisted emulsion–diffusion. J. Supercrit. Fluids 2013, 82, 34–40. [Google Scholar] [CrossRef]
- Trucillo, P.; Campardelli, R. Production of solid lipid nanoparticles with a supercritical fluid assisted process. The J. Supercrit. Fluids 2019, 143, 16–23. [Google Scholar] [CrossRef]
- Acevedo-Morantes, C.Y.; Acevedo-Morantes, M.T.; Suleiman-Rosado, D.; Ramírez-Vick, J.E. Evaluation of the cytotoxic effect of camptothecin solid lipid nanoparticles on MCF7 cells. Drug Deliv. 2013, 20, 338–348. [Google Scholar] [CrossRef]
- Wong, C.Y.; Al-Salami, H.; Dass, C.R. Lyophilisation Improves Bioactivity and Stability of Insulin-Loaded Polymeric-Oligonucleotide Nanoparticles for Diabetes Treatment. AAPS PharmSciTech 2020, 21, 108. [Google Scholar] [CrossRef] [PubMed]
- Castro, F.; Pinto, M.L.; Almeida, R.; Pereira, F.; Silva, A.M.; Pereira, C.L.; Santos, S.G.; Barbosa, M.A.; Gonçalves, R.M.; Oliveira, M.J. Chitosan/poly(γ-glutamic acid) nanoparticles incorporating IFN-γ for immune response modulation in the context of colorectal cancer. Biomater. Sci. 2019, 7, 3386–3403. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, L.; Gallarate, M.; Cavalli, R.; Trotta, M. Solid lipid nanoparticles produced through a coacervation method. J. Microencapsul. 2010, 27, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Chirio, D.; Gallarate, M.; Peira, E.; Battaglia, L.; Muntoni, E.; Riganti, C.; Biasibetti, E.; Capucchio, M.T.; Valazza, A.; Panciani, P.; et al. Positive-charged solid lipid nanoparticles as paclitaxel drug delivery system in glioblastoma treatment. Eur. J. Pharm. Biopharm. 2014, 88, 746–758. [Google Scholar] [CrossRef]
- Battaglia, L.; Muntoni, E.; Chirio, D.; Peira, E.; Annovazzi, L.; Schiffer, D.; Mellai, M.; Riganti, C.; Salaroglio, I.C.; Lanotte, M.; et al. Solid lipid nanoparticles by coacervation loaded with a methotrexate prodrug: Preliminary study for glioma treatment. Nanomedicine 2017, 12, 639–656. [Google Scholar] [CrossRef] [PubMed]
- Clemente, N.; Ferrara, B.; Gigliotti, C.L.; Boggio, E.; Capucchio, M.T.; Biasibetti, E.; Schiffer, D.; Mellai, M.; Annovazzi, L.; Cangemi, L.; et al. Solid lipid nanoparticles carrying temozolomide for melanoma treatment. Preliminary in vitro and in vivo studies. Int. J. Mol. Sci. 2018, 19, 255. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, L.; Gallarate, M.; Peira, E.; Chirio, D.; Solazzi, I.; Giordano, S.M.A.; Gigliotti, C.L.; Riganti, C.; Dianzani, C. Bevacizumab loaded solid lipid nanoparticles prepared by the coacervation technique: Preliminary in vitro studies. Nanotechnology 2015, 26, 255102. [Google Scholar] [CrossRef] [Green Version]
- Muntoni, E.; Marini, E.; Ahmadi, N.; Milla, P.; Ghè, C.; Bargoni, A.; Capucchio, M.T.; Biasibetti, E.; Battaglia, L. Lipid nanoparticles as vehicles for oral delivery of insulin and insulin analogs: Preliminary ex vivo and in vivo studies. Acta Diabetol. 2019, 56, 1283–1292. [Google Scholar] [CrossRef]
- Wang, T.; Wang, N.; Zhang, Y.; Shen, W.; Gao, X.; Li, T. Solvent injection-lyophilization of tert-butyl alcohol/water cosolvent systems for the preparation of drug-loaded solid lipid nanoparticles. Colloids Surf. B Biointerfaces 2010, 79, 254–261. [Google Scholar] [CrossRef]
- Zhang, S.; Yun, J.; Shen, S.; Chen, Z.; Yao, K.; Chen, J.; Chen, B. Formation of solid lipid nanoparticles in a microchannel system with a cross-shaped junction. Chem. Eng. Sci. 2008, 63, 5600–5605. [Google Scholar] [CrossRef]
- Zhang, S.-H.; Shen, S.-C.; Chen, Z.; Yun, J.-X.; Yao, K.-J.; Chen, B.-B.; Chen, J.-Z. Preparation of solid lipid nanoparticles in co-flowing microchannels. Chem. Eng. J. 2008, 144, 324–328. [Google Scholar] [CrossRef]
- Kimura, N.; Maeki, M.; Sato, Y.; Ishida, A.; Tani, H.; Harashima, H.; Tokeshi, M. Development of a Microfluidic-Based Post-Treatment Process for Size-Controlled Lipid Nanoparticles and Application to siRNA Delivery. ACS Appl. Mater. Interfaces 2020, 12, 34011–34020. [Google Scholar] [CrossRef]
- Singh, A.; Ahmad, I.; Akhter, S.; Jain, G.K.; Iqbal, Z.; Talegaonkar, S.; Ahmad, F.J. Nanocarrier based formulation of Thymoquinone improves oral delivery: Stability assessment, in vitro and in vivo studies. Colloids Surf. B Biointerfaces 2013, 102, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Ahmad, I.; Ahmad, S.; Iqbal, Z.; Ahmad, F.J. A novel monolithic controlled delivery system of resveratrol for enhanced hepatoprotection: Nanoformulation development, pharmacokinetics and pharmacodynamics. Drug Dev. Ind. Pharm. 2016, 42, 1524–1536. [Google Scholar] [CrossRef] [PubMed]
- Pandita, D.; Ahuja, A.; Lather, V.; Benjamin, B.; Dutta, T.; Velpandian, T.; Khar, R.K. Development of Lipid-Based Nanoparticles for Enhancing the Oral Bioavailability of Paclitaxel. AAPS PharmSciTech 2011, 12, 712–722. [Google Scholar] [CrossRef]
- Lingling, G.; Yuan, Z.; Weigen, L. Preparation, optimization, characterization and in vivo pharmacokinetic study of asiatic acid tromethamine salt-loaded solid lipid nanoparticles. Drug Dev. Ind. Pharm. 2016, 42, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Dodiya, S.; Chavhan, S.; Korde, A.; Sawant, K.K. Solid lipid nanoparticles and nanosuspension of adefovir dipivoxil for bioavailability improvement: Formulation, characterization, pharmacokinetic and biodistribution studies. Drug Dev. Ind. Pharm. 2013, 39, 733–743. [Google Scholar] [CrossRef]
- Parveen, R.; Ahmad, F.J.; Iqbal, Z.; Samim, M.; Ahmad, S. Solid lipid nanoparticles of anticancer drug andrographolide: Formulation, in vitro and in vivo studies. Drug Dev. Ind. Pharm. 2014, 40, 1206–1212. [Google Scholar] [CrossRef]
- Hashem, F.M.; Nasr, M.; Khairy, A. In vitro cytotoxicity and bioavailability of solid lipid nanoparticles containing tamoxifen citrate. Pharm. Dev. Technol. 2014, 19, 824–832. [Google Scholar] [CrossRef]
- Hansraj, G.P.; Singh, S.K.; Kumar, P. Sumatriptan succinate loaded chitosan solid lipid nanoparticles for enhanced anti-migraine potential. Int. J. Biol. Macromol. 2015, 81, 467–476. [Google Scholar] [CrossRef]
- E Eleraky, N.; M Omar, M.; A Mahmoud, H.; A Abou-Taleb, H. Nanostructured Lipid Carriers to Mediate Brain Delivery of Temazepam: Design and In Vivo Study. Pharmaceutics 2020, 12, 451. [Google Scholar] [CrossRef]
- Sharma, S.; Verma, A.; Singh, J.; Teja, B.V.; Mittapelly, N.; Pandey, G.; Urandur, S.; Shukla, R.P.; Konwar, R.; Mishra, P.R. Vitamin B6 Tethered Endosomal pH Responsive Lipid Nanoparticles for Triggered Intracellular Release of Doxorubicin. ACS Appl. Mater. Interfaces 2016, 8, 30407–30421. [Google Scholar] [CrossRef]
- Sahu, P.K.; Mishra, D.K.; Jain, N.; Rajoriya, V.; Jain, A.K. Mannosylated solid lipid nanoparticles for lung-targeted delivery of Paclitaxel. Drug Dev. Ind. Pharm. 2015, 41, 640–649. [Google Scholar] [CrossRef]
- Bishnoi, M.; Jain, A.; Hurkat, P.; Jain, S.K. Aceclofenac-loaded chondroitin sulfate conjugated SLNs for effective management of osteoarthritis. J. Drug Target. 2014, 22, 805–812. [Google Scholar] [CrossRef]
- Shilpi, S.; Vimal, V.D.; Soni, V. Assessment of lactoferrin-conjugated solid lipid nanoparticles for efficient targeting to the lung. Prog. Biomater. 2015, 4, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, A.; Jain, A.; Jain, A.; Jain, A.; Pandey, V.; Chashoo, G.; Soni, V.; Sharma, P.R. Targeted SLNs for management of HIV-1 associated dementia. Drug Dev. Ind. Pharm. 2015, 41, 1321–1327. [Google Scholar] [CrossRef]
- Dinesh, M.; Himanshu, M.; Pradyumna, K.M.; Manoj, N.; Vaibhav, D.; Narendra, K.J. Evaluation of Solid Lipid Nanoparticles as Carriers for Delivery of Hepatitis B Surface Antigen for Vaccination Using Subcutaneous Route. J. Pharm. Pharm. Sci. 2010, 13, 495–509. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Jain, S.; Khare, P.; Gulbake, A.; Bansal, D.; Jain, S.K. Design and development of solid lipid nanoparticles for topical delivery of an anti-fungal agent. Drug Deliv. 2010, 17, 443–451. [Google Scholar] [CrossRef]
- Jain, A.K.; Jain, A.; Garg, N.K.; Agarwal, A.; Jain, A.; Jain, S.A.; Tyagi, R.K.; Jain, R.K.; Agrawal, H.; Agrawal, G.P. Adapalene loaded solid lipid nanoparticles gel: An effective approach for acne treatment. Colloids Surf. B Biointerfaces 2014, 121, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Bikkad, M.L.; Nathani, A.H.; Mandlik, S.K.; Shrotriya, S.N.; Ranpise, N.S. Halobetasol propionate-loaded solid lipid nanoparticles (SLN) for skin targeting by topical delivery. J. Liposome Res. 2014, 24, 113–123. [Google Scholar] [CrossRef]
- Madan, J.R.; Khude, P.A.; Dua, K. Development and evaluation of solid lipid nanoparticles of mometasone furoate for topical delivery. Int. J. Pharm. Investig. 2014, 4, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Gänger, S.; Schindowski, K. Tailoring Formulations for Intranasal Nose-to-Brain Delivery: A Review on Architecture, Physico-Chemical Characteristics and Mucociliary Clearance of the Nasal Olfactory Mucosa. Pharmaceutics 2018, 10, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandita, D.; Ahuja, A.; Velpandian, T.; Lather, V.; Dutta, T.; Khar, R. Characterization and in vitro assessment of paclitaxel loaded lipid nanoparticles formulated using modified solvent injection technique. Die Pharm. -An Int. J. Pharm. Sci. 2009, 64, 301–310. [Google Scholar]
- Shah, M.; Pathak, K. Development and Statistical Optimization of Solid Lipid Nanoparticles of Simvastatin by Using 23 Full-Factorial Design. AAPS PharmSciTech 2010, 11, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Dube, B.; Sawant, K. Synthesis of cytarabine lipid drug conjugate for treatment of meningeal leukemia: Development, characterization and in vitro cell line studies. J. Biomed. Nanotechnol. 2012, 8, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, R.; Gradzielski, M.; Mehta, S.K. Biomimetic Solid Lipid Nanoparticles of Sophorolipids Designed for Antileprosy Drugs. J. Phys. Chem. B 2018, 122, 6837–6845. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, F.; Faghfouri, L.; Hamidi, M. Preparation, optimization, and in-vitro characterization of α-tocopherol-loaded solid lipid nanoparticles (SLNs). Drug Dev. Ind. Pharm. 2020, 46, 159–171. [Google Scholar] [CrossRef]
- Müller, R.H.; Runge, S.; Ravelli, V.; Mehnert, W.; Thünemann, A.F.; Souto, E.B. Oral bioavailability of cyclosporine: Solid lipid nanoparticles (SLN®) versus drug nanocrystals. Int. J. Pharm. 2006, 317, 82–89. [Google Scholar] [CrossRef]
- Müller, R.H.; Runge, S.A.; Ravelli, V.; Thünemann, A.F.; Mehnert, W.; Souto, E.B. Cyclosporine-loaded solid lipid nanoparticles (SLN®): Drug–lipid physicochemical interactions and characterization of drug incorporation. Eur. J. Pharm. Biopharm. 2008, 68, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, Y.; Smith, E. Experimental Design for the Optimization of Lipid Nanoparticles. J. Pharm. Sci. 2009, 98, 1813–1819. [Google Scholar] [CrossRef]
- Anton, N.; Benoit, J.-P.; Saulnier, P. Design and production of nanoparticles formulated from nano-emulsion templates—A review. J. Control. Release 2008, 128, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Sanjula, B.; Shah, F.M.; Javed, A.; Alka, A. Effect of poloxamer 188 on lymphatic uptake of carvedilol-loaded solid lipid nanoparticles for bioavailability enhancement. J. Drug Target. 2009, 17, 249–256. [Google Scholar] [CrossRef]
- Paliwal, R.; Rai, S.; Vaidya, B.; Khatri, K.; Goyal, A.K.; Mishra, N.; Mehta, A.; Vyas, S.P. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 184–191. [Google Scholar] [CrossRef]
- Liu, D.; Jiang, S.; Shen, H.; Qin, S.; Liu, J.; Zhang, Q.; Li, R.; Xu, Q. Diclofenac sodium-loaded solid lipid nanoparticles prepared by emulsion/solvent evaporation method. J. Nanoparticle Res. 2011, 13, 2375–2386. [Google Scholar] [CrossRef]
- Shafique, H.; Ahad, A.; Khan, W.; Want, M.Y.; Bhatt, P.C.; Ahmad, S.; Panda, B.P.; Mujeeb, M. Ganoderic acid -loaded solid lipid nanoparticles ameliorate d-galactosamine induced hepatotoxicity in Wistar rats. J. Drug Deliv. Sci. Technol. 2019, 50, 48–56. [Google Scholar] [CrossRef]
- Song, C.X.; Labhasetwar, V.; Murphy, H.; Qu, X.; Humphrey, W.R.; Shebuski, R.J.; Levy, R.J. Formulation and characterization of biodegradable nanoparticles for intravascular local drug delivery. J. Control. Release 1997, 43, 197–212. [Google Scholar] [CrossRef]
- Govender, T.; Stolnik, S.; Garnett, M.C.; Illum, L.; Davis, S.S. PLGA nanoparticles prepared by nanoprecipitation: Drug loading and release studies of a water soluble drug. J. Control. Release 1999, 57, 171–185. [Google Scholar] [CrossRef]
- Hu, F.Q.; Hong, Y.; Yuan, H. Preparation and characterization of solid lipid nanoparticles containing peptide. Int. J. Pharm. 2004, 273, 29–35. [Google Scholar] [CrossRef]
- Baek, J.-S.; Pham, C.V.; Myung, C.-S.; Cho, C.-W. Tadalafil-loaded nanostructured lipid carriers using permeation enhancers. Int. J. Pharm. 2015, 495, 701–709. [Google Scholar] [CrossRef]
- Mitri, K.; Shegokar, R.; Gohla, S.; Anselmi, C.; Müller, R.H. Lipid nanocarriers for dermal delivery of lutein: Preparation, characterization, stability and performance. Int. J. Pharm. 2011, 414, 267–275. [Google Scholar] [CrossRef]
- Souto, E.B.; Wissing, S.A.; Barbosa, C.M.; Müller, R.H. Development of a controlled release formulation based on SLN and NLC for topical clotrimazole delivery. Int. J. Pharm. 2004, 278, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Kalam, M.A.; Sultana, Y.; Ali, A.; Aqil, M.; Mishra, A.K.; Chuttani, K. Preparation, characterization, and evaluation of gatifloxacin loaded solid lipid nanoparticles as colloidal ocular drug delivery system. J. Drug Target. 2010, 18, 191–204. [Google Scholar] [CrossRef]
- Peltonen, L.; Aitta, J.; Hyvönen, S.; Karjalainen, M.; Hirvonen, J. Improved entrapment efficiency of hydrophilic drug substance during nanoprecipitation of poly(l)lactide nanoparticles. AAPS PharmSciTech 2009, 5, E16. [Google Scholar] [CrossRef] [Green Version]
- Lamprecht, A.; Ubrich, N.; Hombreiro Pérez, M.; Lehr, C.M.; Hoffman, M.; Maincent, P. Influences of process parameters on nanoparticle preparation performed by a double emulsion pressure homogenization technique. Int. J. Pharm. 2000, 196, 177–182. [Google Scholar] [CrossRef]
- Esposito, E.; Boschi, A.; Ravani, L.; Cortesi, R.; Drechsler, M.; Mariani, P.; Moscatelli, S.; Contado, C.; Di Domenico, G.; Nastruzzi, C.; et al. Biodistribution of nanostructured lipid carriers: A tomographic study. Eur. J. Pharm. Biopharm. 2015, 89, 145–156. [Google Scholar] [CrossRef]
- Esposito, E.; Sguizzato, M.; Drechsler, M.; Mariani, P.; Carducci, F.; Nastruzzi, C.; Valacchi, G.; Cortesi, R. Lipid nanostructures for antioxidant delivery: A comparative preformulation study. Beilstein J. Nanotechnol. 2019, 10, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.; Kumar, A.C.K.; Priya, V.K.; Yadav, C.M.; Raju, P.Y. Formulation and evaluation of chitosan solid lipid nanoparticles of carbamazepine. Lipids in Health Dis. 2012, 11, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amis, T.M.; Renukuntla, J.; Bolla, P.K.; Clark, B.A. Selection of Cryoprotectant in Lyophilization of Progesterone-Loaded Stearic Acid Solid Lipid Nanoparticles. Pharmaceutics 2020, 12, 892. [Google Scholar] [CrossRef]
- Sanchez-Vazquez, B.; Lee, J.B.; Strimaite, M.; Buanz, A.; Bailey, R.; Gershkovich, P.; Pasparakis, G.; Williams, G.R. Solid lipid nanoparticles self-assembled from spray dried microparticles. Int. J. Pharm. 2019, 572, 118784. [Google Scholar] [CrossRef]
- Vicente-Pascual, M.; Gómez-Aguado, I.; Rodríguez-Castejón, J.; Rodríguez-Gascón, A.; Muntoni, E.; Battaglia, L.; del Pozo-Rodríguez, A.; Solinís Aspiazu, M.Á. Topical Administration of SLN-Based Gene Therapy for the Treatment of Corneal Inflammation by De Novo IL-10 Production. Pharmaceutics 2020, 12, 584. [Google Scholar] [CrossRef]
- Kubo, T.; Yanagihara, K.; Sato, Y.; Nishimura, Y.; Kondo, S.; Seyama, T. Gene-Silencing Potency of Symmetric and Asymmetric Lipid-Conjugated siRNAs and Its Correlation with Dicer Recognition. Bioconjugate Chem. 2013, 24, 2045–2057. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Mechanism | Advantage | Disadvantage |
|---|---|---|---|
| Hot high-pressure homogenization | High shear stress and cavitational forces | Speed, straightforward, avoidance of organic solvents, scalability | Drug degradation under high temperature, drug loss into the aqueous phase |
| Cold high-pressure homogenization | High shear stress and cavitational forces | Prevention of drug degradation, applicability to hydrophilic drugs | Large particles, broad size distributions |
| High-speed stirring and ultra-sonication | High shear between two solid adjacent area Formation, growth, and implosive collapse of bubbles in a liquid | straightforward, avoidance of organic solvents, low cost, scalability | Exposure of drugs to high temperatures, metal contamination from sonicator probes, high surfactant concentrations, low lipid concentrations |
| Microemulsion | Spontaneous interfacial tension reduction under dilution | Simplicity, reproducibility, scalability, avoidance of organic solvents | Large amount of water to dilute microemulsions, high concentration of surfactants |
| Solvent emulsification-diffusion | Diffusion of solvent from lipid phase to aqueous phase leading to lipid precipitation | Simplicity, avoidance of heat, small PS, narrow size distribution Scalability, applicability to both hydrophilic and hydrophobic drugs | Residual solvent, additional solvent removal procedures |
| Solvent emulsification-evaporation method | Evaporation of solvent in lipid phase leading to lipid precipitation | Simplicity, avoidance of heat, small PS, narrow size distribution | Residual solvent, additional solvent removal procedure Dilute suspensions, requirement of evaporation or ultra-filtration |
| Double emulsion | Lipid crystallization due to solvent evaporation or low temperature | Applicability to hydrophilic drugs | Low EE and DL Large PS |
| Phase inversion temperature (PIT) | Spontaneous inversion between oil/water and water/oil emulsions with temperature change | Low energy, avoidance of organic solvents, narrow size distribution, good stability | Instability of emulsion |
| Membrane contactor | Formation of small droplets after pressing lipid phase through membrane pores | Scalability, control of size | Clogging of membrane |
| Supercritical fluid-based methods | Quick evaporation or diffusion of solvent with the help of supercritical fluid, resulting in lipid precipitation | Uniform particle size distribution, high solvent extraction efficiency | Use of organic solvent, high expense |
| Coacervation | Precipitation of alkaline salts of fatty acids when decreasing pH | Simplicity, no sophisticated instrument, avoidance of organic solvents | Applicability only to lipids in alkaline salt form and non pH-sensitive drugs |
| Solvent injection | Diffusion of solvent from lipid phase to aqueous phase leading to lipid precipitation | Simplicity, straightforward, fast production process, no sophisticated instrument | Residual solvent, additional solvent removal procedure |
| Lipid(s)/Emulsifier(s) | Drug or Active Ingredient | Solvent | Outcomes | Year, Reference |
|---|---|---|---|---|
| Lipoid®S 100/sucrose fatty acid ester | Paclitaxel | Acetone | PS: 188 nm, PDI: 0.396, and EE: 92.2%SLNs showed sustained-release for 14 days (in vitro) | 2006, [40] |
| Palmitic or stearic acid/phosphatidylcholine | Idebenone | Ethanol | PS: 170–183 nm, PDI: 0.113–0.134, and EE: 83–86% SLNs showed sustained-release for 7 days (in vitro) SLNs increased drug protecting activity against free radical-induced oxidative damage of astrocyte cells | 2006, [41] |
| Stearic acid, soya lecithin/poloxamer 188 | Paclitaxel | Diethyl ether | Drug amount and emulsifier concentration were varied to get the optimized SLNs (PS: 113 nm, PDI: 0.156, and EE: 89.0%) SLNs had lower toxicity on HepG2 cell than free drug SLNs was stable for 90 days | 2009, [165] |
| Tristearin, soya lecithin/polysorbate 80 | Miconazole nitrate | Ethanol | Parameters were varied to obtain optimized NLCs (PS: 206 nm, PDI: 0.21, and EE: 90.9%) NLCs-based hydrogels increased skin retention 10-fold as compared with miconazole suspension and miconazole hydrogel | 2010, [160] |
| Tristearin/polysorbate 80 | Hepatitis B surface antigen (HBsAg) | Acetone | HBsAg was loaded onto SLNs surface Mannosylated SLNs (PS: 96.6 nm, PDI: 0.05, and EE: 64.5%) SLNs showed better cellular uptake, lesser toxicity, and greater immune response Subcutaneous administration of SLNs was potential for vaccine delivery against hepatitis B | 2010, [159] |
| Monostearin/poloxamer 407 | Simvastatin | Isopropanol | A 23 factorial design was performed to optimize SLNs (PS: 259 nm, EE: 75.8%) SLNs showed sustained-release for 55 h (in vitro) | 2010, [166] |
| Tristearin, soya lecithin/polysorbate 80 | Doxorubicin hydrochloride | Acetone + ethanol | Mannosylated SLNs (PS: 360 nm, PDI: 0.135, and EE: 70.3%) Mannosylated SLNs showed sustained-release in mice. They delivered a higher dug concentration to the tumor mass. | 2010, [38] |
| Glycerol monostearate, oleic acid/poloxamer 407 | Simvastatin | Isopropanol | A 23 factorial design was performed to optimize NLCs (PS: 212 nm, PDI: 0.344, and EE: 84%) NLCs showed a higher bioavailability than drug suspension and drug-loaded SLNs | 2011, [42] |
| Monostearin, soya lecithin/poloxamer 188 | Puerarin | Methanol + ethanol | SLNs enhanced oral bioavailability of the drug 3 times Tissue concentration of the drug increased, particularly the hearts and brain after oral administration of SLNs | 2011, [39] |
| Stearylamine, soya lecithin, α-tocopherol/poloxamer 188 | Paclitaxel | Diethyl ether | Drug amount and emulsifier concentration were varied to obtain optimized SLNs (PS: 96 nm, PDI: 0.162, EE: 75.4%, and DL: 31.5%) SLNs increased oral bioavailability of the drug 10 times in mice | 2011, [147] |
| Monostearin/Lecithin + poloxamer 188 | Ondansetron hydrochloride | Ethanol | A 23 factorial design was performed to get the optimized SLNs (PS: 320 nm, PDI: 0.296, and EE: 49.7%) The SLNs effectively delivered the drug to the brain by intranasal administration on rabbits | 2012, [43] |
| Stearic acid/polysorbate 80 | Cytarabine | Isopropanol | Drug and lipid was conjugated prior to SLNs preparation - PS: 137 nm, PDI: 0.151, and EE: 58.4% SLNs showed drug sustained-release (3 days in vitro) and increased toxicity on leukemic EL-4 cells as compared with drug solution | 2012, [167] |
| Glyceryl behenate (Compritol® 888 ATO)/poloxamer 407 | Terbinafine hydrochloride | Isopropanol | A 33 factorial design was performed to get the optimized SLNs (PS: 274 nm, PDI: 0.32, and EE: 74.6%) SLN-based gel was more effective than a commercial product when applying in rats (pharmacodynamics studies) - SLNs were stable for 90 days | 2013, [44] |
| Monostearin/polysorbate 80 + poloxamer 188 | Thymoquinon | Ethanol | Box-Behnken design was used to optimize the SLNs (PS: 166 nm, EE: 71.6%) Oral bioavailability increased 5-fold in rats as compared with drug suspension | 2013, [145] |
| Dynasan 114, soya phosphatidylcholine/poloxamer 407 | Adefovir dipivoxil | Isopropanol | Different process and formulation parameters were evaluated to optimize SLNs (PS: 267 nm, EE: 73.5%, and DL: 2%) SLNs increased oral bioavailability 2- and 1.5-fold as compared with micro-suspension and nanosuspension, respectively SLNs increased drug accumulation in liver, kidneys, intestine, and stomach. | 2013, [149] |
| Cetyl alcohol/polysorbate 80 | Andrographolide | Ethanol | PS: 154 nm, PDI: 0.172, EE: 91.4%, and DL: 18.6% SLNs increased oral bioavailability (3.41-fold) and antitumor activity as compared with the drug suspension | 2014, [150] |
| Tristearin, soya lecithin/polysorbate 80 | Adapalene | Acetone + ethanol | PS: 148 nm, PDI: 0.169, and EE: 89.9% SLNs-based gel showed sustained-release in vitro | 2014, [161] |
| Tristearin, hydrogenated soya phosphatidylcholine/polysorbate 80 | Aceclofenac | Ethanol | SLNs was conjugated with chondroitin sulfate (CS-SLNs) with PS: 154 nm, PDI: 0.403, and EE: 65.4% SLNs and CS-SLNs increased drug amount in the inflammatory knee joint (2- and 10-fold as compared with the drug solution, respectively) after IV administration in rats | 2014, [156] |
| Monostearin/polysorbate 80 | Halobetasol propionate | Isopropanol | A 32 full factorial design was applied to optimize the SLNs (PS: 200 nm and EE: 93%) SLNs-based carbopol gels prolonged drug release up to 12 h on human cadaver skin, reduced systemic uptake, increased drug accumulation in skin, and were nonirritant to rabbit skin | 2014, [162] |
| Monostearin or stearic acid/polysorbate 80 or poloxamer 188 | Tamoxifen | Methanol | Lipid and emulsifier were varied to obtain optimized SLNs (PS: 130 nm, PDI, 0.231, and EE: 86.1%) SLNs increased oral bioavailability (1.6-fold) following oral administration in rats | 2014, [151] |
| Monostearin, Tefose-63/polysorbate 80 | Mometasone furoate | Ethanol | SLNs was optimized (PS: 124 nm, and EE: 55.6%) SLNs-based carbopol gel increased skin deposition 2.67- and 20-fold as compared with a marketed cream and drug-loaded gel SLNs-based gel increased skin permeability 15.2-fold as compared with a marketed cream | 2014, [163] |
| Tristearin, distearoyl-phosphatidyl ethanolamine/polysorbate 80 | Paclitaxel | Acetone + ethanol | Mannosylated SLNs (PS: 254 nm, PDI: 0.312) Mannosylated SLNs improved antiproliferative efficacy in lung cancer cells. They delivered a higher drug concentration to alveolar cells after IV injection in rats | 2015, [155] |
| Tristearin, soya lecithin, stearylamine/polysorbate 80 | Rifampicin | Ethanol | Drug-loaded SLNs was coupled with lactoferrin to enhance SLNS delivery to lung (PS: 271 nm, PDI: 0.124, and EE: 68.4%) In vivo biodistribution study: lactoferrin-coupled SLNs had 47.7% drug uptakes by the lungs (3.05 times higher than uncoupled SLNs) following IV injection in rats | 2015, [157] |
| Tripalmitin/polysorbate 80 | Sumatriptan | Ethanol | A 23 randomized full factorial design was performed to optimize the SLNs (PS: 236 nm, and EE 91.3%) SLNs showed a 4.54-fold increase in brain/blood ratio of drug (2 h after oral administration in rats) SLNs improved anti-migraine potential in behavioral studies | 2015, [152] |
| Tristearin, hydrogenated soya phosphatidylcholine/polysorbate 80 | Nifedipine | Ethanol | SLNs was further coated with polysorbate 80 (PS: 121 nm, PDI: 0.261, and EE: 71.5%) Coated-SLNs increased bioavailability about 5- and 2-fold as compared with free drug and uncoated SLNs, respectively (IV administration in rats) Coated-SLNs increased drug accumulation in brain | 2015, [158] |
| Compritol® 888 ATO, Gelucire® 50/13/polysorbate 80 | Resveratrol | Ethanol | Box–Behnken design was applied to optimize SLNs (PS: 191 nm, PDI: 0.156, and EE: 73.7%) SLNs increased oral bioavailability nearly 5-fold in rats as compared with resveratrol suspension Pharmacodynamic data showed a decrease in the serum biomarker enzymes as compared with control and marketed formulation (against paracetamol-induced liver cirrhosis) | 2016, [146] |
| Monostearin/poloxamer 188 | Asiatic acid | Ethanol | A Box–Behnken design was used to optimize the formulations (PS: 237 nm, EE: 64.4%, and DL: 31.9%) SLN increased oral bioavailability 2.5-fold following oral administration in rats | 2016, [148] |
| Vitamin B6-stearic acid conjugation/polysorbate 80 | Doxorubicin | Ethanol | Vitamin B6 was conjugated with lipid to modify charge of SLNs (PS: 114 nm, PDI: 0.101, and DL: 7.1%) Vitamin B6-modified SLNs had an increased therapeutic efficacy and lower toxicity in tumor-bearing rats as compared with free drug Vitamin B6-modified SLNs prolonged drug circulation in blood and increased drug accumulation to tumor site in rats | 2016, [154] |
| Sophorolipid/poloxamer 407 and 188 | Rifampicin + dapsone | Ethanol | Five different polymers were used to stabilize SLNs, and poloxamer 407 and 188 were the best options - There was no in vivo study | 2018, [168] |
| Tripalmitin, Phosal® 53MCT/polysorbate 80 | Ondansetron hydrochloride | Ethanol | Various parameters were investigated to get the optimized NLCs (PS: 185 nm, PDI: 0.214, EE: 93.2%, and DL: 10.43%) NLCs showed sustained-release in vitro and in vivo for up to 4 days following subcutaneous administration in rats | 2019, [76] |
| YSK05, cholesterol, DMG-PEG2K | siRNA | Ethanol | NLCs were subjected to post-treatment using an integrated baffle device (PS: 33 nm and EE: 90%) The siFVII knocked down the plasma coagulation factor VII at mice liver tissue more than 80% (IV injection, mice) | 2020, [144] |
| Compritol® 888 ATO, oleic acid/poloxamer 407 | Temazepam | Acetone + ethanol | A 42 full factorial design was applied to optimize NLCs (PS: 307 nm, PDIL 0.09, and EE: 75.2%) NLCs increase oral bioavailability nearly 3-fold in rats as compared with temazepam suspension NLCs improved brain uptake of 99mTc-temazepam | 2020, [153] |
| Stearic acid, phosphatidylcholine | Alpha-tocopherol | Ethanol | Various parameters were evaluated to optimize SLNs (PS: 175 nm, EE: 90.9%, and DL: 59.4%)No in vivo study was conducted | 2020, [169] |
| Lipid(s) | Lipid Concentration Changes | Results | Reference |
|---|---|---|---|
| Softisan® 100 | 10–40 mg/mL | Increase in PS (~140–210 nm) | [36] |
| Tripalmitin, Phosal® 53MCT | 20–80 mg/mL | From 20–60 mg/mL: PS (~135–185 nm), PDI (~0.2, no change), EE (81.1–93.2%), and DL (9.20–10.43%) From 60–80 mg/mL: significant increases in PS (to ~235 nm) and PDI (to ~ 0.47), negligible changes in EE and DL | [76] |
| Glycerin monostearate | 10–15 mg/mL | Increase in PS (320–360 nm) | [43] |
| Glycerol monostearate | 50–100 mg/mL | Increase in PS (272–315 nm) and EE (69.7–80.9%) | [166] |
| Stearic acid | 40–50 mg/mL | Increase in PS (282–305 nm) and PDI (0.32–0.69) | [169] |
| Lipid(s) | Change of Liquid Lipid Level | Results | Reference |
|---|---|---|---|
| Tristearin, soya lecithin | 20–50% * | Decrease in PS (426–311 nm) and PDI (0.38–0.24) | [160] |
| Tripalmitin, Phosal® 53MCT | 0–50% * | From 0–40%: decreases in PS (479–185 nm) and PDI (0.441–0.214), increases in EE (81.2–93.2%) and DL (9.21–10.43%) From 40–50%: significant increases in PS (to 213 nm) and PDI (to 0.521) | [76] |
| Glycerin monostearate, oleic acid | 15–30 mg | Decrease in PS (210–194 nm) and PDI (0.355–0.242), increase in EE (83.9–93.3%) | [42] |
| Tristearin, distearoyl-phosphatidyl ethanolamine | 33–67% * | From 33–50%: slight increase in PS (186–195 nm), decrease in PDI (0.388–0.265) From 50–67%: increase in PS (19–233 nm) and PDI (0.265–0.404) | [155] |
| Stearic acid, hosphatidylcholine | 5–15 mg/mL | From 5–10 mg/mL: decreases in PS (283–234 nm) and PDI (0.57–0.41) From 10–15 mg/mL: increases in PS (234–247 nm) and PDI (0.41–0.68) | [169] |
| Lipid(s)/Drug | Change of Initial Drug Amount | Results | Reference |
|---|---|---|---|
| Stearic acid, soya lecithin/Paclitaxel | 0.05–0.25–0.5 mmol | PS (99–113–157 nm), EE (72.2–89.0–66.5%), DL (16.3–25.0–23.6%) | [165] |
| Stearic acid, soya lecithin, α-tocopherol/Paclitaxel | 0.05–0.25–0.5 mmol | PS (90–96–129 nm), EE (58.6–75.4–53.0%), DL (12.0–31.5–18.1%) | [147] |
| Tripalmitin, Phosal® 53MCT/Ondansetron hydrochloride | 6.3–16.7% * | From 6.3–12.5%: PS (172–185 nm), PDI (0.191–0.214), EE (86.7–93.2%), and DL (5.14–10.43%) From 12.5–16.7%: PS (185–254 nm), PDI (0.214–0.509), EE (93.2–83.3%), DL (10.43–12.20%) | [76] |
| Tristearin, soya lecithin/Miconazole nitrate | 2.5–10% * | From 2.5–7.5%: PS (322–328 nm), PDI (0.28–0.26), and EE (87.6–92.7%) From 7.5–10%: PS (328–353 nm), PDI (0.26–0.41), and EE (92.7–84.9%) | [160] |
| Tristearin, distearoyl-phosphatidyl ethanolamine/Paclitaxel | 5–20% * | From 5–10%: PS (209–195 nm) and EE (74.1–84.5%) From 10–20%: PS (195–318 nm) and EE (84.5–75.5%) | [155] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duong, V.-A.; Nguyen, T.-T.-L.; Maeng, H.-J. Preparation of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Drug Delivery and the Effects of Preparation Parameters of Solvent Injection Method. Molecules 2020, 25, 4781. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25204781
Duong V-A, Nguyen T-T-L, Maeng H-J. Preparation of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Drug Delivery and the Effects of Preparation Parameters of Solvent Injection Method. Molecules. 2020; 25(20):4781. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25204781
Chicago/Turabian StyleDuong, Van-An, Thi-Thao-Linh Nguyen, and Han-Joo Maeng. 2020. "Preparation of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Drug Delivery and the Effects of Preparation Parameters of Solvent Injection Method" Molecules 25, no. 20: 4781. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25204781