
In Vitro and In Silico Evaluation of Anticancer Activity of New Indole-Based 1,3,4-Oxadiazoles as EGFR and COX-2 Inhibitors

 ,
,  ,
,  ,
,  , ,
, ,  , ,
, ,  and
and 
Abstract
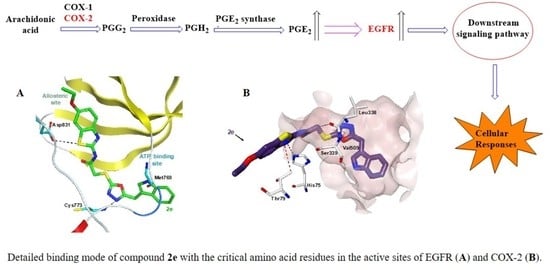
:
1. Introduction
2. Results and Discussion
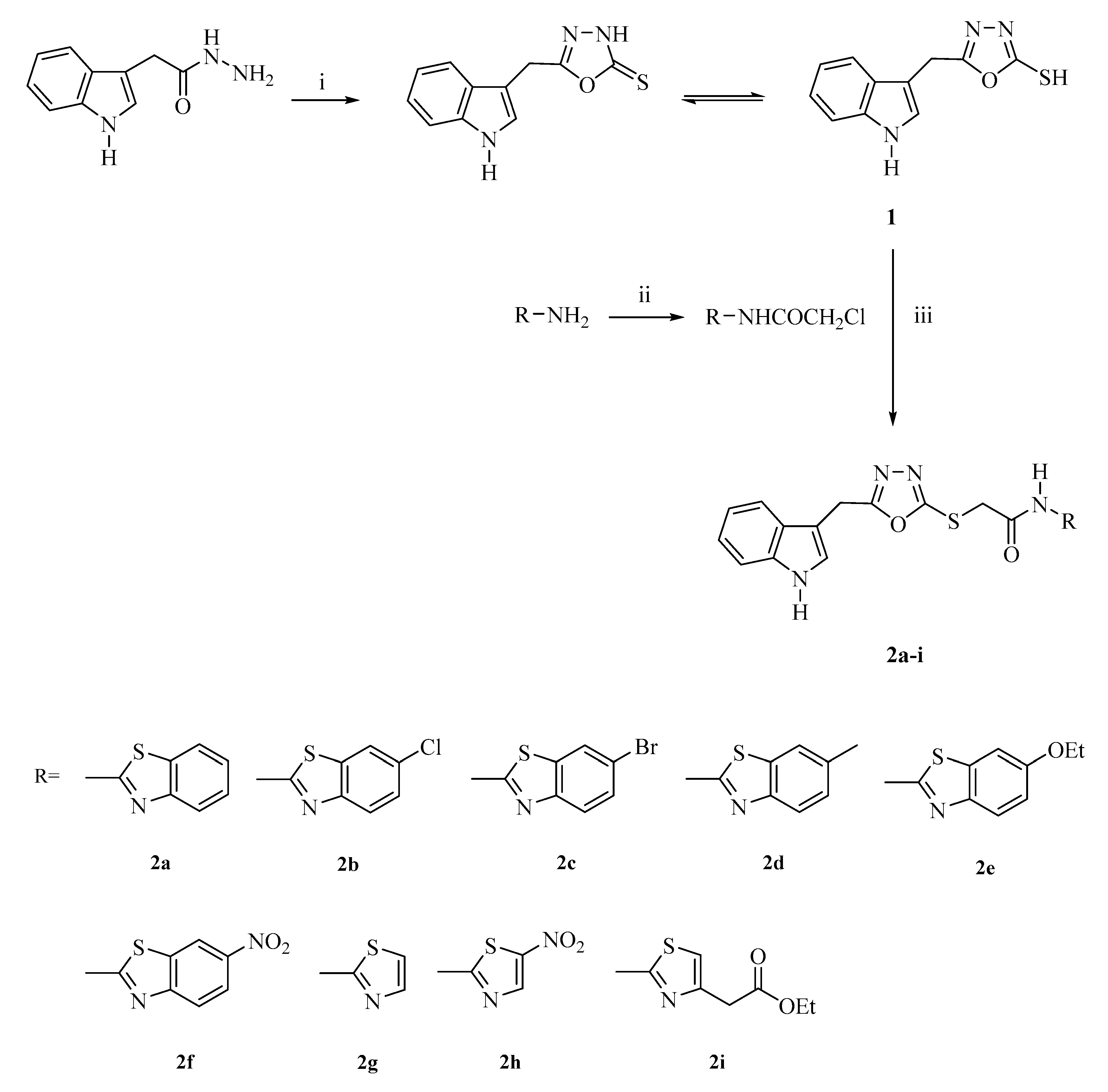
2.1. Chemistry
2.2. Cytotoxicity
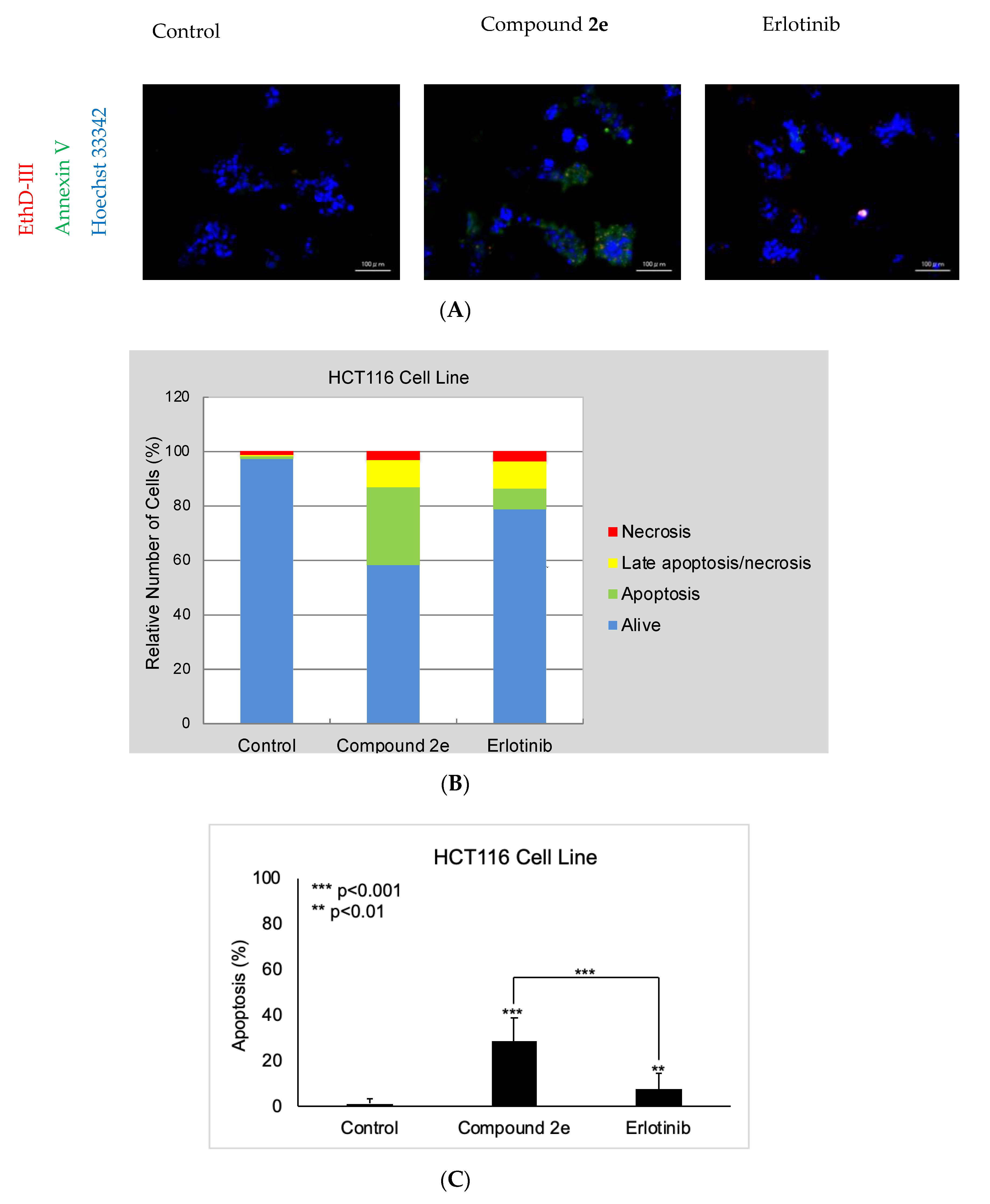
2.3. Apoptosis
2.4. Kinase and COX Inhibition
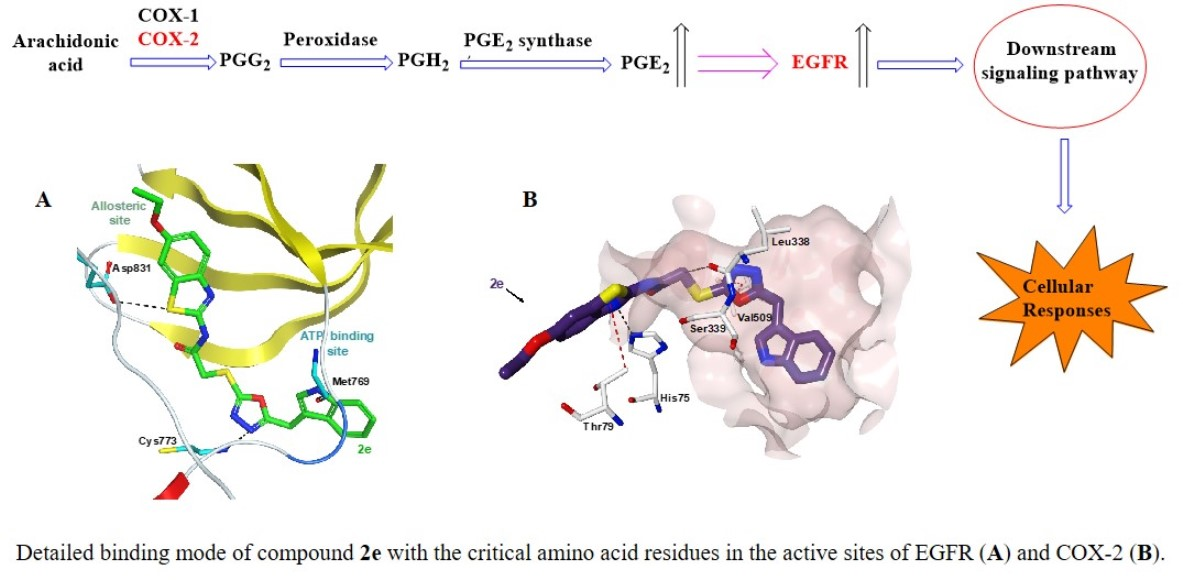
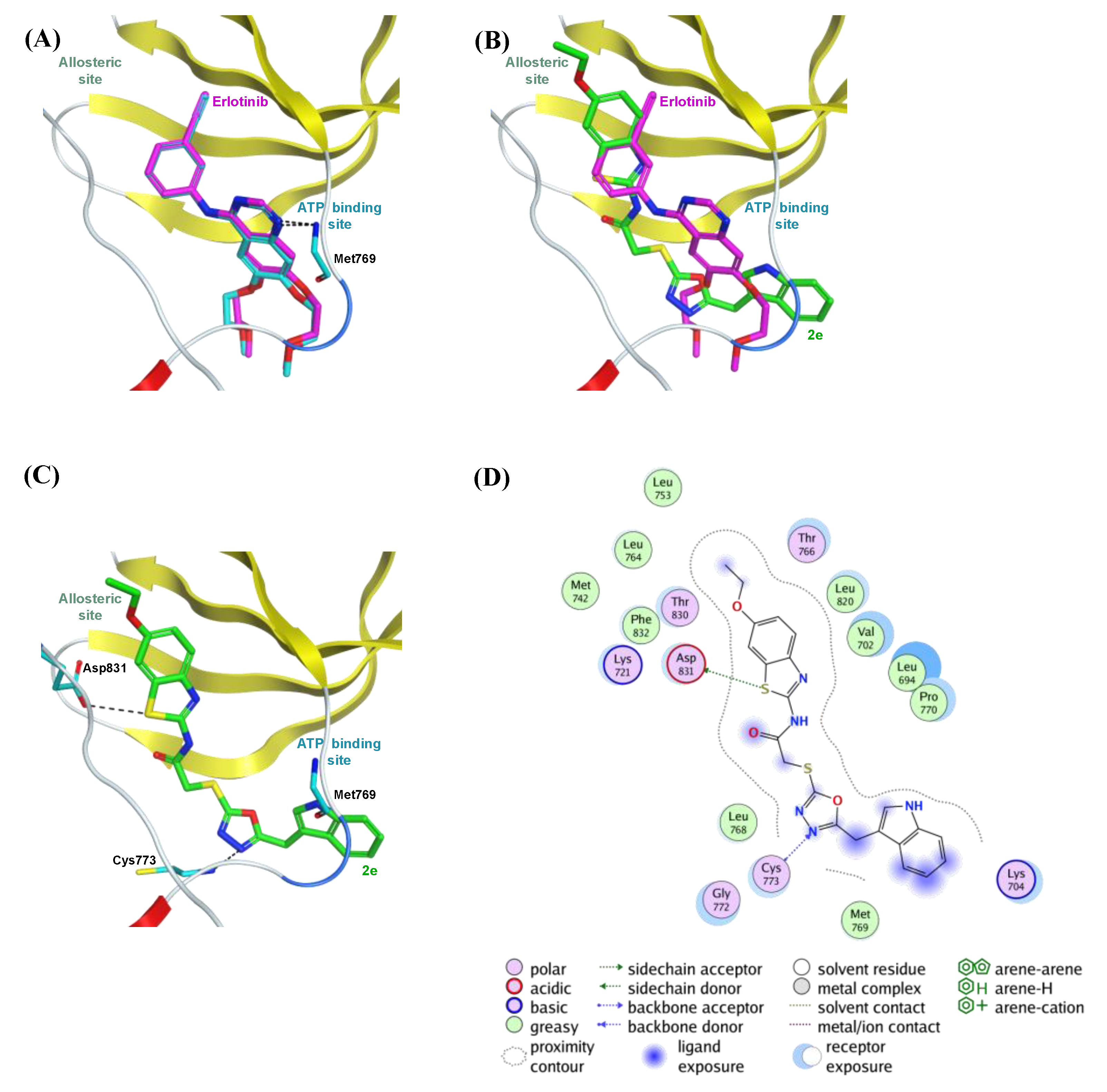
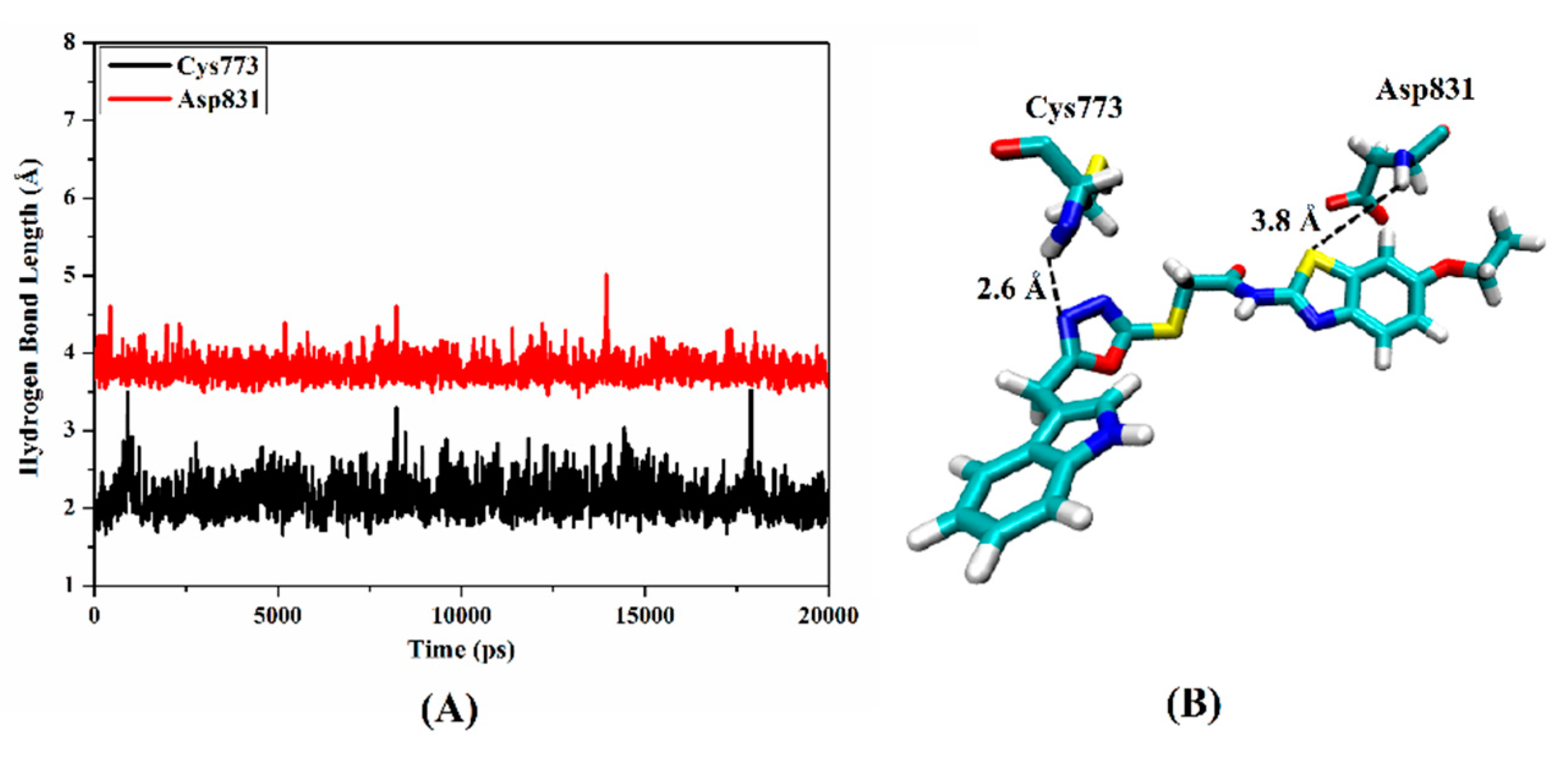
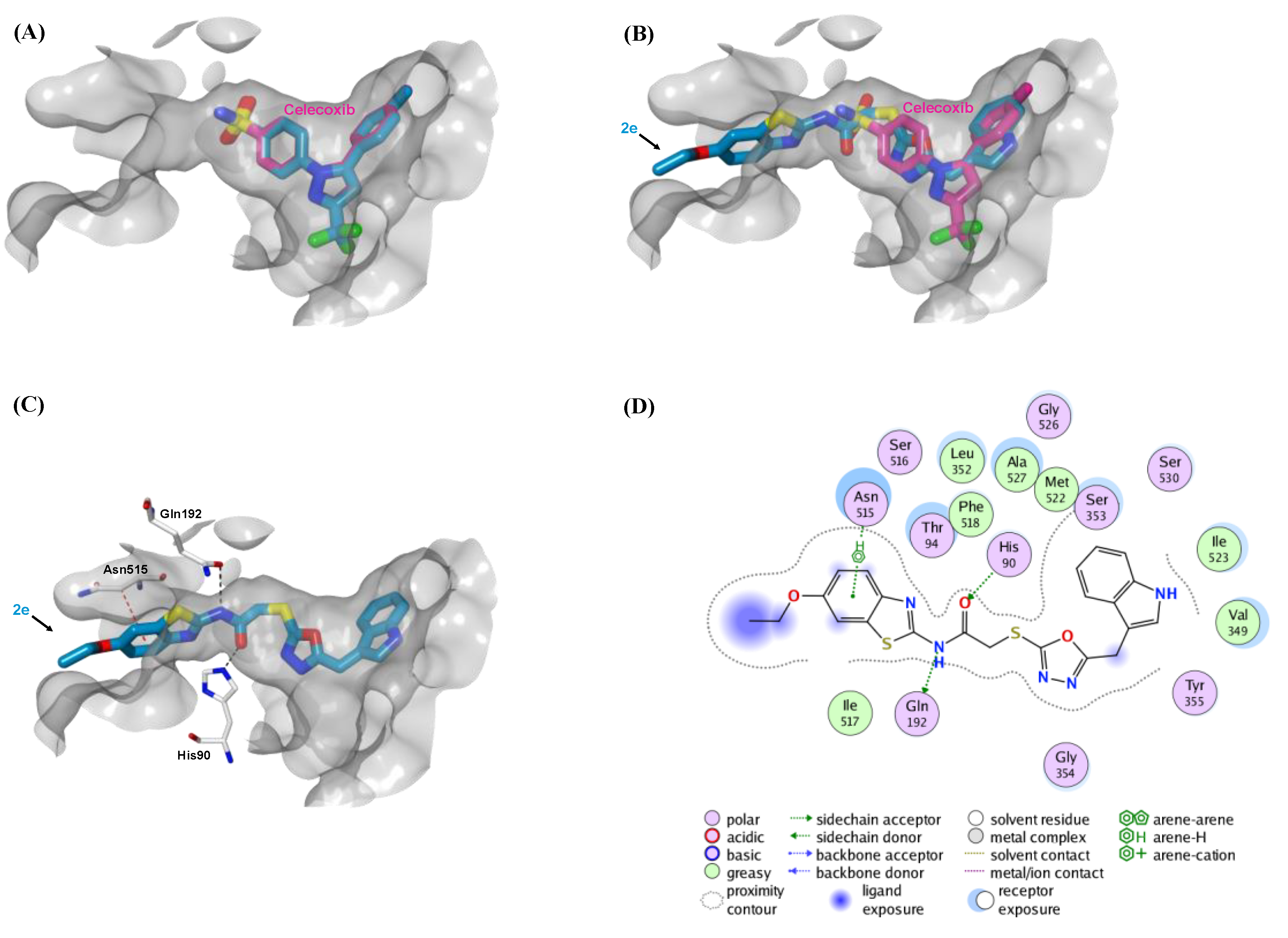
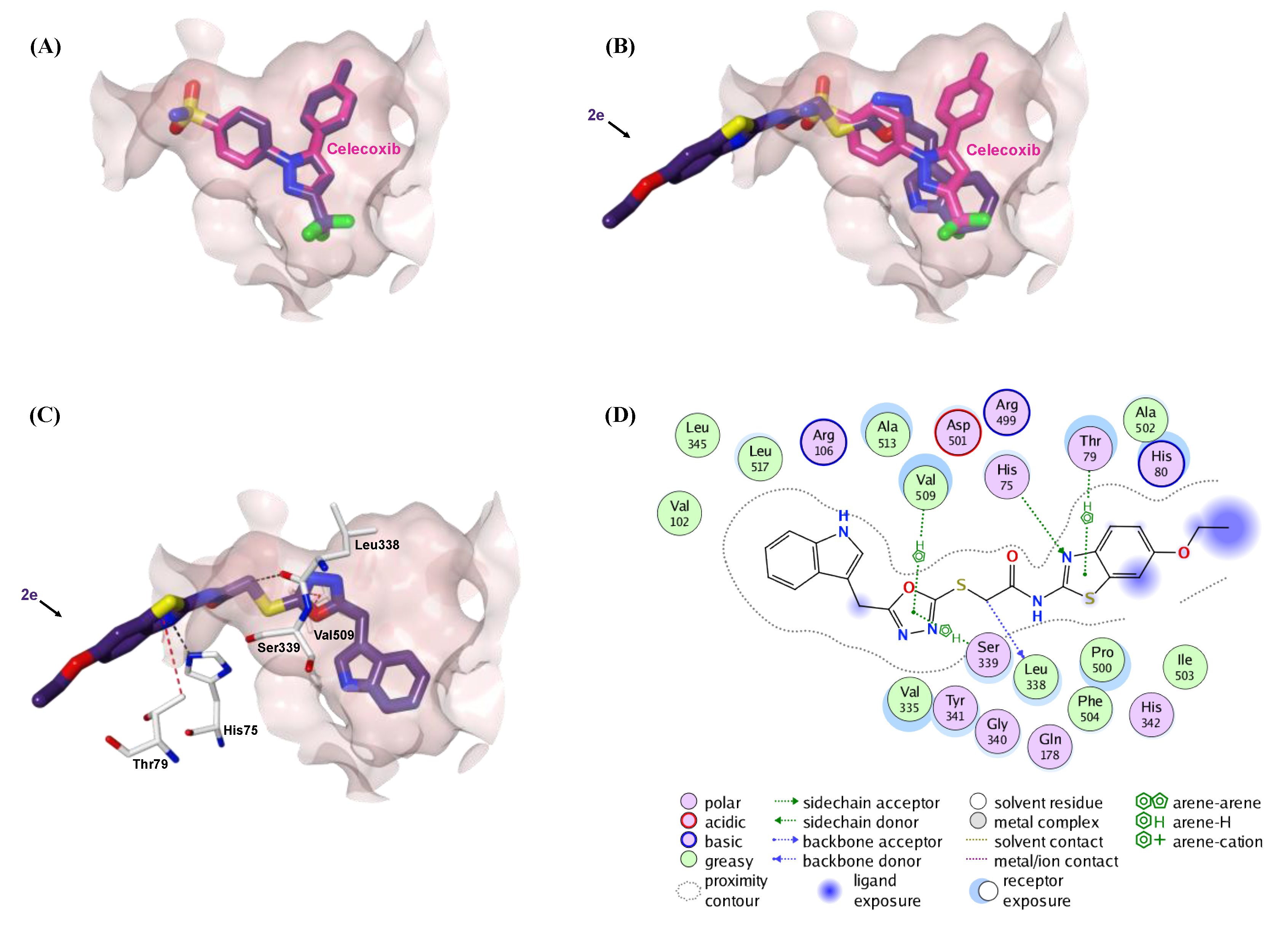
2.5. Molecular Docking
3. Materials and Methods
3.1. Chemistry
General Procedure for the Synthesis of the Compounds
5-((1H-Indol-3-yl)methyl)-1,3,4-oxadiazole-2(3H)-thione (1)
2-Chloro-N-(aryl)acetamides
2-[(5-((1H-Indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl)thio]-N-(thiazol/benzothiazol-2-yl)acetamides (2a–i)
2-[(5-((1H-Indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl)thio]-N-(benzothiazol-2-yl)acetamide (2a)
2-[(5-((1H-Indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl)thio]-N-(6-chlorobenzothiazol-2-yl)acetamide (2b)
2-[(5-((1H-Indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl)thio]-N-(6-bromobenzothiazol-2-yl)acetamide (2c)
2-[(5-((1H-Indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl)thio]-N-(6-methylbenzothiazol-2-yl)acetamide (2d)
2-[(5-((1H-Indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl)thio]-N-(6-ethoxybenzothiazol-2-yl)acetamide (2e)
2-[(5-((1H-Indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl)thio]-N-(6-nitrobenzothiazol-2-yl)acetamide (2f)
2-[(5-((1H-Indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl)thio]-N-(thiazol-2-yl)acetamide (2g)
2-[(5-((1H-Indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl)thio]-N-[5-nitrothiazol-2-yl]acetamide (2h)
2-[(5-((1H-Indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl)thio]-N-[4-(ethoxycarbonylmethyl)thiazol-2-yl]acetamide (2i)
3.2. Biochemistry
3.2.1. Cell Culture and Drug Treatment
3.2.2. Cytotoxicity Assay
3.2.3. Detection of Cell Death
3.2.4. Kinase Inhibition Assay
3.2.5. In Vitro COX Inhibition Assay
3.3. In Silico Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.I.; Ishida, H.; Kubota, Y.; Sasaki, Y. Toxicities of receptor tyrosine kinase inhibitors in cancer pharmacotherapy: Management with clinical pharmacology. Curr. Drug Metab. 2017, 18, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Roberts, T.M. Epigenetic regulation of RTK signaling. J. Mol. Med. 2017, 95, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.D.; Hristova, K. The RTK interactome: Overview and perspective on RTK heterointeractions. Chem. Rev. 2019, 119, 5881–5921. [Google Scholar] [CrossRef] [PubMed]
- Neben, C.L.; Lo, M.; Jura, N.; Klein, O.D. Feedback regulation of RTK signaling in development. Dev. Biol. 2019, 447, 71–89. [Google Scholar] [CrossRef]
- Sever, B.; Altıntop, M.D.; Radwan, M.O.; Özdemir, A.; Otsuka, M.; Fujita, M.; Ciftci, H.I. Design, synthesis and biological evaluation of a new series of thiazolyl-pyrazolines as dual EGFR and HER2 inhibitors. Eur. J. Med. Chem. 2019, 182, 111648. [Google Scholar] [CrossRef]
- Choura, M.; Rebaï, A. Receptor tyrosine kinases: From biology to pathology. J. Recept. Signal Transduct. 2011, 31, 387–394. [Google Scholar] [CrossRef]
- Pellat, A.; Vaquero, J.; Fouassier, L. Role of ErbB/HER family of receptor tyrosine kinases in cholangiocyte biology. Hepatology 2018, 67, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Zhang, G.; Haura, E.B. Targeting epidermal growth factor receptor: Central signaling kinase in lung cancer. Biochem. Pharmacol. 2010, 80, 613–623. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets. 2012, 16, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Ismail, R.S.M.; Ismail, N.S.M.; Abuserii, S.; Abou, D.A.; Ella, E. Recent advances in 4-aminoquinazoline based scaffold derivatives targeting EGFR kinases as anticancer agents. Future J. Pharm. Sci. 2016, 2, 9–19. [Google Scholar] [CrossRef]
- Concu, R.; Cordeiro, M.N.D.S. Looking for new inhibitors for the epidermal growth factor receptor. Curr. Top. Med. Chem. 2018, 18, 219–232. [Google Scholar] [CrossRef]
- Shah, R.R.; Shah, D.R. Safety and tolerability of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in oncology. Drug Saf. 2019, 42, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Kris, M.G. ‘Targeting’ the epidermal growth factor receptor tyrosine kinase with gefitinib (Iressa) in non-small cell lung cancer (NSCLC). Semin. Cancer Biol. 2004, 14, 33–40. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Li, L.; Fan, P.; Chou, H.; Li, J.; Wang, K.; Li, H. Herbacetin suppressed MMP9 mediated angiogenesis of malignant melanoma through blocking EGFR-ERK/AKT signaling pathway. Biochimie 2019, 162, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Lung cancers: Molecular characterization, clonal heterogeneity and evolution, and cancer stem cells. Cancers 2018, 10, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchiarelli, S.; Bennati, C. Oncogene addicted non-small-cell lung cancer: Current standard and hot topics. Future Oncol. 2018, 14, 3–17. [Google Scholar] [CrossRef]
- Yoda, S.; Dagogo-Jack, I.; Hata, A.N. Targeting oncogenic drivers in lung cancer: Recent progress, current challenges and future opportunities. Pharmacol. Ther. 2019, 193, 20–30. [Google Scholar] [CrossRef]
- Finocchiaro, G.; Toschi, L.; Garassino, I.; De Vincenzo, F.; Campagnoli, E.; Zucali, P.; Cavina, R.; Ceresoli, G.L.; Santoro, A.; Cappuzzo, F. EGFR tyrosine kinase inhibitors: A therapy for a few, for the majority or for all non-small cell lung cancer patients? Expert Opin. Med. Diagn. 2007, 1, 183–191. [Google Scholar] [CrossRef]
- Schettino, C.; Bareschino, M.A.; Ricci, V.; Ciardiello, F. Erlotinib: An EGF receptor tyrosine kinase inhibitor in non-small-cell lung cancer treatment. Expert Rev. Respir. Med. 2008, 2, 167–178. [Google Scholar] [CrossRef]
- Masood, A.; Kancha, R.K.; Subramanian, J. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in non-small cell lung cancer harboring uncommon EGFR mutations: Focus on afatinib. Semin. Oncol. 2019, 46, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Modest, D.P.; Pant, S.; Sartore-Bianchi, A. Treatment sequencing in metastatic colorectal cancer. Eur. J. Cancer 2019, 109, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Chu, E. An update on the current and emerging targeted agents in metastatic colorectal cancer. Clin. Colorectal Cancer 2012, 11, 1–13. [Google Scholar] [CrossRef]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef]
- Ma, L.; Dong, L.; Chang, P. CD44v6 engages in colorectal cancer progression. Cell Death Dis. 2019, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, C.; Jiménez-Cervantes, C.; Sánchez-Laorden, B.; García-Borrón, J.C. Functional interplay between secreted ligands and receptors in melanoma. Semin. Cell Dev. Biol. 2018, 78, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Garay, T.; Molnár, E.; Juhász, É.; László, V.; Barbai, T.; Dobos, J.; Schelch, K.; Pirker, C.; Grusch, M.; Berger, W.; et al. Sensitivity of melanoma cells to EGFR and FGFR activation but not inhibition is influenced by oncogenic BRAF and NRAS mutations. Pathol. Oncol. Res. 2015, 21, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Katunarić, M.; Jurišić, D.; Petković, M.; Grahovac, M.; Grahovac, B.; Zamolo, G. EGFR and cyclin D1 in nodular melanoma: Correlation with pathohistological parameters and overall survival. Melanoma Res. 2014, 24, 584–591. [Google Scholar] [CrossRef]
- Wang, J.; Huang, S.K.; Marzese, D.M.; Hsu, S.C.; Kawas, N.P.; Chong, K.K.; Long, G.V.; Menzies, A.M.; Scolyer, R.A.; Izraely, S.; et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J. Investig. Dermatol. 2015, 135, 532–541. [Google Scholar] [CrossRef] [Green Version]
- Rákosy, Z.; Vízkeleti, L.; Ecsedi, S.; Vokó, Z.; Bégány, A.; Barok, M.; Krekk, Z.; Gallai, M.; Szentirmay, Z.; Ádány, R.; et al. EGFR gene copy number alterations in primary cutaneous malignant melanomas are associated with poor prognosis. Int. J. Cancer 2007, 121, 1729–1737. [Google Scholar] [CrossRef]
- Sandler, A.B.; Dubinett, S.M. COX-2 inhibition and lung cancer. Semin. Oncol. 2004, 31, 45–52. [Google Scholar] [CrossRef]
- Shao, J.; Evers, B.M.; Sheng, H. Prostaglandin E2 synergistically enhances receptor tyrosine kinase-dependent signaling system in colon cancer cells. J. Biol Chem. 2004, 279, 14287–14293. [Google Scholar] [CrossRef] [Green Version]
- Reckamp, K.L.; Krysan, K.; Morrow, J.D.; Milne, G.L.; Newman, R.A.; Tucker, C.; Elashoff, R.M.; Dubinett, S.M.; Figlin, R.A. A phase I trial to determine the optimal biological dose of celecoxib when combined with erlotinib in advanced non-small cell lung cancer. Clin. Cancer Res. 2006, 12, 3381–3388. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, N.; Chaki, R.; Mandal, V.; Mandal, S.C. COX-2 as a target for cancer chemotherapy. Pharmacol. Rep. 2010, 62, 233–244. [Google Scholar] [CrossRef]
- Liu, R.; Xu, K.P.; Tan, G.S. Cyclooxygenase-2 inhibitors in lung cancer treatment: Bench to bed. Eur. J. Pharmacol. 2015, 769, 127–133. [Google Scholar] [CrossRef]
- Chadha, N.; Silakari, O. Indoles: As Multitarget Directed Ligands in Medicinal Chemistry. In Key Heterocycle Cores for Designing Multitargeting Molecules; Silakari, O., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 285–321. [Google Scholar]
- Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Indole: A privileged scaffold for the design of anti-cancer agents. Eur. J. Med. Chem. 2019, 183, 111691. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru, J.; Skouta, R. Role of indole scaffolds as pharmacophores in the development of anti-lung cancer agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef] [Green Version]
- Chennamaneni, S.; Zhong, B.; Lama, R.; Su, B. COX inhibitors indomethacin and sulindac derivatives as antiproliferative agents: Synthesis, biological evaluation, and mechanism investigation. Eur. J. Med. Chem. 2012, 56, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Setia, S.; Vaish, V.; Sanyal, S.N. Chemopreventive effects of NSAIDs as inhibitors of cyclooxygenase-2 and inducers of apoptosis in experimental lung carcinogenesis. Mol. Cell Biochem. 2012, 366, 89–99. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Uddin, M.d.J.; Felts, A.S.; Crews, B.C.; Rouzer, C.A.; Marnett, L.J. The 2′-trifluoromethyl analogue of indomethacin is a potent and selective COX-2 inhibitor. A.C.S. Med. Chem. Lett. 2013, 4, 486–490. [Google Scholar] [CrossRef]
- Cheng, Y.L.; Zhang, G.Y.; Li, C.; Lin, J. Screening for novel protein targets of indomethacin in HCT116 human colon cancer cells using proteomics. Oncol. Lett. 2013, 6, 1222–1228. [Google Scholar] [CrossRef] [Green Version]
- Sever, B.; Altıntop, M.D.; Kuş, G.; Özkurt, M.; Özdemir, A.; Kaplancıklı, Z.A. Indomethacin based new triazolothiadiazine derivatives: Synthesis, evaluation of their anticancer effects on T98 human glioma cell line related to COX-2 inhibition and docking studies. Eur. J. Med. Chem. 2016, 113, 179–186. [Google Scholar] [CrossRef]
- Bajaj, S.; Asati, V.; Singh, J.; Roy, P.P. 1,3,4-Oxadiazoles: An emerging scaffold to target growth factors, enzymes and kinases as anticancer agents. Eur. J. Med. Chem. 2015, 97, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Altıntop, M.D.; Sever, B.; Akalın Çiftçi, G.; Turan-Zitouni, G.; Kaplancıklı, Z.A.; Özdemir, A. Design, synthesis, in vitro and in silico evaluation of a new series of oxadiazole-based anticancer agents as potential Akt and FAK inhibitors. Eur. J. Med. Chem. 2018, 155, 905–924. [Google Scholar] [CrossRef]
- Glomb, T.; Szymankiewicz, K.; Świątek, P. Anti-cancer activity of derivatives of 1,3,4-oxadiazole. Molecules 2018, 23, 3361. [Google Scholar] [CrossRef] [Green Version]
- Sever, B.; Altıntop, M.D.; Akalın Çiftçi, G. In vitro and in silico assessment of antiproliferative activity of new acetamides bearing 1,3,4-oxadiazole and pyrimidine cores via COX inhibition. J. Res. Pharm. 2020, 24, 656–669. [Google Scholar] [CrossRef]
- El-Sayed, N.A.; Nour, M.S.; Salem, M.A.; Arafa, R.K. New oxadiazoles with selective-COX-2 and EGFR dual inhibitory activity: Design, synthesis, cytotoxicity evaluation and in silico studies. Eur. J. Med. Chem. 2019, 183, 111693. [Google Scholar] [CrossRef]
- Morigi, R.; Locatelli, A.; Leoni, A.; Rambaldi, M. Recent patents on thiazole derivatives endowed with antitumor activity. Recent Pat. Anticancer Drug Discov. 2015, 10, 280–297. [Google Scholar] [CrossRef]
- Pathak, N.; Rathi, E.; Kumar, N.; Kini, S.G.; Rao, C.M. A review on anticancer potentials of benzothiazole derivatives. Mini Rev. Med. Chem. 2020, 20, 12–23. [Google Scholar] [CrossRef]
- Abdelazeem, A.H.; El-Saadi, M.T.; Said, E.G.; Youssif, B.G.M.; Omar, H.A.; El-Moghazy, S.M. Novel diphenylthiazole derivatives with multi-target mechanism: Synthesis, docking study, anticancer and anti-inflammatory activities. Bioorg. Chem. 2017, 75, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Abdelgawad, M.A.; Bakr, R.B.; Omar, H.A. Design, synthesis and biological evaluation of some novel benzothiazole/benzoxazole and/or benzimidazole derivatives incorporating a pyrazole scaffold as antiproliferative agents. Bioorg. Chem. 2017, 74, 82–90. [Google Scholar] [CrossRef]
- Mubeen, M.; Kini, S.G.; Kumar, A.; Pai, K.S.R. Design, synthesis, biological evaluation and in silico studies of few novel 2-substituted benzothiazole derivatives as potential EGFR inhibitors. Lett. Drug Des. Discov. 2019, 16, 961–971. [Google Scholar] [CrossRef]
- Abdelbaset, M.S.; Abdel-Aziz, M.; Ramadan, M.; Abdelrahman, M.H.; Abbas Bukhari, S.N.; Ali, T.F.S.; Abuo-Rahma, G.E.A. Discovery of novel thienoquinoline-2-carboxamide chalcone derivatives as antiproliferative EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. 2019, 27, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Hammerman, P.S.; Jänne, P.A.; Johnson, B.E. Resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res. 2009, 15, 7502–7509. [Google Scholar] [CrossRef] [Green Version]
- Ciftci, H.I.; Can, M.; Ellakwa, D.E.; Suner, S.C.; Ibrahim, M.A.; Oral, A.; Sekeroglu, N.; Özalp, B.; Otsuka, M.; Fujita, M.; et al. Anticancer activity of Turkish marine extracts: A purple sponge extract induces apoptosis with multitarget kinase inhibition activity. Inv. New Drugs 2020, 38, 1326–1333. [Google Scholar] [CrossRef]
- Ciftci, H.I. Design, Antiproliferative activity of α-tomatine and molecular target identification. Turk. J. Agric. Nat. Sci. 2020, 7, 290–300. [Google Scholar]
- Bayrak, N.; Yıldırım, H.; Yıldız, M.; Radwan, M.O.; Otsuka, M.; Fujita, M.; Ciftci, H.I.; Tuyun, A.F. A novel series of chlorinated plastoquinone analogs: Design, synthesis, and evaluation of anticancer activity. Chem. Biol. Drug Des. 2019, 95, 343–354. [Google Scholar] [CrossRef]
- Ali, T.F.S.; Ciftci, H.I.; Radwan, M.O.; Koga, R.; Ohsugi, T.; Okiyama, Y.; Honma, T.; Nakata, A.; Ito, A.; Yoshida, M.; et al. New SIRT2 inhibitors: Histidine-based bleomycin spin-off. Bioorg. Med. Chem. 2019, 27, 1767–1775. [Google Scholar] [CrossRef]
- Radwan, M.O.; Ciftci, H.I.; Ali, T.F.S.; Ellakwa, D.E.; Koga, R.; Tateishi, H.; Nakata, A.; Ito, A.; Yoshida, M.; Okamoto, Y.; et al. Antiproliferative S-trityl-l-cysteine-derived compounds as SIRT2 Inhibitors: Repurposing and Solubility Enhancement. Molecules 2019, 24, 3295. [Google Scholar] [CrossRef] [Green Version]
- Ciftci, H.I.; Radwan, M.O.; Ozturk, S.E.; Gokce Ulusoy, N.; Sozer, E.; Ellakwa, D.E.; Ocak, Z.; Can, M.; Ali, T.F.S.; Abd-Alla, H.I.; et al. Design, synthesis and biological evaluation of pentacyclic triterpene derivatives: Optimization of anti-ABL kinase activity. Molecules 2019, 24, 3535. [Google Scholar] [CrossRef] [Green Version]
- Radwan, M.O.; Ciftci, H.I.; Ali, T.F.S.; Koga, R.; Tateishi, H.; Nakata, A.; Ito, A.; Yoshida, M.; Fujita, M.; Otsuka, M. Structure activity study of S-trityl-cysteamine dimethylaminopyridine derivatives as SIRT2 inhibitors: Improvement of SIRT2 binding and inhibition. Bioorg. Med. Chem. Lett. 2020, 30, 127458. [Google Scholar] [CrossRef]
- Ciftci, H. Effects of glycyrrhetic acid on human chronic myelogenous leukemia cells. Turk. J. Pharm. Sci. 2020, 17, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H.I.; Bayrak, N.; Yıldırım, H.; Yıldız, M.; Radwan, M.O.; Otsuka, M.; Fujita, M.; Tuyun, A.F. Discovery and structure–activity relationship of plastoquinone analogs as anticancer agents against chronic myelogenous leukemia cells. Arch. Pharm. 2019, 352, e1900170. [Google Scholar] [CrossRef]
- Tateishi, H.; Monde, K.; Anraku, K.; Koga, R.; Hayashi, Y.; Ciftci, H.I.; DeMirci, H.; Higashi, T.; Motoyama, K.; Arima, H.; et al. A clue to unprecedented strategy to HIV eradication: “Lock-in and apoptosis”. Sci. Rep. 2017, 7, 8957. [Google Scholar] [CrossRef] [PubMed]
- Bayrak, N.; Yıldırım, H.; Yıldız, M.; Radwan, M.O.; Otsuka, M.; Fujita, M.; Tuyun, A.F.; Ciftci, H.I. Design, synthesis, and biological activity of Plastoquinone analogs as a new class of anticancer agents. Bioorg. Chem. 2019, 92, 103255. [Google Scholar] [CrossRef]
- Rimon, G.; Sidhu, R.S.; Lauver, D.A.; Lee, J.Y.; Sharma, N.P.; Yuan, C.; Frieler, R.A.; Trievel, R.C.; Lucchesi, B.R.; Smith, W.L. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc. Natl. Acad. Sci. USA 2010, 107, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.L.; Limburg, D.; Graneto, M.J.; Springer, J.; Hamper, J.R.; Liao, S.; Pawlitz, J.L.; Kurumbail, R.G.; Maziasz, T.; Talley, J.J.; et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 2010, 20, 7159–7163. [Google Scholar] [CrossRef]
- Abdelhafez, O.H.; Ali, T.F.S.; Fahim, J.R.; Desoukey, S.Y.; Ahmed, S.; Behery, F.A.; Kamel, M.S.; Gulder, T.A.M.; Abdelmohsen, U.R. Anti-inflammatory potential of green synthesized silver nanoparticles of the soft coral Nephthea sp. supported by metabolomics analysis and docking studies. Int. J. Nanomedicine 2020, 15, 5345–5360. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Hegazy, M.F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Abdeljawaad, K.A.A.; Abdelrahman, A.H.M.; Hegazy, M.F. Natural-like products as potential SARS-CoV-2 Mpro inhibitors: In-silico drug discovery. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
Sample Availability: Samples of compounds 2a–i are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) | ||
|---|---|---|---|
| HCT-116 Cell Line | A549 Cell Line | A375 Cell Line | |
| 2a | 26.25 ± 1.75 | 40.39 ± 7.74 | 65.12 ± 10.11 |
| 2b | >100 | 60.67 ± 9.48 | 37.55 ± 5.62 |
| 2c | >100 | 24.88 ± 1.96 | 48.53 ± 5.78 |
| 2d | 18.23 ± 1.19 | 36.86 ± 5.43 | 30.19 ± 3.12 |
| 2e | 6.43 ± 0.72 | 9.62 ± 1.14 | 8.07 ± 1.36 |
| 2f | >100 | >100 | >100 |
| 2g | >100 | >100 | >100 |
| 2h | >100 | >100 | >100 |
| 2i | >100 | 71.89 ± 10.27 | 95.86 ± 12.41 |
| Erlotinib | 17.86 ± 3.22 | 19.41 ± 2.38 | 23.81 ± 4.17 |
| Compound | IC50 (μM) | SI * | |
|---|---|---|---|
| Jurkat Cells | PBMCs | ||
| 2a | 17.68 ± 3.59 | >300 | >16.97 |
| 2d | 21.06 ± 1.85 | >300 | >14.25 |
| 2e | 6.45 ± 1.02 | >300 | >46.51 |
| Erlotinib | 9.47 ± 2.15 | 45.71 ± 8.88 | 4.83 |
| Kinase | IC50 (μM) | |
|---|---|---|
| Compound 2e | Erlotinib | |
| EGFR | 2.80 ± 0.52 | 0.04 ± 0.01 |
| HER2 | >30 | >30 |
| HER4 | 8.22 ± 1.64 | 2.18 ± 0.73 |
| IGF1R | >30 | >30 |
| InsR | 29.10 ± 4.66 | >30 |
| KDR | >30 | 4.48 ± 1.02 |
| PDGFRα | >30 | 6.85 ± 1.82 |
| PDGFRβ | 6.16 ± 1.58 | >30 |
| Compound | IC50 (µM) | SI * | |
|---|---|---|---|
| COX-1 | COX-2 | ||
| 2e | 73.5 ± 2.12 | 37.5 ± 3.54 | 1.96 |
| Celecoxib | 8.875 ± 0.375 | 2.75 ± 0.05 | 3.23 |
| Indomethacin | 0.12 ± 0.01 | 0.575 ± 0.075 | 0.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sever, B.; Altıntop, M.D.; Özdemir, A.; Akalın Çiftçi, G.; Ellakwa, D.E.; Tateishi, H.; Radwan, M.O.; Ibrahim, M.A.A.; Otsuka, M.; Fujita, M.; et al. In Vitro and In Silico Evaluation of Anticancer Activity of New Indole-Based 1,3,4-Oxadiazoles as EGFR and COX-2 Inhibitors. Molecules 2020, 25, 5190. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25215190
Sever B, Altıntop MD, Özdemir A, Akalın Çiftçi G, Ellakwa DE, Tateishi H, Radwan MO, Ibrahim MAA, Otsuka M, Fujita M, et al. In Vitro and In Silico Evaluation of Anticancer Activity of New Indole-Based 1,3,4-Oxadiazoles as EGFR and COX-2 Inhibitors. Molecules. 2020; 25(21):5190. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25215190
Chicago/Turabian StyleSever, Belgin, Mehlika Dilek Altıntop, Ahmet Özdemir, Gülşen Akalın Çiftçi, Doha E. Ellakwa, Hiroshi Tateishi, Mohamed O. Radwan, Mahmoud A. A. Ibrahim, Masami Otsuka, Mikako Fujita, and et al. 2020. "In Vitro and In Silico Evaluation of Anticancer Activity of New Indole-Based 1,3,4-Oxadiazoles as EGFR and COX-2 Inhibitors" Molecules 25, no. 21: 5190. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25215190