
Synthesis, In Silico Prediction and In Vitro Evaluation of Antitumor Activities of Novel Pyrido[2,3-d]pyrimidine, Xanthine and Lumazine Derivatives


Abstract
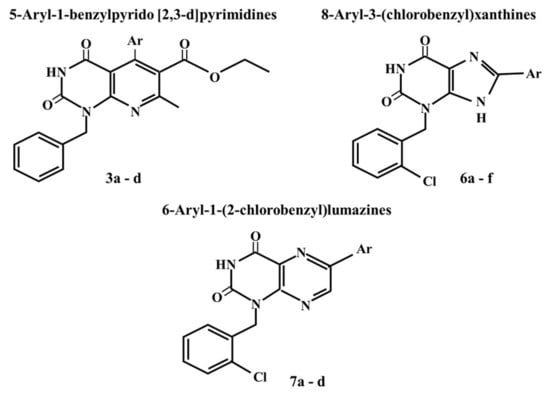
:
1. Introduction
2. Results
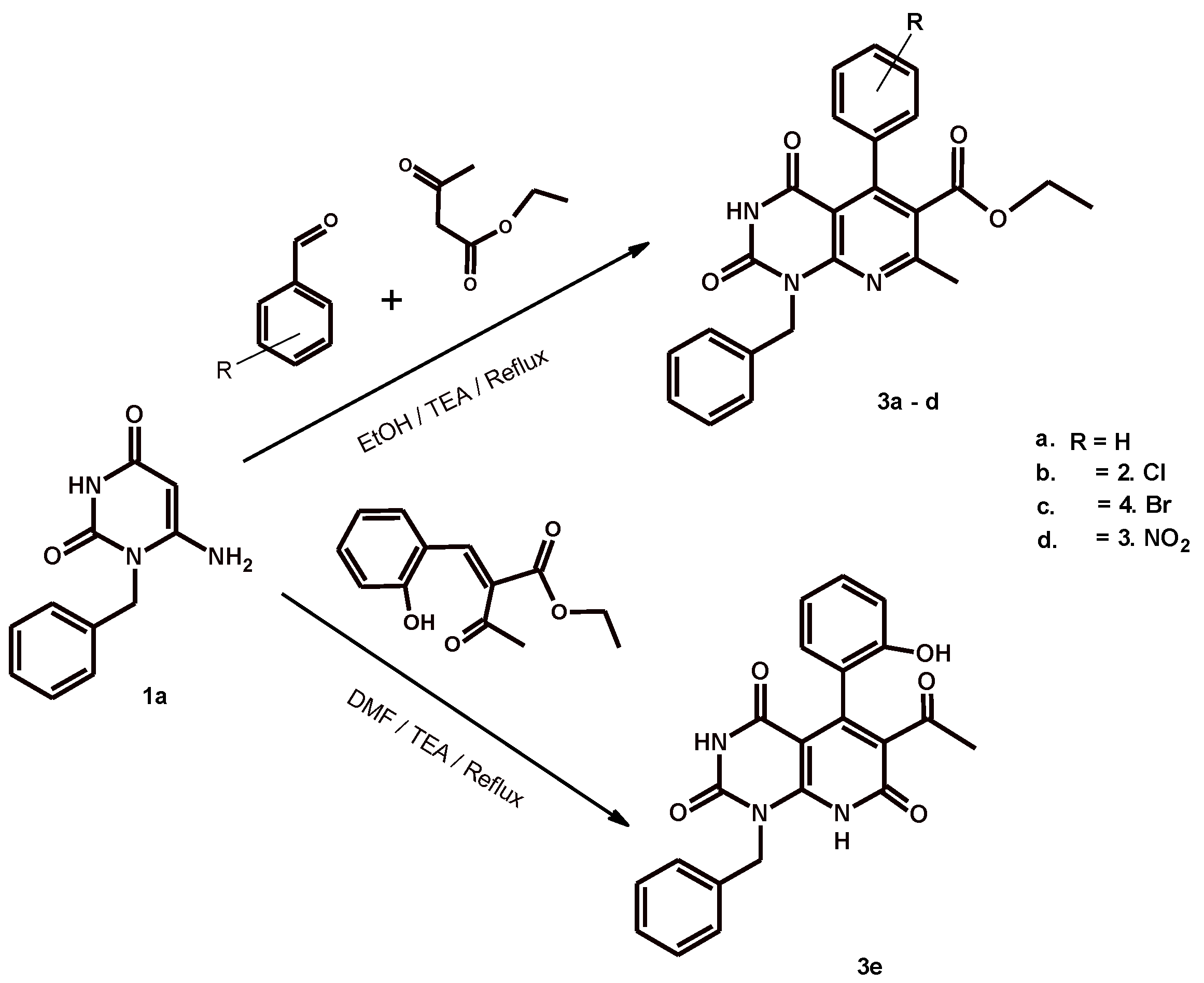
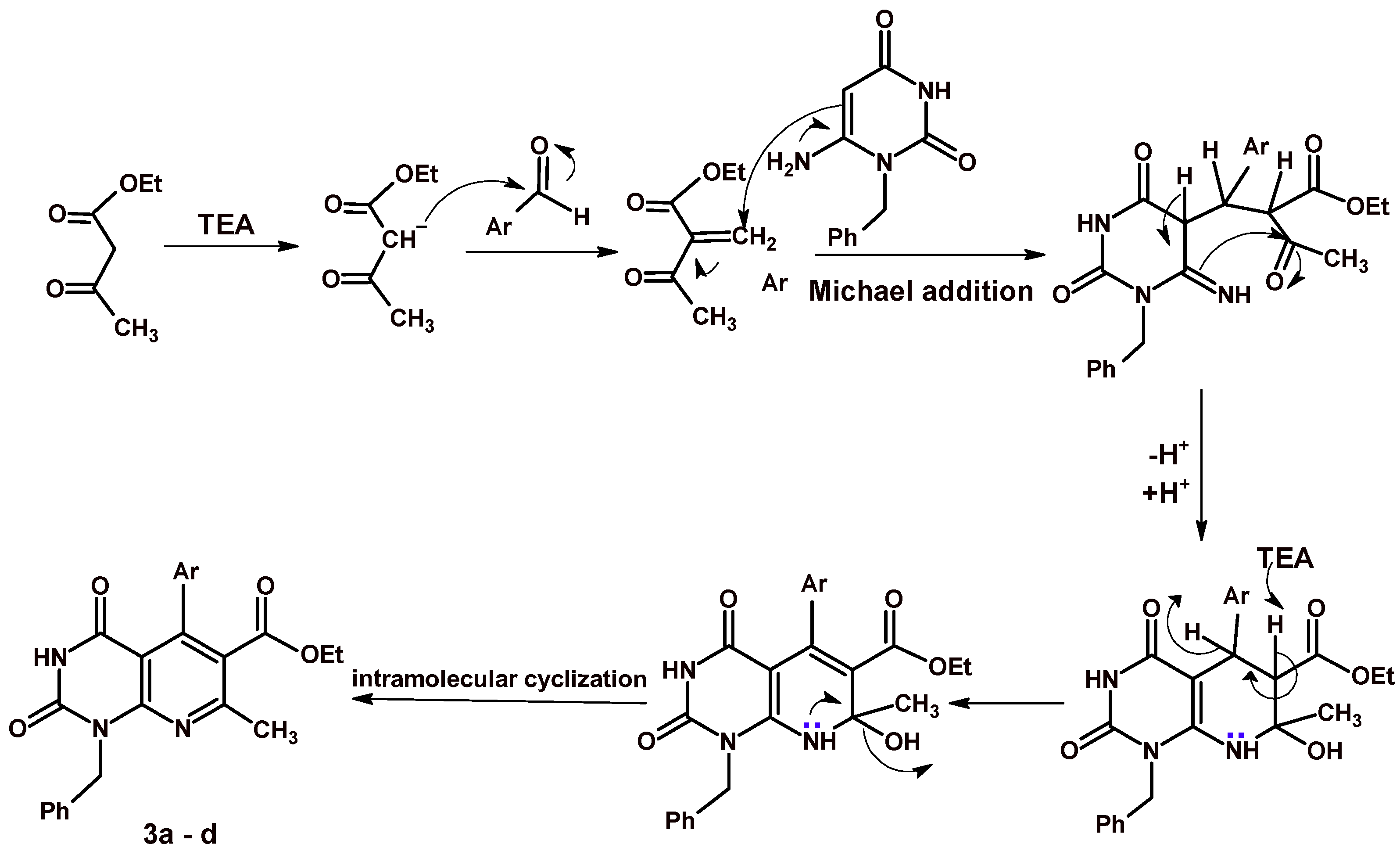
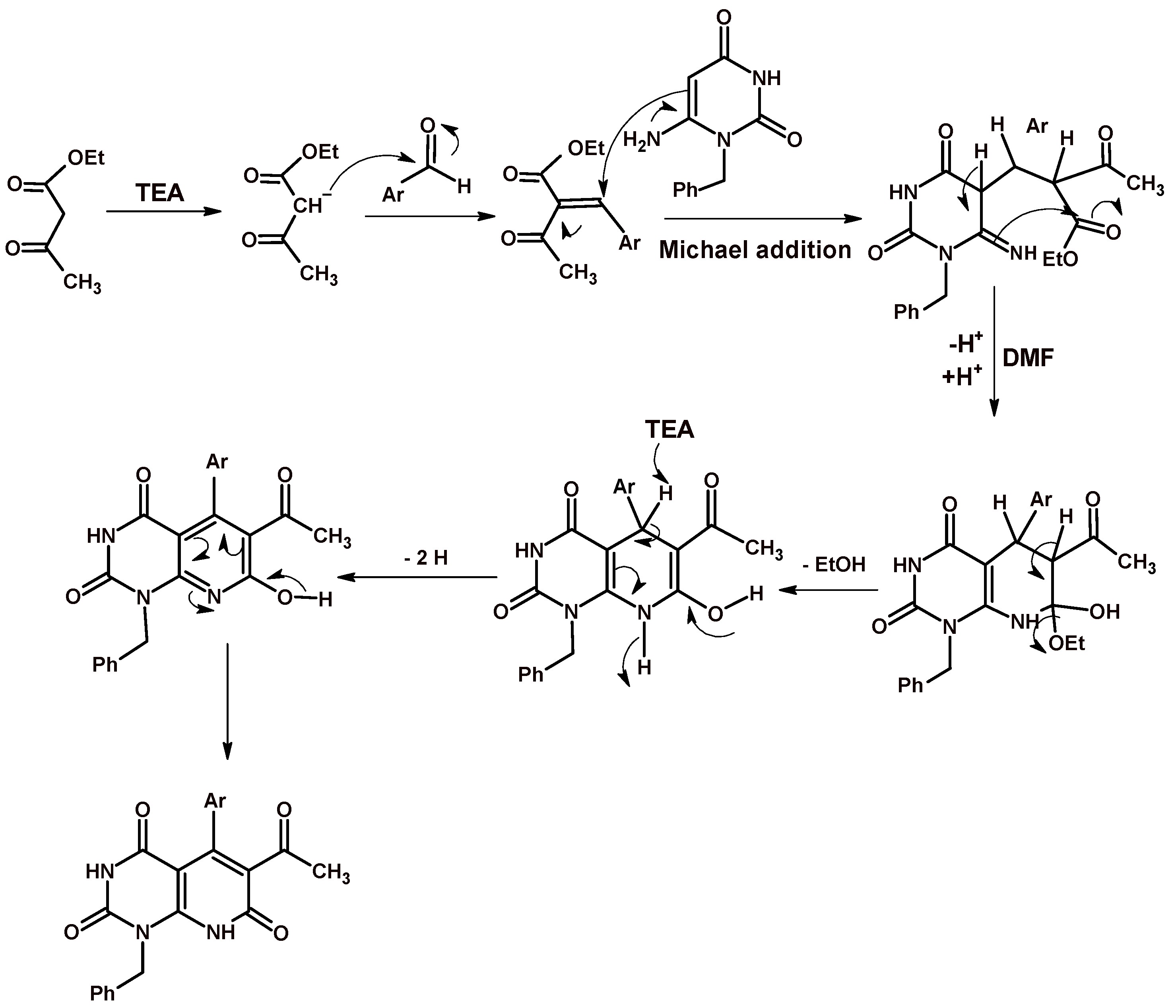
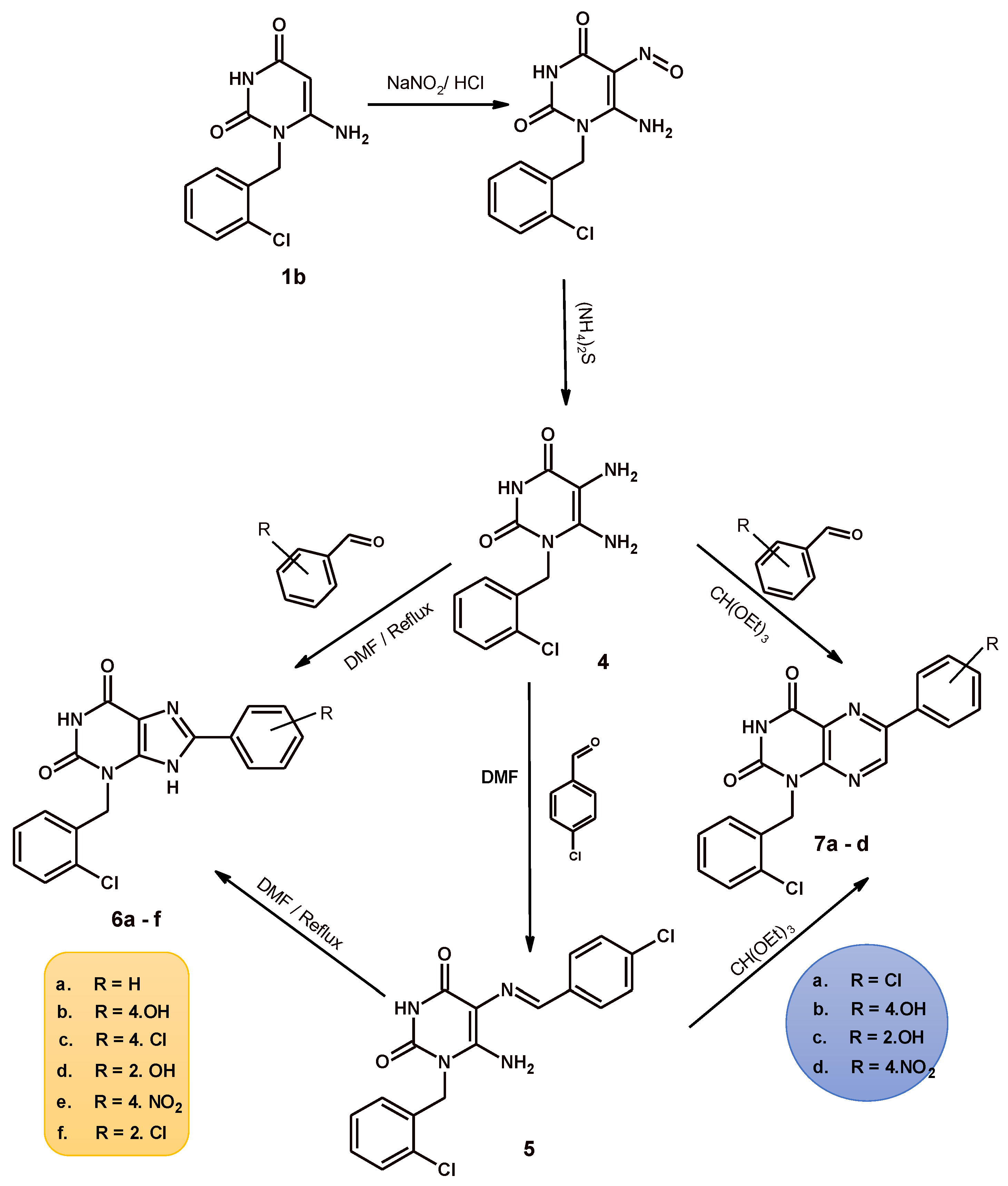
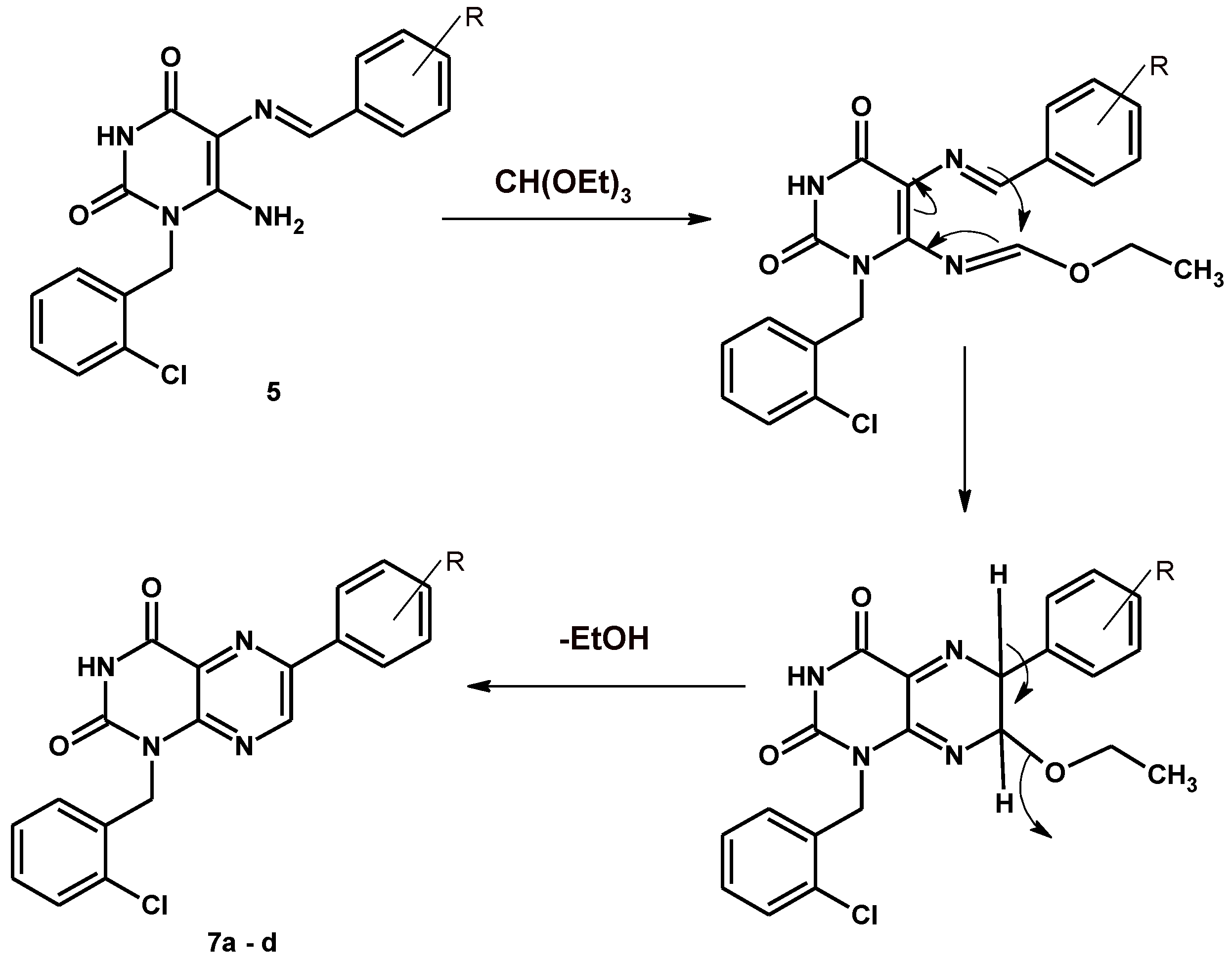
2.1. Chemistry
2.2. Biological Activity
Anticancer Evaluation
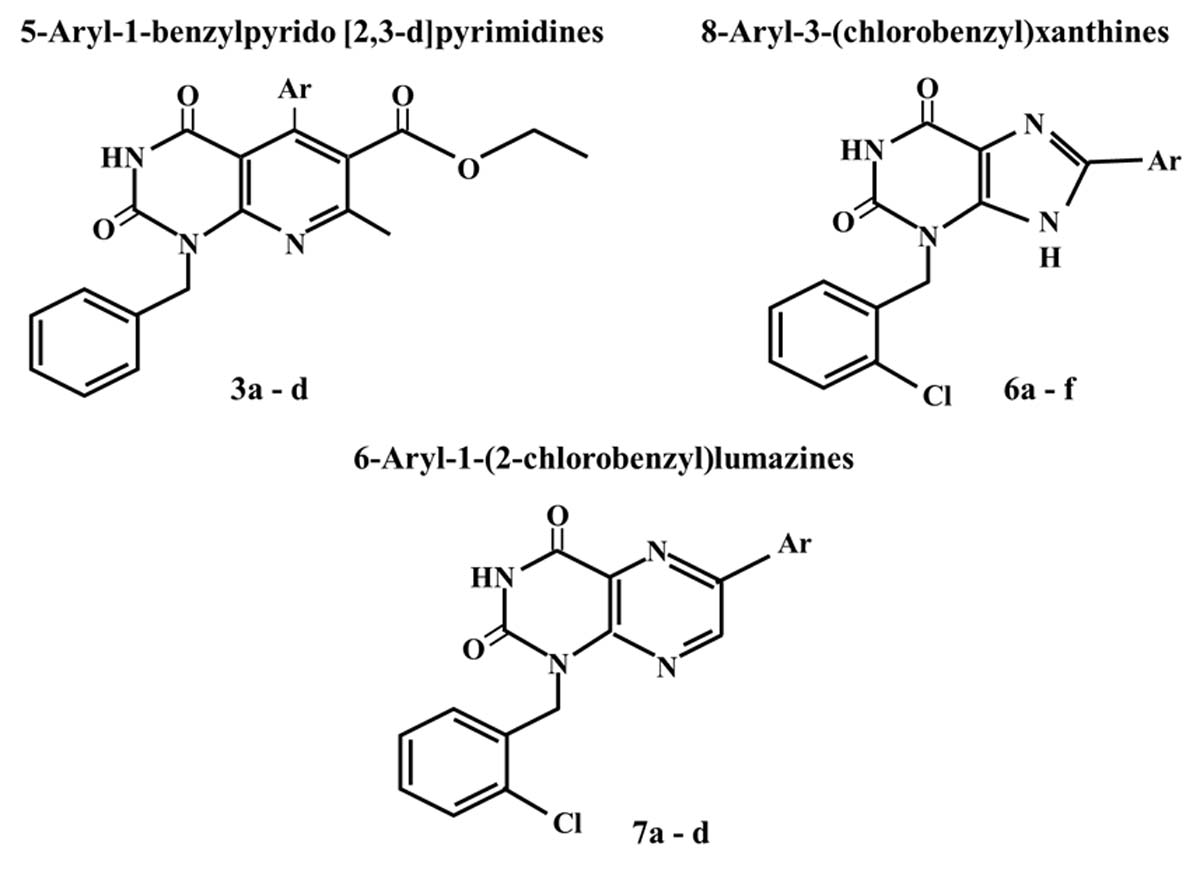
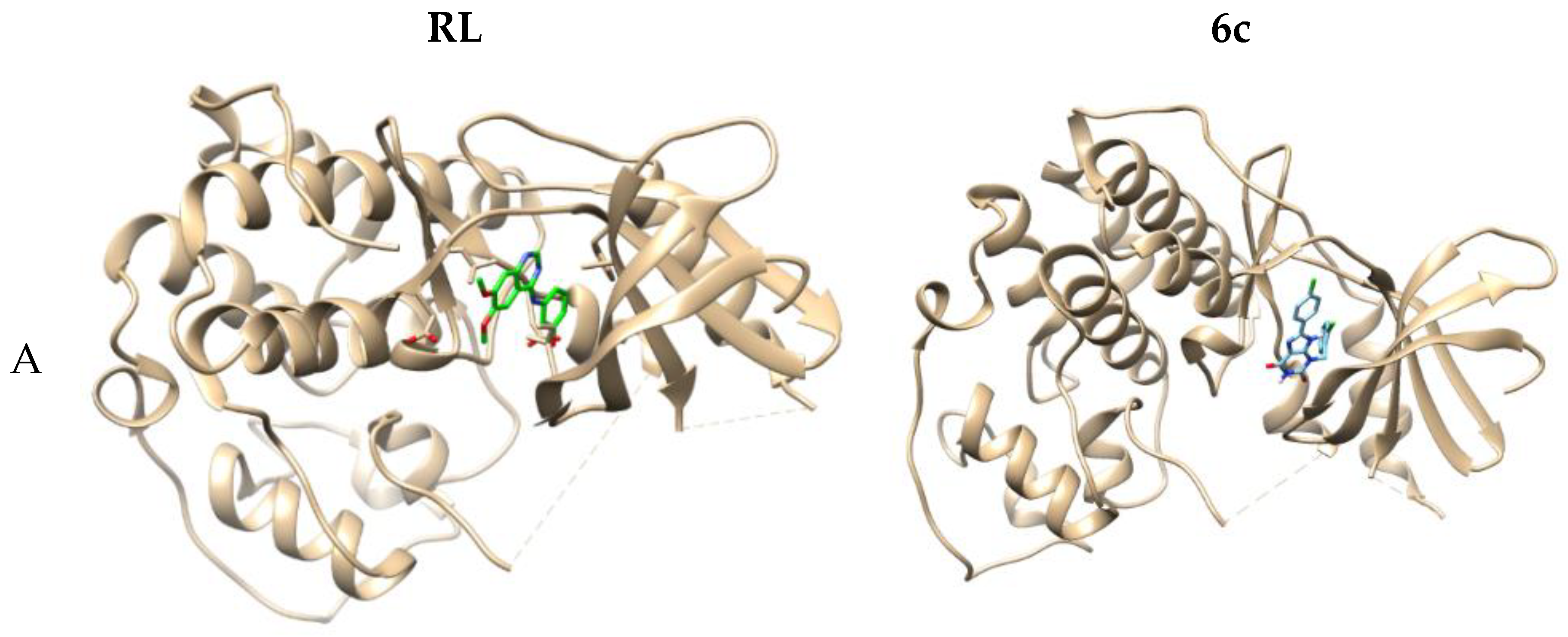
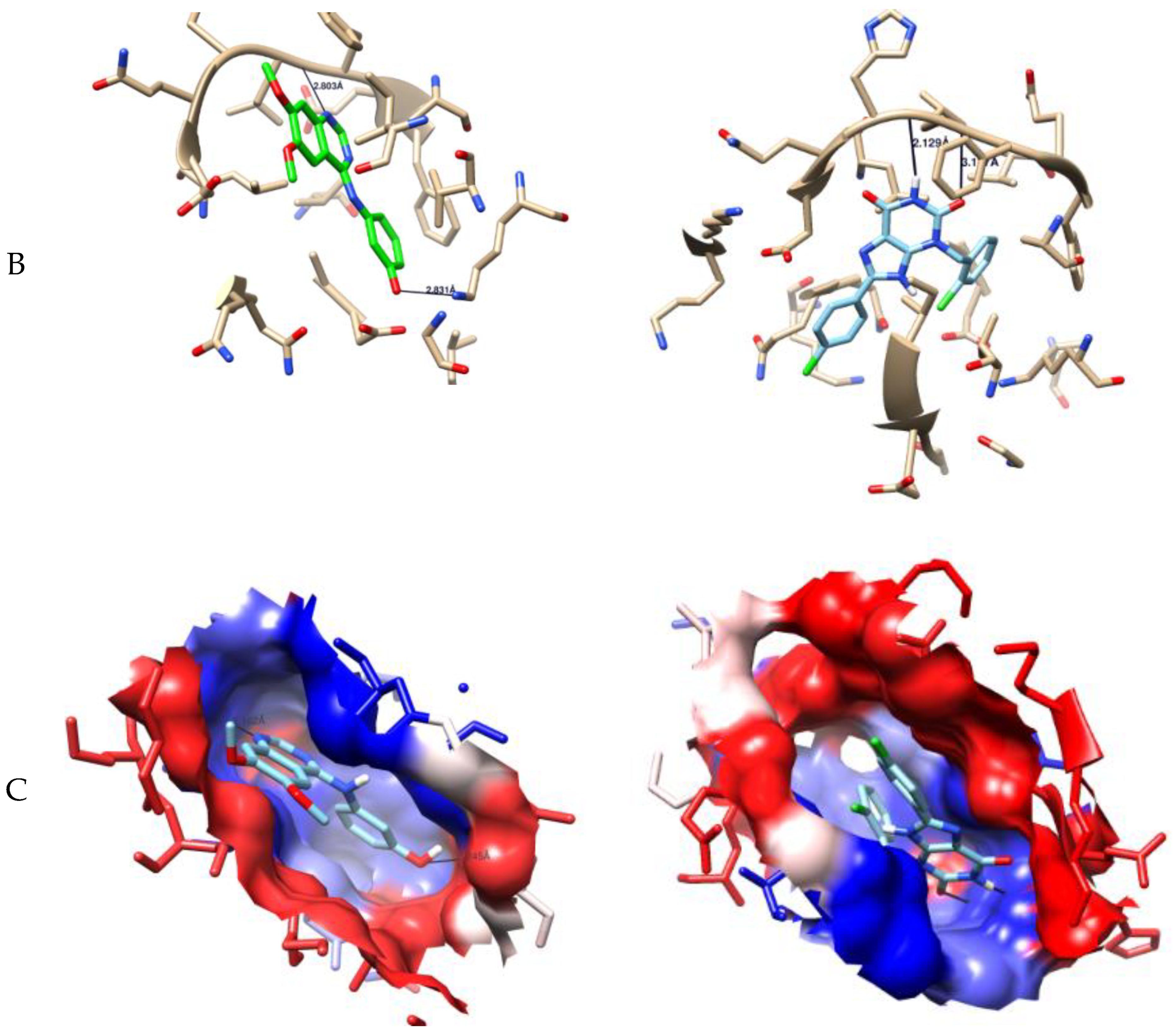
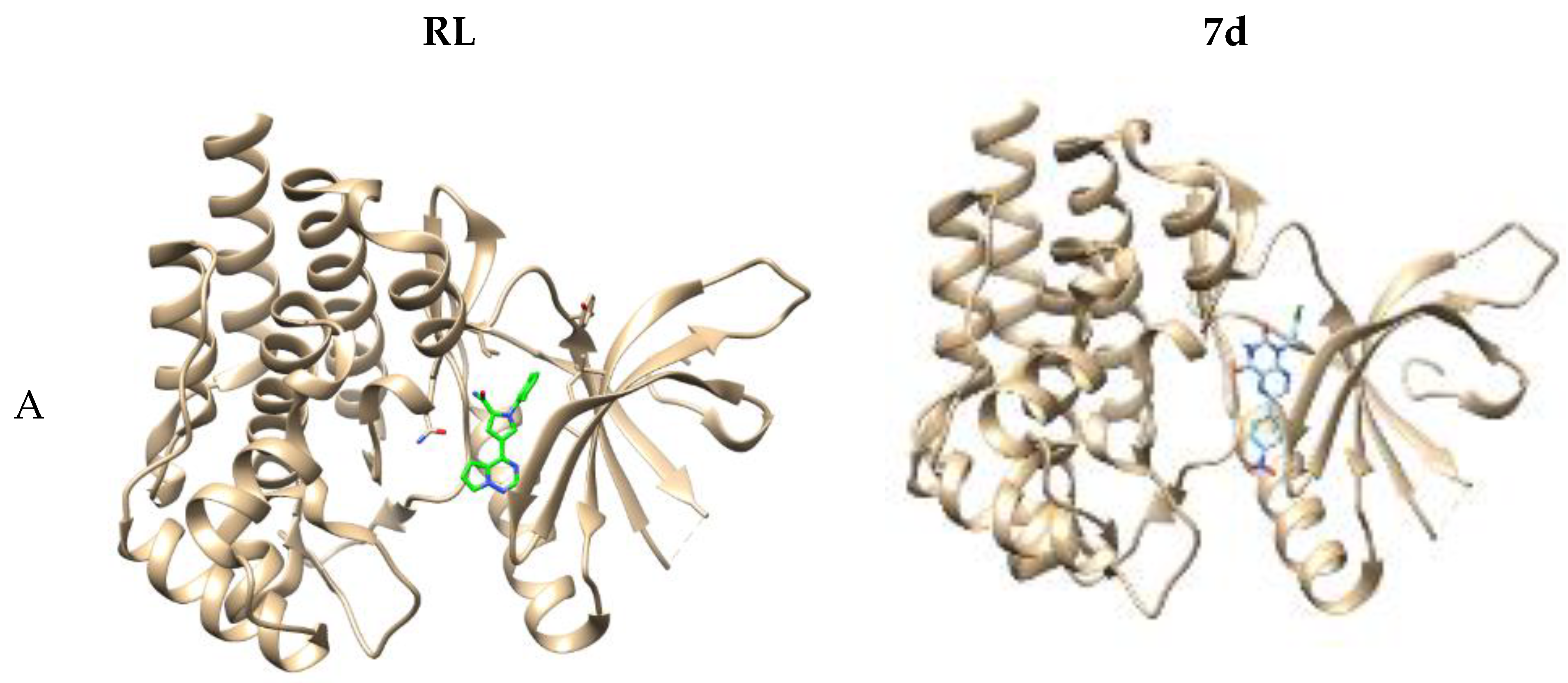
2.3. Computer-Aided Docking
3. Experimental Part
3.1. Chemistry
3.1.1. Ethyl 5-Aryl-1-benzyl-7-methyl-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carboxylate 3a–d
3.1.2. 8-Aryl-3-(2-chlorobenzyl)-xanthine
8-Aryl-3-(2-chlorobenzyl)-3,9-dihydro-1H-purine-2,6-dione 6a–f
3.1.3. 3-(2-Chlorobenzyl)-8-phenylxanthine
3-[(2-Chlorophenyl)methyl]-8-(4-hydroxyphenyl)-3,9-dihydro-1H-purine-2,6-dione
3-[(2-Chlorophenyl)methyl]-8-(4-chlorophenyl)-3,9-dihydro-1H-purine-2,6-dione
3-[(2-Chlorophenyl)methyl]-8-(2-hydroxyphenyl)-3,9-dihydro-1H-purine-2,6-dione
3-[(2-Chlorophenyl)methyl]-8-(4-nitrophenyl)-3,9-dihydro-1H-purine-2,6-dione
8-(4-Bromophenyl)-3-[(2-chlorophenyl)methyl]-3,9-dihydro-1H-purine-2,6-dione
6-Aryl-1-[(2-chlorophenyl)methyl]pteridine-2,4(1H,3H)-dione
6-(4-Chlorophenyl)-1-[(2-chlorophenyl)methyl]pteridine-2,4(1H,3H)-dione
1-[(2-Chlorophenyl)methyl]-6-(4-hydroxyphenyl)pteridine-2,4(1H,3H)-dione
1-(2-Chlorobenzyl)-6-(2-hydroxyphenyl)pteridine-2,4(1H,3H)-dione
1-(2-Chlorobenzyl)-6-(4-nitrophenyl)pteridine-2,4(1H,3H)-dione
3.2. Biological Activity
3.2.1. Anticancer Evaluation
Evaluation of Cytotoxic Effects of the Prepared Compounds
3.2.2. Molecular Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poofery, J.; Khaw-On, P.; Subhawa, S.; Sripanidkulchai, B.; Tantraworasin, A.; Saeteng, S.; Siwachat, S.; Lertprasertsuk, N.; Banjerdpongchai, R. Potential of Thai Herbal Extracts on Lung Cancer Treatment by Inducing Apoptosis and Synergizing Chemotherapy. Molecules 2020, 25, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global Epidemiology of Lung Cancer. Ann. Glob. Health 2019, 85, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Zhu, Y.; Lu, B.; Zhao, W.; Zhao, X. Survivin expression modulates the sensitivity of A549 lung cancer cells resistance to vincristine. Oncol. Lett. 2018, 16, 5466–5472. [Google Scholar] [CrossRef]
- Jiao, X.-Q.; Qian, X.-L.; Wu, L.; Li, B.; Wang, Y.; Kong, X.; Xiong, L.-X. microRNA: The Impact on Cancer Stemness and Therapeutic Resistance. Cells 2019, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.; Malvezzi, M.; Negri, E.; La Vecchia, C.; Boffetta, P. Risk factors for lung cancer worldwide. Eur. Respir. J. 2016, 48, 889–902. [Google Scholar] [CrossRef] [Green Version]
- Alkhatib, M.H.; Alyamani, S.A.; Abdu, F. Incorporation of methotrexate into coconut oil nanoemulsion potentiates its antiproliferation activity and attenuates its oxidative stress. Drug Deliv. 2020, 27, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Alghamdi, H.I.; AlShehri, A.F.; Farhat, G.N. An overview of mortality & predictors of small-cell and non-small cell lung cancer among Saudi patients. J. Epidemiol. Glob. Health 2017, 7, S1–S6. [Google Scholar] [CrossRef]
- Okuyama, A. Lung cancer incidence rates in the world from the Cancer Incidence in Five Continents XI. Jpn. J. Clin. Oncol. 2018, 48, 300–301. [Google Scholar] [CrossRef]
- Matsuda, T.; Machii, R. Morphological distribution of lung cancer from Cancer Incidence in Five Continents Vol. X. Jpn. J. Clin. Oncol. 2015, 45, 404. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Qin, Q.; Li, B.; Wang, J.; Zhang, K. Magnetic resonance imaging for N staging in non-small cell lung cancer: A systematic review and meta-analysis. Thorac. Cancer 2015, 6, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, S.; Zhu, F.; Wang, F.; Tian, H.; Fan, Z.; Ke, S.; Hou, Z.; Li, Y. Watson–Crick GC”-inspired supramolecular nanodrug of methotrexate and 5-fluorouracil for tumor microenvironment-activatable self-recognizing synergistic chemotherapy. J. Mater. Chem. B 2020, 8, 3829–3841. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Chen, Z.G.; Shin, D.M. Advances of Cancer Therapy by Nanotechnology. Cancer Res. Treat. 2009, 41, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [Green Version]
- Weissman, I.L. Stem Cells: Biology, Transplantation, and Political Ethics1. Proc. Am. Philos. Soc. 2006, 150, 121–147. [Google Scholar]
- Ye, Q.; Liu, K.; Shen, Q.; Li, Q.; Hao, J.; Han, F.; Jiang, R.-W. Reversal of Multidrug Resistance in Cancer by Multi-Functional Flavonoids. Front. Oncol. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.-H.; Lee, L.-M.; Hsieh, M.-C.; Yan, M.-D.; Yao, C.-J.; Chang, P.-Y.; Chen, T.-L.; Chang, H.-Y.; Cheng, A.-L.; Lai, G.-M.; et al. Aspirin antagonizes the cytotoxic effect of methotrexate in lung cancer cells. Oncol. Rep. 2013, 30, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Fotoohi, A.K.; Albertioni, F. Mechanisms of antifolate resistance and methotrexate efficacy in leukemia cells. Leuk. Lymphoma 2008, 49, 410–426. [Google Scholar] [CrossRef]
- Duthie, S.J. Folic-acid-mediated inhibition of human colon-cancer cell growth. Nutrition 2001, 17, 736–737. [Google Scholar] [CrossRef]
- Hider, S.L.; A Bruce, I.; Thomson, W. The pharmacogenetics of methotrexate. Rheumatology 2007, 46, 1520–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashkes, P.J.; Becker, M.L.; Cabral, D.A.; Laxer, R.; Paller, A.S.; Rabinovich, C.E.; Turner, D.; Zulian, F. Methotrexate: New Uses for an Old Drug. J. Pediatr. 2014, 164, 231–236. [Google Scholar] [CrossRef]
- Levêque, D.; Becker, G.; Toussaint, E.; Fornecker, L.-M.; Paillard, C. Clinical pharmacokinetics of methotrexate in oncology. Int. J. Pharmacokinet. 2017, 2, 137–147. [Google Scholar] [CrossRef]
- Yoon, S.-A.; Choi, J.R.; Kim, J.-O.; Shin, J.-Y.; Zhang, X.; Kang, J.-H. Influence of reduced folate carrier and dihydrofolate reducatse genes on methotrexate-induced cytotoxicity. Cancer Res. Treat. 2010, 42, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Lee, J.-E.; Jang, W.-S.; Lee, S.-J.; Park, S.; Kang, H.J.; Lee, S.-S. A combination of methotrexate and irradiation promotes cell death in NK/T-cell lymphoma cells via down-regulation of NF-κB signaling. Leuk. Res. 2012, 36, 350–357. [Google Scholar] [CrossRef]
- Pan, Z.; Yang, G.; He, H.; Zhao, G.; Yuan, T.; Li, Y.; Shi, W.; Gao, P.; Dong, L.; Li, Y. Concurrent radiotherapy and intrathecal methotrexate for treating leptomeningeal metastasis from solid tumors with adverse prognostic factors: A prospective and single-arm study. Int. J. Cancer 2016, 139, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Naidu, M.U.R.; Ramana, G.V.; Rani, P.U.; Mohan, L.K.; Suman, A.; Roy, P. Chemotherapy-Induced and/or Radiation Therapy-Induced Oral Mucositis-Complicating the Treatment of Cancer. Neoplasia 2004, 6, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronstein, B.N. Low-Dose Methotrexate: A Mainstay in the Treatment of Rheumatoid Arthritis. Pharmacol. Rev. 2005, 57, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; Cronstein, B.N. Understanding the mechanisms of action of methotrexate: Implications for the treatment of rheumatoid arthritis. Bull. NYU Hosp. Jt. Dis. 2007, 65, 168–173. [Google Scholar]
- Watanabe, K.; Arakawa, Y.; Oguma, E.; Uehara, T.; Yanagi, M.; Oyama, C.; Ikeda, Y.; Sasaki, K.; Isobe, K.; Mori, M.; et al. Characteristics of methotrexate-induced stroke-like neurotoxicity. Int. J. Hematol. 2018, 108, 630–636. [Google Scholar] [CrossRef]
- Howard, S.C.; McCormick, J.; Pui, C.; Buddington, R.K.; Harvey, R.D. Preventing and Managing Toxicities of High-Dose Methotrexate. Oncologist 2016, 21, 1471–1482. [Google Scholar] [CrossRef] [Green Version]
- Saygin, M.; Ozturk, O.; Ozmen, O.; Ilhan, I.; Gonca, T.; Gumral, N.; Orhan, H.; Aslankoc, R. The impact of methotrexate on lung inflammatory and apoptotic pathway biomarkers—The role of gallic acid. Biomed. Pharmacother. 2016, 84, 1689–1696. [Google Scholar] [CrossRef]
- Guinan, M.; Benckendorff, C.; Smith, M.; Miller, G. Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues. Molecules 2020, 25, 2050. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.A.; Agili, F.A. A Novel Synthesis of Fused Uracils: Indenopyrimidopyridazines, Pyrimidopyridazines, and Pyrazolopyrimidines for Antimicrobial and Antitumor Evalution. Molecules 2016, 21, 1714. [Google Scholar] [CrossRef] [Green Version]
- El-Kalyoubi, S.; Fayed, E. Synthesis and evaluation of antitumor activities of novel fused tri- and tetracyclic uracil derivatives. J. Chem. Res. 2016, 40, 771–777. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.A.; Fayed, E.A.; Abdel-Razek, A.S. One pot synthesis, antimicrobial and antioxidant activities of fused uracils: Pyrimidodiazepines, lumazines, triazolouracil and xanthines. Chem. Cent. J. 2017, 11, 1–13. [Google Scholar] [CrossRef]
- Youssif, S.; Agili, F. ChemInform Abstract: One-Pot Synthesis of Fused 2-Thiouracils: Pyrimidopyrimidines, Pyridopyrimidines and Imidazolopyrimidines. Z. Nat. B 2008, 63, 860–864. [Google Scholar] [CrossRef]
- Marx, D.; Wingen, L.M.; Schnakenburg, G.; Müller, C.E.; Scholz, M.S. Fast, Efficient, and Versatile Synthesis of 6-amino-5-carboxamidouracils as Precursors for 8-Substituted Xanthines. Front. Chem. 2019, 7, 56. [Google Scholar] [CrossRef]
- Tantawy, M.A.; El-Sherbeeny, N.A.; Helmi, N.; Alazragi, R.; Salem, N.; Elaidy, S.M. Synthetic antiprotozoal thiazolide drug induced apoptosis in colorectal cancer cells: Implications of IL-6/JAK2/STAT3 and p53/caspases-dependent signaling pathways based on molecular docking and in vitro study. Mol. Cell. Biochem. 2020, 469, 143–157. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Gomha, S.M.; Riyadh, S.M.; Mahmmoud, E.A.; Elaasser, M.M. Synthesis and Anticancer Activities of Thiazoles, 1,3-Thiazines, and Thiazolidine Using Chitosan-Grafted-Poly(vinylpyridine) as Basic Catalyst. Heterocycles 2015, 91, 1227–1243. [Google Scholar]
- El-Sayed, A.S.A.; Yassin, M.A.; Ali, G.S. Transcriptional and Proteomic Profiling of Aspergillus flavipes in Response to Sulfur Starvation. PLoS ONE 2015, 10, e0144304. [Google Scholar] [CrossRef] [Green Version]
- Ali, G.S.; Norman, D.; El-Sayed, A.S. Soluble and Volatile Metabolites of Plant Growth-Promoting Rhizobacteria (PGPRs): Role and Practical Applications in Inhibiting Pathogens and Activating Induced Systemic Resistance (ISR). Adv. Bot. Res. 2015, 75, 241–281. [Google Scholar]
- El-Sayed, A.S.A.; Ruff, L.E.; Ghany, S.E.A.; Ali, G.S.; Esener, S. Molecular and Spectroscopic Characterization of Aspergillus flavipes and Pseudomonas putida L-Methionine γ-Lyase in Vitro. Appl. Biochem. Biotechnol. 2017, 181, 1513–1532. [Google Scholar] [CrossRef]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA—An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef]
- Kattan, S.W.; Nafie, M.S.; Elmgeed, G.A.; Alelwani, W.; Badar, M.; Tantawy, M.A. Molecular docking, anti-proliferative activity and induction of apoptosis in human liver cancer cells treated with androstane derivatives: Implication of PI3K/AKT/mTOR pathway. J. Steroid Biochem. Mol. Biol. 2020, 198, 105604. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Tantawy, M.A.; Sroor, F.M.; Mohamed, M.F.; El-Naggar, M.E.; Saleh, F.M.; Hassaneen, H.M.; Abdelhamid, I.A. Molecular Docking Study, Cytotoxicity, Cell Cycle Arrest and Apoptotic Induction of Novel Chalcones Incorporating Thiadiazolyl Isoquinoline in Cervical Cancer. Anti-Cancer Agents Med. Chem. 2020, 20, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Nafie, M.S.; Tantawy, M.A.; Elmgeed, G.A. Screening of different drug design tools to predict the mode of action of steroidal derivatives as anti-cancer agents. Steroids 2019, 152, 108485. [Google Scholar] [CrossRef] [PubMed]
- El-Far, A.H.; Tantawy, M.A.; Al Jaouni, S.K.; Mousa, S.A. Thymoquinone-chemotherapeutic combinations: New regimen to combat cancer and cancer stem cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1581–1598. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tested Compounds | IC50 Values (μM) |
|---|---|
| 3a | 237 ± 6.3 |
| 3b | 10.3 ± 0.2 |
| 3c | 349 ± 7.8 |
| 3d | 59.5 ± 2.5 |
| 3e | 246 ± 7.1 |
| 5 | 62.0 ± 2.4 |
| 6a | 54.0 ± 1.8 |
| 6b | 58.5 ± 1.7 |
| 6c | 27.0 ± 1.1 |
| 6d | 23.1 ± 0.6 |
| 6e | 26.3 ± 1.3 |
| 6f | 141 ± 3.9 |
| 7a | 86.1 ± 2.8 |
| 7b | 84.8 ± 3.4 |
| 7c | 24.9 ± 1.2 |
| 7d | 12.2 ± 0.3 |
| Methotrexate | 36.3 ± 3.9 |
| Comp. No | CDK2 | BCL2 | Jak2 | MDM2-P53 | DHFR |
|---|---|---|---|---|---|
| PDB: 1DI8 | PDB: 2O2F | PDB: 5AEP | PDB: 2LZG | PDB: 4DFR | |
| ΔGb a | ΔGb a | ΔGb a | ΔGb a | ΔGb a | |
| 3a | −8.5 | −7.8 | −7.6 | −7.6 | −8.7 |
| 3b | −8.7 | −7.7 | −7.7 | −7.0 | −8.8 |
| 3c | −8.9 | −7.8 | −7.7 | −7.0 | −8.6 |
| 3d | −9.3 | −7.1 | −8.1 | −7.0 | −8.5 |
| 3e | −8.6 | −7.7 | −8.7 | −7.7 | −8.7 |
| 5 | −8.8 | −8.5 | −9.5 | −7.8 | −8.2 |
| 6a | -9.7 | -8.5 | -9.3 | -7.8 | -8.5 |
| 6b | −9.6 | −8.5 | −9.6 | −7.9 | −8.6 |
| 6c | −9.3 | −8.9 | −9.6 | −7.9 | −8.8 |
| 6d | −10.1 | −8.5 | −9.4 | −8.4 | −8.6 |
| 6e | −9.8 | −8.9 | −9.7 | −7.8 | −9.1 |
| 6f | −9.0 | −8.9 | −9.6 | −8.0 | −8.6 |
| 7a | −9.7 | −8.5 | −9.8 | −8.1 | −8.8 |
| 7b | −9.2 | −8.5 | −9.7 | −7.8 | −8.6 |
| 7c | −9.2 | −8.5 | −9.7 | −8.3 | −9.2 |
| 7d | −9.5 | −8.8 | −9.8 | −7.6 | −8.7 |
| Methotrexate | −7.6 | −8.0 | −9.0 | −6.9 | −7.8 |
| Reference ligand | −8.3 | −10.6 | −9.1 | −8.2 | −7.8 |
| Comp. No | Types of Interactions | |||
|---|---|---|---|---|
| Hydrogen Bonding | Hydrophobic | |||
| No | Length Å | AA a | AA a | |
| 3b | 1 | 2.040 | ASP86 | ILE10, VAL18, VAL64, LEU298, LEU134, LEU83, LEU133, PHE80, PHE82 |
| 6c | 1 | 2.129 | LEU83 | ILE10, VAL18, VAL64, LEU134, LEU83, LEU148, PHE80, PHE82 |
| 3.137 | LEU83 | |||
| 6d | 2 | 3.245 | LYS33 | ILE10, VAL18, VAL64, LEU134, LEU83, LEU148, PHE80, PHE82 |
| 2.329 | ASP145 | |||
| 6e | 1 | 2.802 | LYS33 | ILE10, VAL18, VAL64, LEU134, LEU83, PHE80, PHE82 |
| 7c | 2 | 3.350 | LEU83 | ILE10, VAL18, VAL64, LEU134, LEU83, LEU148, PHE80, PHE82 |
| 1.998 | LEU83 | |||
| 7d | 1 | 2.499 | GLU12 | ILE10, VAL18, VAL64, LEU134, LEU83, LEU298, PHE80, PHE82 |
| RL b | 2 | 2.831 | LYS33 | ILE10, VAL18, VAL64, LEU148, LEU83, LEU134, PHE82 |
| 2.803 | LEU83 | |||
| Comp. No | Types of Interactions | |||
|---|---|---|---|---|
| Hydrogen Bonding | Hydrophobic | |||
| No | Length Å | AA a | AA a | |
| 3b | 0 | - | - | LEU855, LEU983, LEU932, VAL863, VAL911, PHE860, ILE982 |
| 6c | 1 | 2.940 | ARG980 | LEU855, LEU997, LEU983, LEU932, VAL863, PHE860 |
| 6d | 1 | 3.179 | LEU932 | LEU855, LEU997, LEU982, LEU932, VAL863, VAL 911, ILE982 |
| 6e | 1 | 2.849 | ARG980 | LEU855, LEU997, LEU983, LEU932, VAL863, VAL 911, ILE982 |
| 7c | 2 | 3.181 | PHE860 | LEU884, LEU997, LEU983, LEU932, VAL863, VAL911, PHE860, PHE 895, ILE982 |
| 2.264 | ASP976 | |||
| 7d | 1 | 2.228 | ARG980 | LEU855, LEU997, LEU983, LEU932, VAL863, VAL911, PHE860, ILE982 |
| RL b | 2 | 3.631 | ARG980 | LEU855, LEU997, LEU983, LEU932, VAL863, VAL911, PHE860, ILE982 |
| 3.142 | LEU932 | |||
| Comp. No | Types of Interactions | |||
|---|---|---|---|---|
| Hydrogen Bonding | Hydrophobic | |||
| No | Length Å | AA a | AA a | |
| 3b | 1 | 1.950 | ILE94 | ILE14, ILE5, ILE94, ILE50, LEU24, LEU28, LEU54, PHE31 |
| 6c | 1 | 2.482 | TRP22 | ILE14, ILE5, ILE94, ILE50, LEU24, LEU28, LEU54, LEU8, PHE31 |
| 6d | 2 | 2.482 | MET20 | ILE115, ILE14, ILE5, ILE94, ILE50, LEU24, LEU28, LEU54, LEU8, PHE31 |
| 2.200 | SER49 | |||
| 6e | 1 | 2.476 | SER49 | ILE14, ILE5, ILE94, ILE50, LEU24, LEU28, LEU54, LEU8, PHE31 |
| 7c | 1 | 2.098 | TRY100 | ILE115, ILE14, ILE5, ILE94, ILE50, LEU24, LEU28, LEU54, LEU8, PHE31 |
| 7d | 0 | - | - | ILE14, ILE5, ILE94, ILE50, LEU24, LEU28, LEU54, LEU8, PHE31 |
| Methotrexate | 3 | 2.066 | SER49 | ILE115, ILE14, ILE5, ILE94, ILE50, LEU24, LEU28, LEU54, LEU8, VAL13, PHE31 |
| 2.135 | SER49 | |||
| 2.002 | THR46 | |||
Sample Availability: Samples of the compounds are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Kalyoubi, S.; Agili, F. Synthesis, In Silico Prediction and In Vitro Evaluation of Antitumor Activities of Novel Pyrido[2,3-d]pyrimidine, Xanthine and Lumazine Derivatives. Molecules 2020, 25, 5205. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25215205
El-Kalyoubi S, Agili F. Synthesis, In Silico Prediction and In Vitro Evaluation of Antitumor Activities of Novel Pyrido[2,3-d]pyrimidine, Xanthine and Lumazine Derivatives. Molecules. 2020; 25(21):5205. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25215205
Chicago/Turabian StyleEl-Kalyoubi, Samar, and Fatimah Agili. 2020. "Synthesis, In Silico Prediction and In Vitro Evaluation of Antitumor Activities of Novel Pyrido[2,3-d]pyrimidine, Xanthine and Lumazine Derivatives" Molecules 25, no. 21: 5205. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25215205