A General and Scalable Synthesis of Polysubstituted Indoles



1
Instituto de Productos Naturales y Agrobiología, Consejo Superior de Investigaciones Científicas, Astrofísico Francisco Sánchez 3, 38206 La Laguna, Spain
2
Doctoral and Postgraduate School, Universidad de La Laguna, Apartado Postal 456, 38200 La Laguna, Spain
*
Authors to whom correspondence should be addressed.
Molecules 2020, 25(23), 5595; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235595
Submission received: 3 November 2020
/
Revised: 26 November 2020
/
Accepted: 26 November 2020
/
Published: 28 November 2020
(This article belongs to the Collection Heterocyclic Compounds)
Abstract
:A consecutive 2-step synthesis of N-unprotected polysubstituted indoles bearing an electron-withdrawing group at the C-3 position from readily available nitroarenes is reported. The protocol is based on the [3,3]-sigmatropic rearrangement of N-oxyenamines generated by the DABCO-catalyzed reaction of N-arylhydroxylamines and conjugated terminal alkynes, and delivers indoles endowed with a wide array of substitution patterns and topologies.
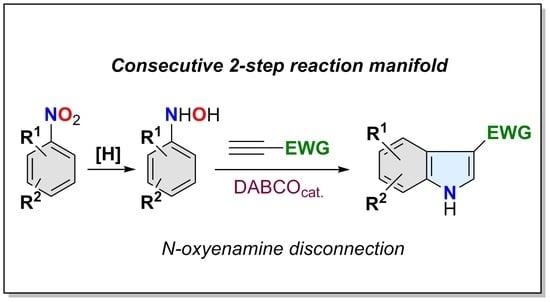
1. Introduction
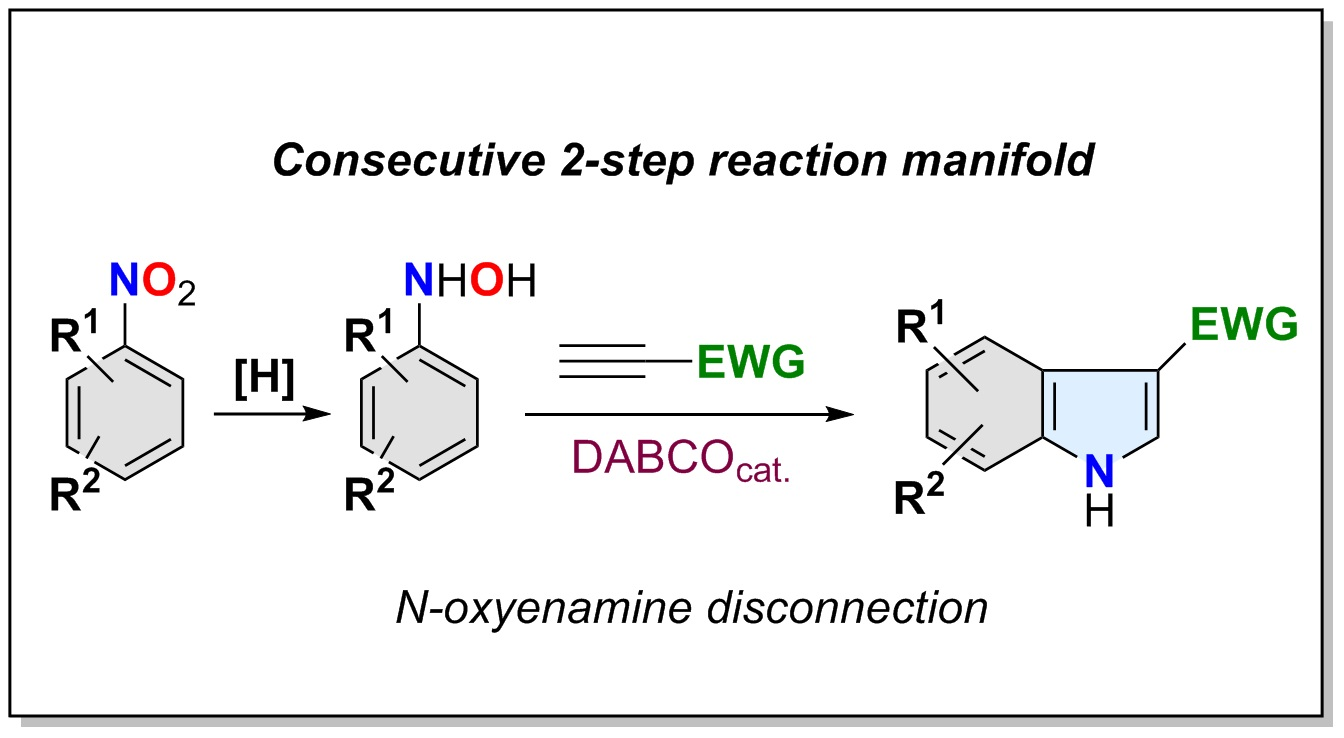
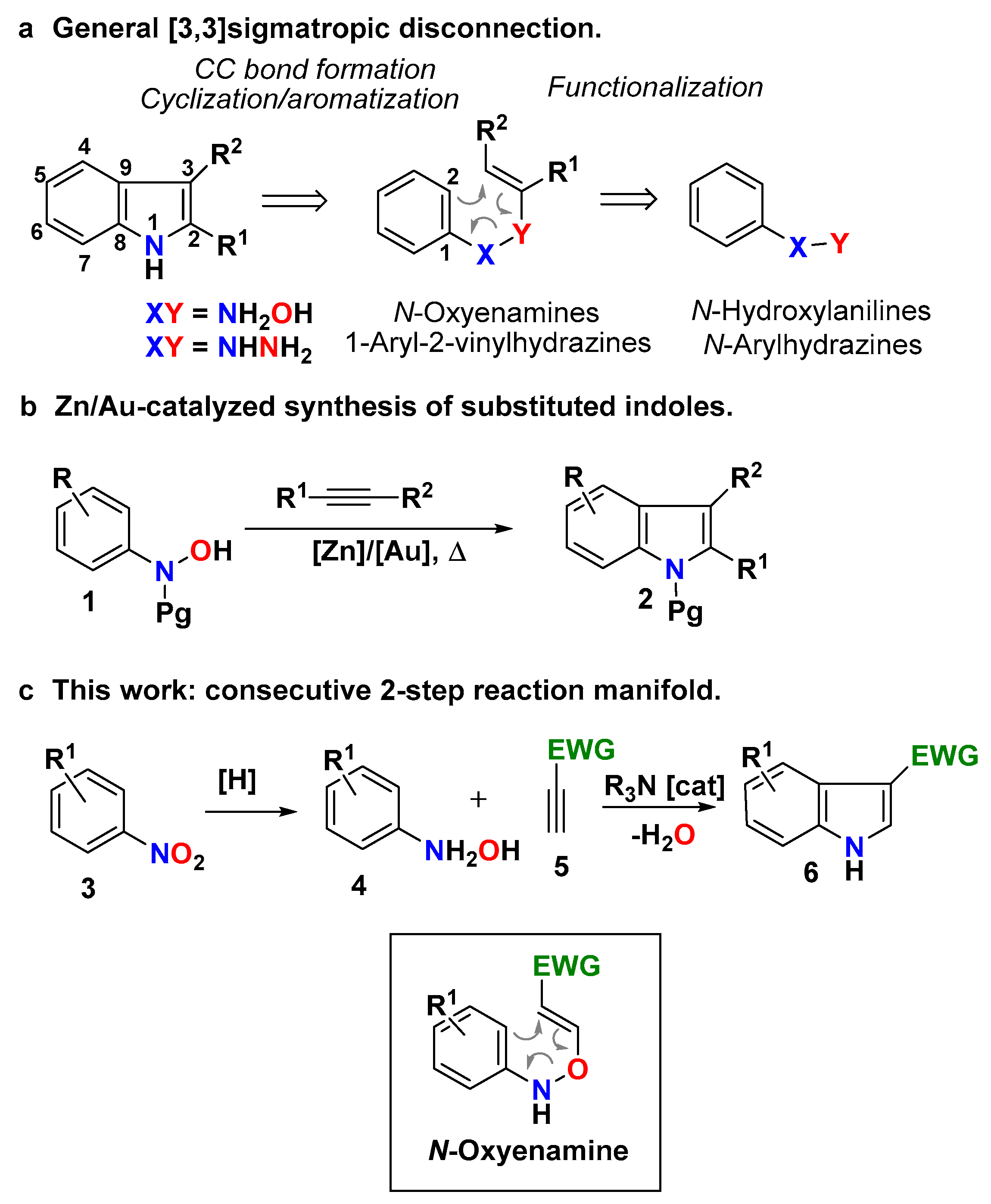
The indole moiety occupies a privileged place in the realm of heterocyclic chemistry [1]. It constitutes the core structure of many natural products and synthetic bioactive compounds exhibiting a wide array of biological activities [2,3,4,5,6], and has also found use as structural parts of material science devices [7,8,9] and chiral catalysts [10,11,12]. Recent studies have shown that the substitution at the C-3 position with electron-withdrawing groups (3-EWG-indoles) not only endows these structures with important pharmaceutical/therapeutic activities [13,14,15,16,17,18,19,20,21], but also it converts them into versatile synthetic molecular platforms in heterocyclic chemistry, such as, in dearomatization-based synthetic disconnections [22]. The catalytic asymmetric dearomatization of 3-nitroindoles constitutes an iconic example of this synthetic potential [23,24,25,26,27,28,29,30,31,32,33,34]. The most traditional synthetic approach to these functionalized indoles relies on the Friedel–Crafts acylation of preformed indoles [35,36,37]. This transformation suffers important drawbacks such as mandatory nitrogen protection, regioselectivity control, stoichiometric use of metal salts and Lewis acid promoters, and strict exclusion of moisture. Although modern alternatives using metal-catalysis [38,39,40,41,42] or photocatalysis [43,44,45,46] have overcome some of these difficulties, the implementation of real-world strategies to prepare these heterocycles from simple and widely accessible starting materials remains a synthetic challenge in medicinal and synthetic chemistry. In spite of the many advances achieved in the synthesis of indoles during the last 100 years [47], the de novo synthesis of 3-EWG-indoles remains largely unexplored. A large number of strategies for the synthesis of indoles use alkynes and a convenient nitrogen source as substrates [48] and the annelation of the five-membered ring to an existing functionalized benzene ring as the main synthetic disconnection [49]. The inherent alkyne reactivity and the broad access to nitrogen sources constitute practical advantages for these strategies. Among them, the [3,3]-sigmatropic rearrangement of N-oxyenamines [50] or 1-aryl-2-vinylhydrazines [51,52] have been objects of profuse research (Scheme 1a). Prototypical examples of these strategies constitute the Bartoli [53,54] (N-oxyenamines) and the Fischer [55] (1-aryl-2-vinylhydrazines) syntheses of indoles. Although both reactions have been largely used in the preparation of indoles [47], they present important drawbacks for general preparative synthetic use. Whereas those based on the Fischer disconnection suffer the limitations associated with the hydrazine synthesis and drastic reaction conditions, the Bartoli method, although synthetically powerful, requires the use of ortho-substituted nitroarenes and functional tolerance to Grignard reagents. Recently, an elegant example of the use of the N-oxyenamine disconnection to gain access to polysusbtituted indoles has been reported by Zhang and co-workers (Scheme 1b) [56]. The methodology uses the Zn/Au-catalyzed reaction of N-protected N-arylhydroxamic acids (or N-aryl-N-hydroxycarbamates) 1 and alkynes to construct the indole core with a diverse pattern of substitution at both rings. Although the methodology was effective for the synthesis of alkyl indole-3-carboxylates (2, the unique reported example: Pg = Ac; R1 = CH2CH2Ph, R2 = CO2Me), its extension to other 3-EWG-indoles remains to be assessed [57].
We envisioned that a simpler and general methodology using the N-oxyenamine disconnection could be arranged on the basis of the well-established addition of nucleophiles onto terminal conjugated alkynes catalyzed by tertiary amines (Scheme 1c) [58,59,60,61]. When searching for precedents for this disconnection, we found a reduced number of reported examples, all of them circumscribed to the synthesis of alkyl indole-3-carboxyates [62,63,64,65,66], and with important drawbacks for a practical and general application. They mostly use N-arylhydroxylamine salts or N-protected N-arylhydroxylamines to force the O-addition reaction, and require large excesses of alkyne and long reaction times to deliver the indole-3-carboxylates in low to moderate yields. Therefore, a general and practical protocol for accessing NH-free 3-EWG-indoles based on this disconnection remains to be established. In terms of sustainability, such protocol should be direct, operationally simple and scalable, avoiding non-productive N-protection/N-deprotection steps. We report herein our results on the implementation of a strategy fulfilling these requirements and overcoming the practical shortcomings found in previous reports. The strategy delivers NH-free multisubstituted 3-EWG-indoles through a consecutive 2-step reaction manifold, and it uses nitroarenes and activated terminal alkynes as building blocks (Scheme 1c). The reaction manifold takes advantage of the O-reactivity of N-arylhydroxylamines toward terminal conjugated alkynes and of the rich arsenal of commercially available functionalized nitroarenes. Whereas the first one avoids unnecessary N-protection/N-deprotection steps (step-economy, reagent-economy), the second one offers an almost countless catalogue of functional diversity on the benzene ring (general outcome). These properties make this reaction manifold a convenient, instrumentally simple, robust and reliable synthetic tool for the exploration/annotation of the chemical space of 3-EWG-indoles.
2. Results and Discussion
2.1. Initial Experiments and Optimization of the Reaction Conditions for a Consecutive 2-Step Protocol
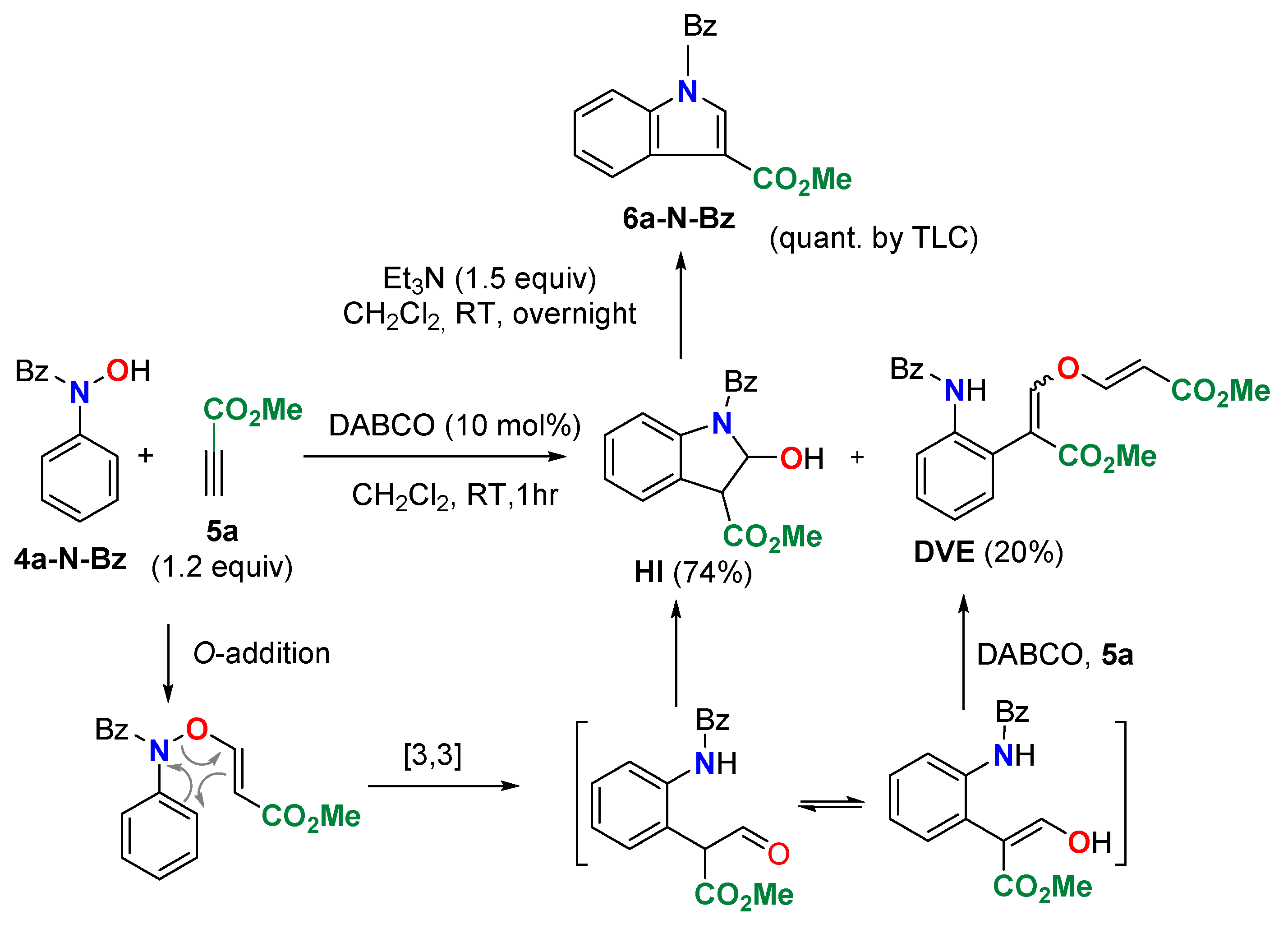
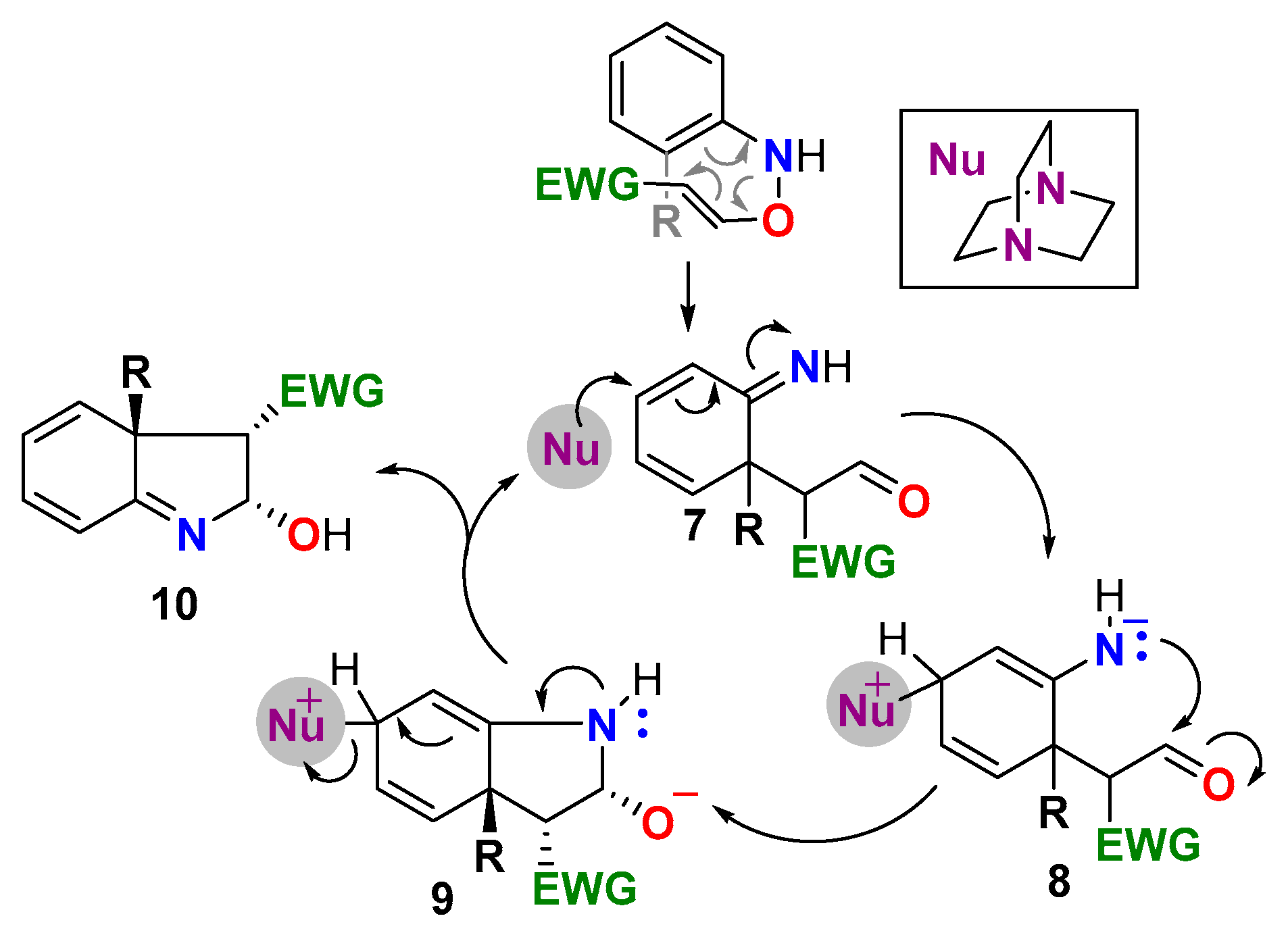
The initial studies began using commercially available N-benzoyl-N-phenylhydroxylamine (4a-N-Bz) as the nitrogen species needed for the synthesis of indoles and the standard conditions reported by our group for the DABCO-catalyzed synthesis of propargyl vinyl ethers [59] (Scheme 2). Although the addition of the hydroxylamine to methyl propiolate (5a) took place in the presence of DABCO, and this was followed by the desired cyclization, the hydroxyindoline intermediate HI was isolated instead of the expected indole 6a-N-Bz. Furthermore, a side-product incorporating a second unit of methyl propiolate was also obtained (DVE). It forms from the DABCO-catalyzed addition of methyl propiolate (5a) on the enol ether generated on the [3,3]sigmatropic rearrangement of the intermediate N-oxyenamine. The overnight reaction of intermediate HI with Et3N afforded the indole 6a-N-Bz in quantitative yield.
At this time, we decided to question the need for the protecting group on the nitrogen atom. Indeed, when starting from N-phenylhydroxylamine (4a) and methyl propiolate (5a) (1.2 equiv) as the reactants, the reaction proceeded smoothly to the desired unprotected indole 6a (Table 1, entry 1). The observed outcome was different to that reported by Hwu et al. [64] in which N-vinyl protected indoles were obtained when N-phenylhydroxylamine (4a) reacted with an excess of methyl propiolate (5a) in the presence of a catalytic amount of DMAP. Strikingly, the report by Hwu et al. claims that DABCO and Et3N did not give significant amounts of the desired indoles under their reaction conditions.
Once the feasibility of the reaction using free N-phenylhydroxylamine (4a) was established, we next undertook the optimization of the reaction (Table 1). Under all assayed conditions, the formation of the desired indole 6a was accompanied by the formation of very small amounts of side-product DVE. Based on these results, and taking into consideration the simplicity of the experimental procedure, we chose the reaction conditions depicted in entry 4 as the standardized conditions for the formation of the desired indoles 6 from the appropriate N-arylhydroxylamines 4.
Once this reaction was optimized, we coupled this reaction with the reduction of the nitrobenzene (3a) to set up a consecutive 2-step reaction manifold (Scheme 3). Thus, reduction of nitrobenzene (3a) with hydrazine monohydrate and Rh on activated charcoal (5%) in THF gave N-phenylhydroxylamine (4a), which, after filtration of the reaction mixture and concentration, was directly submitted to the next reaction step to deliver indole 6a in 80% overall yield. Under these conditions, the overall yield of indole 6a was comparable to that obtained directly from 4a (80% vs 82%).
2.2. Scope of the Consecutive 2-Step Protocol
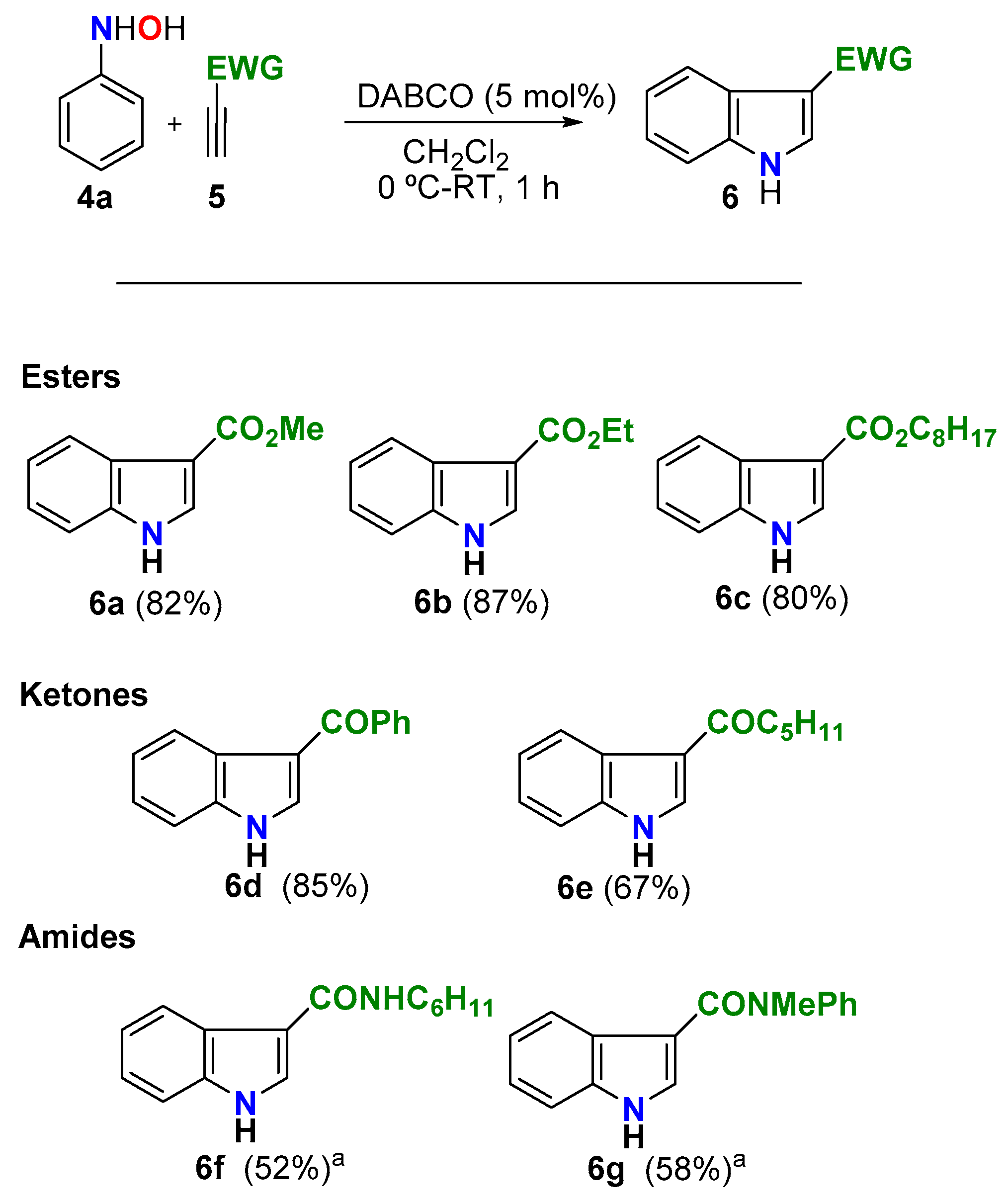
With a convenient protocol in hand, we next explored its scope with regard to the alkyne using N-phenylhydroxylamine (4a) as the nitrogen component (Scheme 4). The reaction was well suited to prepare 3-EWG-indoles incorporating carboxylates (6a–c), aroyl (acyl) groups (6d,e), or carboxamides (6f,g). In the case of N,N-disubstituted propionamides 5f and 5g, the poorest Michael acceptors of the series, the reaction needed more time (overnight) to deliver the corresponding N,N-disubstituted indole-3-carboxamides 6f and 6g in synthetically relevant 52% and 58% yields respectively. The efficiency and simplicity of this transformation are emphasized when compared with the current multistep methodologies to prepare these indole-3-carboxamides [67].
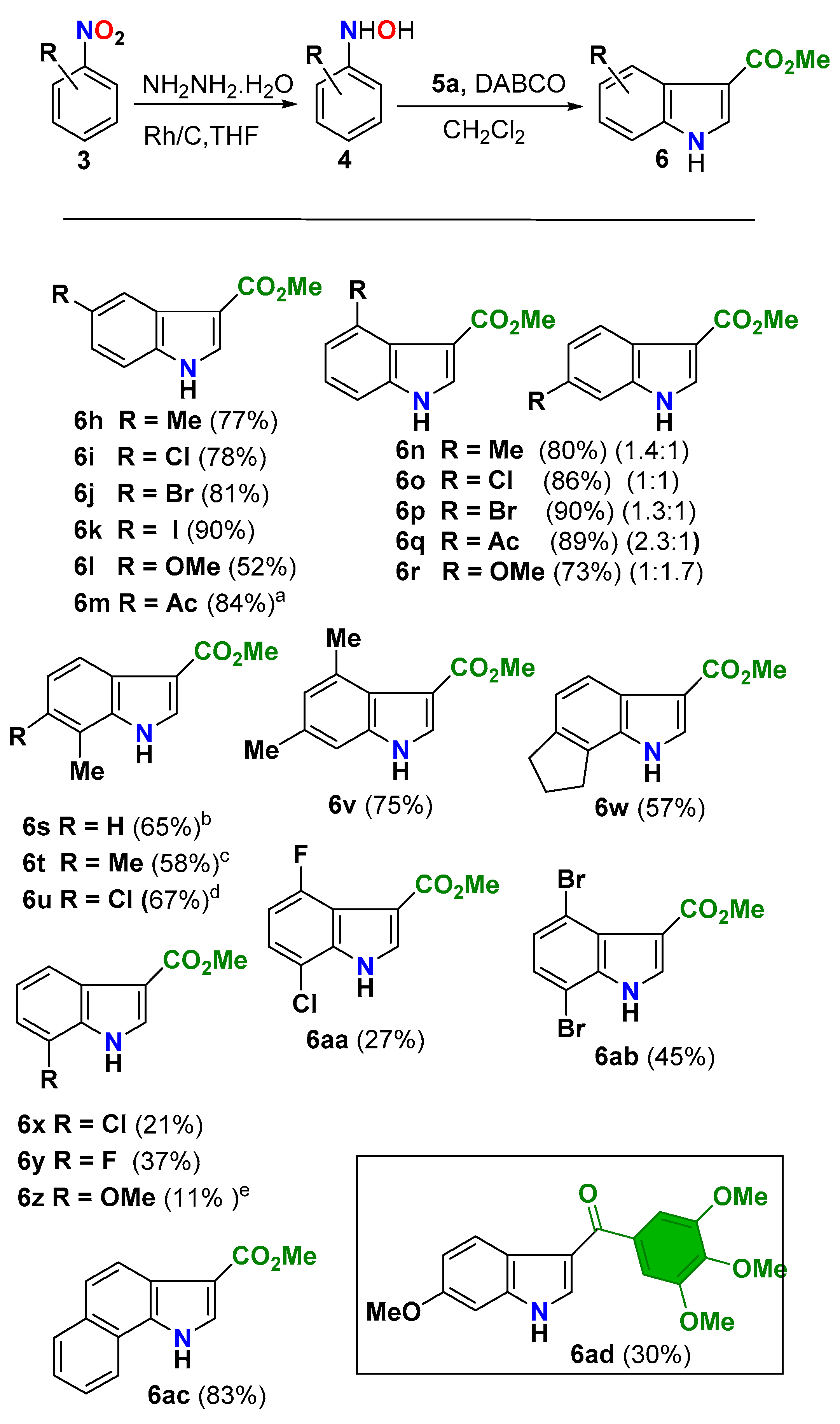
The scope of the consecutive 2-step manifold with regard to the substitution at the benzene ring was explored using methyl propiolate (5a) as the alkyne component and differently substituted nitroarenes 3 (Scheme 5). The manifold tolerated a diverse substitution pattern on the benzene ring to deliver the corresponding methyl indole-3-carboxylates 6 with overall good efficiency and broad functional/structural diversity. A convenient grade of topological diversity could be generated, spanning from the simple monosubstituted derivatives (e.g., 6h–r, 52–90% yield) to more complex polycyclic (6w, 57%) or fused aromatic systems (6ac, 83%). Meta-substituted N-arylhydroxylamines afforded separable mixtures of the corresponding C-4/C-6 regioisomers in excellent yields (80–90%) and with a slight substituent- dependent regioselectivity (Scheme 5, compounds 6n–r). A balance between steric and electronic effects of the substituent seems to steer the outcome of the [3,3]-sigmatropic rearrangement, in line with the reported results in the Fischer synthesis of indoles endowed with this substitution pattern [52,65,68]. Thus, electron-donating substituents (R = OMe) unbalanced the chemical outcome towards the less crowed isomer (6r, 1.7:1 / C-6: C-4 ratio) whereas electron-withdrawing groups did it in the opposite direction (6o–q; from 1:1 to 2.3:1 / C-4:C-6 ratio). It is remarkable that the [3,3]-sigmatropic rearrangement of benzyl ketene acetals bearing a meta-OMe substituent on the aromatic ring preponderantly delivers the C-4 isomer. Theoretical studies pointed to a stabilizing interaction between the lone pair of the substituent and the LUMO orbital of the transition state as being responsible for the observed regioselectivity [69]. The observed inverted regioselectivity in our reaction seems to rule out the existence of this kind of stabilizing interactions.
As expected, substitution at the ortho-position posed a regioselectivity problem in the [3,3]-sigmatropic rearrangement, since the C-C bond formation at the most substituted ortho-position becomes a competitive pathway (Scheme 6). Indeed, in some cases, we were able to isolate the product 10 obtained from this pathway. The effect of this mechanistic divergence was mainly reflected in a diminished efficiency in the reactions of nitroarenes bearing a good leaving group at that position (6x–z, 6aa–ab). In the case of simple alkyl substituents (6s–u) or fused bicycles (6w), the manifold mostly afforded the expected C-7 substituted indole derivatives in moderate to good yields. The versatility of this methodology is highlighted when compared with Zhangs’ protocol [56] (Scheme 1B), which is sensitive to steric impediments and shows a reduced tolerance to alkyl substitution at this position [70]. However, the easy access to ortho-substituted nitroarenes together with the simplicity of the 2-step reaction manifold compensate for these deficiencies and make this methodology a viable synthetic alternative to prepare C-7 substituted 3-EWG indoles.
The reaction manifold was well suited to scale up. As a proof of concept, we carried out the synthesis of methyl 6-acetylindole-3-carboxylate (6m) at 50 mmol scale from 4-acetylnitrobenzene (3m) and methyl propiolate (5a). The scaled manifold afforded indole 6m in an excellent 84% yield (9.08 g) (Scheme 5).
Finally, the anticancer activity of some 3-aroyl indoles [17,19] led us to evaluate our methodology as a novel and practical access to the chemical/therapeutic space of these derivatives. Among the huge number of possible 3-aroylindoles, we chose (6-methoxy-1H-indole-3-yl) (3,4,5-trimethoxyphenyl) methanone, the so-called BPRL0T075 (6ad, box in Scheme 5), an extremely potent cytotoxic compound with good pharmacological properties [21]. The manifold delivered it in a 30% yield along with its C-4 isomer (25%).
3. Materials and Methods
3.1. General Information
1H-NMR and 13C-NMR spectra (see supplementary materials) of DMSO-d6 or CDCl3 solutions were recorded either at 400 and 100 MHz or at 500 and 125 MHz (Bruker Ac 200 and AMX2-500 respectively). Mass spectra (low resolution) (EI/CI) were obtained with a Hewlett-Packard 5995 gas chromatograph/mass spectrometer. High-resolution mass spectra were recorded with a Micromass Autospec mass spectrometer. Analytical thin-layer chromatography plates used were E. Merck Brinkman UV-active silica gel (Kieselgel 60 F254) on aluminum. Flash column chromatography was carried out with E. Merck silica gel 60 (particle size less than 0.020 mm) using appropriate mixtures of ethyl acetate and hexanes, or ethyl acetate and dichloromethane as eluents. All reactions were performed in oven-dried glassware. All materials were obtained from commercial suppliers and used as received. N-phenylhydroxylamine (4a) was prepared on a multigram scale following a standard experimental procedure [71].
3.2. General Procedure for the Synthesis of Indoles 6 Directly from N-phenylhydroxylamine (4a). Preparation of Methyl Indol-3-carboxylate (6a)
To a solution of N-phenylhydroxylamine (4a) (436 mg, 4.0 mmol) in dichloromethane (20 mL) was added the activated alkyne 5 (4.2 mmol) and the resulting solution was cooled to 0 °C using an ice bath. DABCO (22 mg, 0.2 mmol) was added all at once and the reaction mixture was stirred for 1 h. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography (silica gel; n-hexane/ethyl acetate: 70/30 v/v) to give indole 6a (565.2 mg, 80%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 3.93 (s, 3H), 7.25–7.28 (m, 2H), 7.40–7.42 (m, 1H), 7.91 (d, 1H, J = 3.0 Hz), 8.18-8.20 (m, 1H), 8.82 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 51.1, 108.7, 111.5, 121.5, 122.0, 123.2, 125.6, 131.1, 136.1, 165.8 ppm. The values are in accordance with the literature [72].
3.3. General Procedure for the Consecutive 2-Step Synthesis of Indoles 6 from Nitroarenes 3. Preparation of Methyl Indol-3-carboxylate (6a)
Under a nitrogen atmosphere, the nitrobenzene (3a) (4 mmol) was dissolved in dry tetrahydrofuran (20 mL) and Rh/C (5%) (20 mg) was added to the solution. The resulting mixture was cooled to 0 °C, hydrazine monohydrate (250 mg, 5 mmol) was then added dropwise and the reaction was stirred at 0 °C for 3 h. The reaction mixture was filtered through a short pad of celite (aided with the use of dichloromethane) and the solvents evaporated under reduced pressure. The resulting crude product was dissolved in dichloromethane (20 mL) and methyl propiolate (5a) (344 mg, 4.2 mmol) was added. The resulting solution was cooled to 0 °C using an ice bath and DABCO (22 mg, 0.2 mmol) was added at once. The reaction mixture was stirred for 1 h and the solvent was removed under reduced pressure. The crude residue was purified by flash column chromatography (silica gel; n-hexane/ethyl acetate: 70/30 v/v) to give indole 6a [72].
3.4. Characterization Data for Indoles 6 and 3,3a-Dihydro-2H-Indoles 10
Ethyl 1H-indole-3-carboxylate (6b) (330.2 mg, 87%). Off-white solid: 1H-NMR (DMSO-d6, 500 MHz): δ = 1.33 (t, 3H, J = 7.1 Hz), 4.28 (q, 2H, J = 7.1 Hz), 7.16–7.21 (m, 2H), 7.47 (dd, 1H, J = 6.5 and 1.6 Hz), 8.00 (dd, 1H, J = 6.8 and 2.0 Hz), 8.06 (d, 1H, J = 2.3 Hz), 11.90 (bs, 1H). 13C-NMR (DMSO-d6, 125 MHz): δ = 14.5, 59.0, 106.6, 112.3, 120.5, 121.2, 122.3, 125.6, 132.4, 136.4, 164.4 ppm. The values are in accordance with the literature [73].
Octyl 1H-indole-3-carboxylate (6c) (217.5 mg, 80%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 0.88 (t, 3H, J = 6.8 Hz), 1.28–1.39 (m, 8H), 1.43–1.47 (m, 2H), 1.76–1.83 (m, 2H), 4.34 (t, 2H, J = 6.6 Hz), 7.24–7.29 (m, 2H), 7.39–7.42 (m, 1Hz), 7.91 (d, 1H, J = 2.1 Hz), 8.16–8.20 (m, 1H), 8.80 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 14.0, 22.6, 26.2, 29.0, 29.2, 29.3, 31.8, 64.1, 109.2, 111.5, 121.6, 122.0, 123.1, 125.8, 131.0, 136.2, 165.5 ppm. HRMS (ESI-) m/z [M-H]− calculated for C17H22NO2 272.1651, found 272.1650.
(1H-Indol-3-yl)(phenyl)methanone (6d) (351.4 mg, 83%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 7.22–7.28 (m, 2H), 7.52–7.62 (m, 4H), 7.78–7.80 (m, 2H), 7.93 (s, 1H), 8.26 (dd, 1H, J = 6.7 and 1.7 Hz), 12.10 (bs, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 112.2, 115.0, 121.4, 121.9, 123.1, 126.2, 128.3, 128.4, 131.0, 136.7, 140.5, 190.0 ppm. The values are in accordance with the literature [74].
1-(1H-Indol-3-yl)hexan-1-one (6e) (144.5 mg, 67%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 0.87 (t, 3H, J = 6.8 Hz), 1.29–1.34 (m, 4H), 1.60–1.68 (m, 2H), 2.82 (t, 2H, J = 7.4 Hz), 7.14–7.22 (m, 2H), 7.45–7.47 (m, 1H), 8.20 (dd, 1H, J = 6.9 and 1.5 Hz), 8.30 (d, 1H, J = 3.0 Hz), 11.88 (bs, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 13.9, 22.0, 24.6, 31.1, 38.7, 112.0, 116.4, 121.4, 121.5, 122.6, 125.4, 133.6, 136.6, 195.5 ppm. The values are in accordance with the literature [75].
N-cyclohexyl-1H-indole-3-carboxamide (6f) (127.1 mg, 52%). The reaction time was increased to overnight. Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 1.12–1.36 (m, 5H), 1.60–1.63 (m, 1H), 1.70–1.77 (m, 2H), 1.82–1.88 (m, 2H), 3.86–4.08 (m, 1H), 7.06–7.14 (m, 2H), 7.40 (d, 1H, J = 8.0 Hz), 7.60 (d, 1H, J = 6.1 Hz), 8.05 (s, 1H), 8.14 (d, 1H, J = 7.8 Hz), 11.5 (bs, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 25.0 (2C), 25.4, 32.9 (2C), 47.4, 110.8, 111.6, 120.1, 121.1, 121.7, 126.3, 127.5, 136.1, 163.7 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C15H18N2ONa 265.1317, found 265.1314.The product had been previously described in the literature but in CDCl3 [76].
N-Methyl-N-phenyl-1H-indole-3-carboxamide (6g) (290.4 mg, 58%). The reaction time was increased to overnight. Off-white solid: 1H-NMR (DMSO-d6, 500 MHz): δ = 3.36 (s, 3H), 6.36 (d, 1H, J = 3.0 Hz), 7.07 (dd, 1H, J = 7.4 and 1.2 Hz), 7.11 (dd, 1H, J = 7.5 and 1.5 Hz), 7.28–7.32 (m, 4H), 7.37–7.40 (m, 2H), 8.04 (d, 1H, J = 8.0 Hz), 11.28 (bs, 1H). 13C-NMR (DMSO-d6, 125 MHz): δ = 37.9, 109.6, 111.5, 120.3, 121.3, 121.9, 126.8, 127.2, 127.5 (2C), 128.9, 129.4 (2C), 135.1, 145.5, 165.1 ppm. The values are in accordance with the literature [77].
Methyl 5-methyl-1H-indole-3-carboxylate (6h) (533.5 mg, 77%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 2.48 (s, 3H), 3.92 (s, 3H), 7.08 (dd, 1H, J = 8.3 and 1.2 Hz), 7.29 (d, 1H, J = 8.3 Hz), 7.86 (d, 1H, J = 3.0 Hz), 7.98 (d, 1H, J = 1.2 Hz), 8.72 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 21.5, 51.0, 108.2, 111.1, 121.1, 124.8, 126.0, 131.1, 131.6, 134.4, 165.8 ppm. The values are in accordance with the literature [78].
Methyl 5-chloro-1H-indole-3-carboxylate (6i) (309.3 mg, 78%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 4.36 (s, 3H), 7.74 (d, 1H, J = 8.6 Hz), 8.05 (d, 1H, J = 8.6 Hz), 8.55 (s, 1H), 8.69 (s, 1H), 12.65 (s, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 51.6, 107.2, 114.8, 120.6, 123.4, 127.1, 127.8, 134.7, 135.9, 165.4. The values are in accordance with the literature [73].
Methyl 5-bromo-1H-indole-3-carboxylate (6j) (820.0 mg, 81%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 3.90 (s, 3H), 7.28 (d, 1H, J = 8.6 Hz), 7.36 (dd, 1H, J = 8.6 and 1.8 Hz), 7.91 (d, 1H, J = 2.9 Hz), 8.32 (d, 1H, J = 1.8 Hz), 8.61 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 51.2, 108.8, 112.9, 115.7, 124.3, 126.3, 127.4, 131.7, 134.7, 165.1 ppm. The values are in accordance with the literature [79].
Methyl 5-iodo-1H-indole-3-carboxylate (6k) (542.9 mg, 90%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 3.81 (s, 3H), 7.34 (d, 1H, J = 8.5 Hz), 7.48 (dd, 1H, J = 8.5 and 1.5 Hz), 8.09 (d, 1H, J = 2.9 Hz), 8.33 (d, 1H, J = 1.3 Hz), 12.09 (bs, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 50.8, 85.7, 105.7, 114.8, 128.1, 128.7, 130.4, 133.2, 135.5, 164.4. ppm. The values are in accordance with the literature [80].
Methyl 5-methoxy-1H-indole-3-carboxylate (6l). (214.2 mg, 52%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 3.87 (s, 3H), 3.91 (s, 3H), 6.90 (dd, 1H, J = 8.8 and 2.5 Hz), 7.28 (d, 1H, J = 8.8 Hz), 7.66 (d, 1H, J = 2.5 Hz), 7.85 (d, 1H, J = 3.0 Hz), 8.74 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 51.0, 55.8, 102.8, 108.3, 112.3, 113.7, 126.7, 131.0, 131.2, 155.9, 165.8 ppm. The values are in accordance with the literature [79].
Methyl 5-acetyl-1H-indole-3-carboxylate (6m). (9.083 g, 84%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 2.63 (s, 3H), 3.84 (s, 3H), 7.56 (d, 1H, J = 8.6 Hz), 7.84 (dd, 1H, J = 8.6 and 1.5 Hz), 8.22 (d, 1H, J = 3.0 Hz), 8.65 (d, 1H, J = 1.5 Hz), 12.25 (bs, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 26.7, 50.8, 107.7, 112.3, 122.0, 122.5, 125.2, 130.8, 134.2, 138.9, 164.4, 197.4 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C12H11NO3Na 240.0637, found 240.0634.
Methyl 4-methyl-1H-indole-3-carboxylate (6n; C-4 isomer) (348.7 mg, 46%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 2.87 (s, 3H), 3.87 (s, 3H), 7.03 (d, 1H, J = 7.1 Hz), 7.15 (t, 1H, J = 7.6 Hz), 7.22 (d, 1H, J = 8.0 Hz), 7.89 (d, 1H, J = 3.0 Hz), 8.76 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 22.3, 51.1, 109.2, 109.5, 123.4, 124.0, 124.4, 132.1, 132.4, 136.9, 165.4 ppm. The values are in accordance with the literature [81].
Methyl 6-methyl-1H-indole-3-carboxylate (6n; C-6 isomer) (257.7 mg, 34%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 2.45 (s, 3H), 3.91 (s, 3H), 7.10 (d, 1H, J = 8.0 Hz), 7.18 (s, 1H), 7.83 (d, 1H, J = 3.0 Hz), 8.04 (d, 1H, J = 8.0 Hz), 8.70 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 21.6, 51.0, 108.5, 111.4, 121.0, 123.6, 123.8, 130.5, 133.1, 136.5, 165.8 ppm. The values are in accordance with the literature [81].
Methyl 4-chloro-1H-indole-3-carboxylate (6o; C-4 isomer) (186.0 mg, 44%). Pale yellow solid: 1H-NMR (DMSO-d6, 400 MHz) δ = 3.76 (s, 3H), 7.13 – 7.26 (m, 2H), 7.46 (dd, 1H, J = 6.7 and 2.3 Hz), 8.13 (d, 1H, J = 2.6 Hz), 12.18 (s, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ = 51.8, 107.8, 112.4, 123.3, 123.6, 124.1, 125.8, 135.0, 139.3, 164.4. HRMS (ESI-) m/z [M − H]− calculated for C10H7NO2Cl 208.0165, found 208.0163.
Methyl 6-chloro-1H-indole-3-carboxylate (6o; C-6 isomer) (175.6 mg, 42%). Pale yellow solid: 1H-NMR (CDCl3, 400 MHz) δ = 3.94 (s, 3H), 7.26 (d, 1H, J = 9.4 Hz), 7.43 (s, 1H), 7.92 (s, 1H), 8.11 (d, 1H, J = 8.6 Hz), 8.60 (s, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ = 52.7, 108.3, 113.8, 123.4, 123.5, 125.9, 129.0, 135.1, 138.4, 166.7. The values are in accordance with the literature [62].
Methyl 4-bromo-1H-indole-3-carboxylate (6p; C-4 isomer) (258.8 mg, 51%). Pale yellow solid: 1H-NMR (DMSO-d6, 400 MHz) δ = 3.77 (s, 3H), 7.11 (t, 1H, J = 7.9 Hz), 7.39 (d, 1H, J = 7.5 Hz), 7.51 (d, 1H, J = 7.8 Hz), 8.12 (s, 1H), 12.16 (s, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ = 51.7, 108.4, 112.9, 113.7, 124.4, 125.0, 127.1, 134.7, 139.0, 164.5. HRMS (ESI+) m/z [M + Na]+ calculated for C10H8NO2BrNa 275.9636, found 275.9642.
Methyl 6-bromo-1H-indole-3-carboxylate (6p; C-6 isomer) (200.1 mg, 39%). Pale yellow solid: 1H-NMR (DMSO-d6, 400 MHz) δ = 3.80 (s, 3H), 7.32 (dd, 1H, J = 8.5 and 1.8 Hz), 7.67 (s, 1H), 7.92 (d, 1H, J = 8.5 Hz), 8.11 (d, 1H, J = 2.8 Hz), 12.03 (s, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ = 51.7, 107.5, 115.9, 115.9, 123, 125.1, 125.6, 134.2, 138.1, 165.3. HRMS (ESI-) m/z [M − H]− calculated for C10H7NO2Br 251.9660, found 251.9657.
Methyl 4-acetyl-1H-indole-3-carboxylate (6q; C-4 isomer) (539.8 mg, 62%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 2.45 (s, 3H), 3.72 (s, 3H), 7.18 (d, 1H, J = 7.2 Hz), 7.26 (t, 1H, J = 7.5 Hz), 7.60 (d, 1H, J = 7.9 Hz), 8.14 (s, 1H), 12.17 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 31.1, 51.0, 108.2, 114.3, 119.8, 120.7, 122.5, 132.6, 135.3, 137.0, 165.2, 205.2 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C12H11NO3Na 240.0637, found 240.0634.
Methyl 6-acetyl-1H-indole-3-carboxylate (6q; C-6 isomer) (237.7 mg, 27%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 2.66 (s, 3H), 3.86 (s, 3H), 7.84 (dd, 1H, J = 8.4 and 1.5 Hz), 8.10 (d, 1H, J = 8.4 Hz), 8.15 (d, 1H, J = 1.5 Hz), 8.33 (d, 1H, J = 3.0 Hz), 12.39 (bs, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 26.7, 50.8, 106.7, 113.4, 120.1, 121.1, 129.9, 131.5, 135.7, 135.9, 164.4, 197.3 ppm. HRMS (ESI+) m/z [M+Na]+ calculated for C12H11NO3Na 240.0637, found 240.0640.
Methyl 4-methoxy-1H-indole-3-carboxylate (6r; C-4 isomer) (111.8 mg, 27%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 3.86 (s, 3H), 3.93 (s, 3H), 6.66 (d, 1H, J = 7.8 Hz), 7.00 (d, 1H, J = 8.0 Hz), 7.16 (t, 1H, J = 7.0 Hz), 7.81 (d, 1H, J = 2.8 Hz), 9.11 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 51.3, 55.9, 102.9, 105.0, 108.6, 115.4, 124.2, 131.3, 138.4, 154.2, 165.0 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C11H11NO3Na 228.0637, found 228.0634.
Methyl 6-methoxy-1H-indole-3-carboxylate (6r; C-6 isomer) (189.5 mg, 46%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 3.78 (s, 6H), 6.83 (dd, 1H, J = 8.6 and 2.2 Hz), 6.96 (d, 1H, J = 2.2 Hz), 7.84 (d, 1H, J = 8.6 Hz), 7.94 (d, 1H, J = 3.0 Hz), 11.70 (bs, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 50.5, 55.2, 95.2, 106.3, 111.3, 119.7, 121.0, 131.2, 137.1, 156.1, 164.7 ppm. The values are in accordance with the literature [82].
Methyl 7-methyl-1H-indole-3-carboxylate (6s) (245.9.5 mg, 65%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 2.50 (s, 3H), 3.92 (s, 3H), 7.06 (d, 1H, J = 7.1 Hz), 7.19 (t, 1H, J = 7.6 Hz), 7.90 (d, 1H, J = 2.8 Hz), 8.02 (d, 1H, J = 8.0 Hz), 8.71 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 16.5, 51.1, 109.2, 119.2, 120.7, 122.2, 123.8, 125.4, 130.7, 135.7, 165.8 ppm. The values are in accordance with the literature [83].
Methyl 6,7-dimethyl-1H-indole-3-carboxylate (6t) (235.0 mg, 58%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 2.32 (s, 3H), 2.39 (s, 3H), 3.79 (s, 3H), 6.99 (d, 1H, J = 8.0 Hz), 7.71 (d, 1H, J = 8.0 Hz), 7.99 (d, 1H, J = 3.0 Hz), 11.78 (bs, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 13.1, 18.9, 50.5, 106.6, 117.4, 119.4, 123.8, 123.9, 129.5, 131.6, 136.5, 164.8 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C12H13NO2Na 226.0844, found 226.0845.
Methyl 6-chloro-7-methyl-1H-indole-3-carboxylate (6u) (299.8 mg, 67%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 2.52 (s, 3H), 3.91 (s, 3H), 7.26 (d, 1H, J = 8.6 Hz), 7.90 (d, 1H, J = 2.9 Hz), 7.93 (d, 1H, J = 8.6 Hz), 8.56 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 13.8, 51.2, 109.6, 118.6, 119.8, 123.5, 124.1, 128.9, 131.0, 136.2, 165.3 ppm. HRMS (ESI-) m/z [M − H]− calculated for C11H9ClNO2 222.0322, found 222.0314.
Methyl 4,6-dimethyl-1H-indole-3-carboxylate (6v) (607.7 mg, 75%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 2.33 (s, 3H), 2.70 (s, 3H), 3.73 (s, 3H), 6.74 (s, 1H), 7.07 (s, 1H), 7.96 (d, 1H, J = 3.0 Hz), 11.71 (bs, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 20.9, 21.9, 50.6, 107.2, 109.7, 122.1, 124.9, 130.3, 131.6, 132.7, 137.6, 164.6 ppm. HRMS (ESI-) m/z [M − H]− calculated for C12H12NO2 202.0868, found 202.0872.
Methyl 1,6,7,8-tetrahydrocyclopenta[g]indole-3-carboxylate (6w) (305.6 mg, 71%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 2.08–2.15 (m, 2H), 2.95 (t, 2H, J = 7.3 Hz), 3.04 (t, 2H, J = 7.3 Hz), 3.79 (s, 3H), 7.08 (d, 1H, J = 8.0 Hz), 7.79 (d, 1H, J = 8.0 Hz), 7.99 (d, 1H, J = 3.0 Hz), 11.88 (bs, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 25.0, 29.9, 32.6, 50.6, 106.9, 118.1, 118.5, 124.4, 126.5, 131.6, 133.5, 138.3, 164.9 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C13H13NO2Na 238.0844, found 238.0848.
Methyl 7-chloro-1H-indole-3-carboxylate (6x) (175.7 mg, 21%). Pale yellow solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 3.82 (s, 3H), 7.18 (t, 1H, J = 7.8 Hz), 7.29 (dd, 1H, J = 7.7 and 1.1 Hz), 7.98 (d, 1H, J = 7.9 Hz), 8.11 (d, 1H, J = 3.1 Hz), 12.36 (s, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ = 51.7, 108.6, 117.6, 120.4, 122.9, 123.3, 128.4, 134.1, 134.2, 165.3. HRMS (ESI-) m/z [M − H]− calculated for C10H7NO2Cl 208.0165, found 208.0158.
Methyl 7-fluoro-1H-indole-3-carboxylate (6y) (285.6 mg, 37%). Off-white solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 3.81 (s, 3H), 7.05 (dd, 1H, J = 11.5 and 8.0 Hz), 7.13–7.18 (m, 1H), 7.81 (d, 1H, J = 8.0 Hz), 8.12 (s, 1H), 12.49 (bs, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 50.8, 107.3 (d, J = 16 Hz), 107.4, 116.5 (d, J = 3.5 Hz), 121.8 (d, J = 6.3 Hz), 124.2 (d, J = 13.5 Hz), 129.3 (d, J = 5.3 Hz), 133.1, 149.2 (d, J = 244 Hz), 164.4 ppm. HRMS (ESI-) m/z [M − H]− calculated for C10H7FNO2 192.0461, found 192.0457.
Methyl 7-methoxy-1H-indole-3-carboxylate (6z) (90.4 mg, 11%). Off-white solid: 1H-NMR (CDCl3, 500 MHz): δ = 3.91 (s, 3H), 3.94 (s, 3H), 6.70 (d, 1H, J = 7.8 Hz), 7.17 (t, 1H, J = 8.0 Hz), 7.75 (d, 1H, J = 8.0 Hz), 7.87 (d, 1H, J = 3.0 Hz), 8.90 (bs, 1H). 13C-NMR (CDCl3, 125 MHz): δ = 51.0, 55.4, 102.9, 109.2, 113.9, 122.5, 126.7, 127.1, 130.3, 146.1, 165.7 ppm. The values are in accordance with the literature [84].
Methyl 7-chloro-4-fluoro-1H-indole-3-carboxylate (6aa) (245.2 mg, 27%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 3.90 (s, 3H), 6.87 (dd, 1H, J = 10.5 and 8.5 Hz), 7.16 (dd, 1H, J = 8.5 and 3.6 Hz), 7.96 (d, 1H, J = 3.0 Hz), 9.13 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 51.8, 108.8 (d, J = 23 Hz), 109.4 (d, J = 3.5 Hz), 112.1 (d, J = 4.1 Hz), 115.1 (d, J = 22 Hz), 123.0 (d, J = 8.0 Hz), 132.2, 135.6 (d, J = 10.0 Hz), 155.1 (d, J = 253 Hz), 164.0 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C10H7ClFNO2Na 250.0047, found 250.0051.
Methyl 4,7-dibromo-1H-indole-3-carboxylate (6ab) (598.0 mg, 45%). Off-white solid: 1H-NMR (DMSO-d6, 500 MHz): δ = 3.78 (s, 3H), 7.33 (d, 1H, J = 8.0 Hz), 7.37 (d, 1H, J = 8.0 Hz), 8.09 (d, 1H, J = 3.1 Hz), 12.40 (bs, 1H). 13C-NMR (DMSO-d6, 125 MHz): δ = 51.1, 104.6, 109.1, 112.3, 125.3, 126.1, 127.3, 134.3, 136.0, 163.3 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C10H7Br2NO2Na 357.8700, found 357.8698.
Methyl 1H-benzo[g]indole-3-carboxylate (6ac) (745.8 mg, 83%). Yellow-brown solid: 1H-NMR (DMSO-d6, 400 MHz): δ = 3.85 (s, 3H), 7.48 (t, 1H, J = 7.5 Hz), 7.60 (t, 1H, J = 7.5 Hz), 7.65 (d, 1H, J = 8.7 Hz), 7.97 (d, 1H, J = 8.2 Hz), 8.10–8.19 (m, 2H), 8.44 (d, 1H, J = 8.2 Hz), 12.84 (s, 1H). 13C-NMR (DMSO-d6, 100 MHz): δ = 51.6, 109.1, 121.0, 121.6, 122.8, 122.9, 122.9, 125.3, 126.8, 129.3, 130.9, 130.9, 132.1, 165.8. The values are in accordance with the literature [85].
(6-Methoxy-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)methanone (6ad; C-6 isomer) (257.3 mg, 30%). Off-white solid: 1H-NMR (CDCl3, 500 MHz): δ = 3.79 (s, 3H), 3.83 (s, 6H), 3.91 (s, 3H), 6.87 (d, 1H, J = 2.2 Hz), 6.93 (dd, 1H, J = 8.7 and 2.2 Hz), 7.07 (s, 2H), 7.59 (d, 1H, J = 3.0 Hz), 8.21 (d, 1H, J = 8.7 Hz), 9.48 (bs, 1H). 13C-NMR (CDCl3, 125 MHz): δ = 55.5, 56.2 (2C), 60.9, 95.1, 106.4 (2C), 112.2, 116.7, 120.4, 122.9, 132.9, 135.9, 137.5, 140.9, 152.9 (2C), 157.5, 190.6 ppm. The values are in accordance with the literature [20].
(4-Methoxy-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)methanone (6ad; C-4 isomer) (212.1 mg, 25%). Off-white solid: 1H-NMR (DMSO-d6, 500 MHz): δ = 3.65 (s, 3H), 3.75 (s, 3H), 3.77 (s, 6H), 6.63 (d, 1H, J = 7.8 Hz), 7.09 (s, 2H), 7.10 (d, 1H, J = 7.8 Hz), 7.16 (t, 1H, J = 7.9 Hz), 7.75 (d, 1H, J = 3.0 Hz), 11.86 (bs, 1H). 13C-NMR (DMSO-d6, 125 MHz): δ = 55.0, 55.9 (2C), 60.1, 101.9, 105.2, 107.0 (2C), 115.7, 115.9, 123.7, 131.6, 135.3, 138.2, 140.7, 152.3 (2C), 153.6, 188.3 ppm. The values are in accordance with the literature [20].
(±)-Methyl (2R,3R,3aS)-2-hydroxy-3a-methyl-3,3a-dihydro-2H-indole-3-carboxylate (10s) (144.1 mg, 21%). Off-white solid: 1H-NMR (CDCl3, 400 MHz): δ = 1.00 (s, 3H), 2.95 (d, 1H, J = 7.4 Hz), 3.79 (s, 3H), 5.7 (bs, 1H), 5.89 (d, 1H, J = 7.4 Hz), 6.09–6.13 (m, 1H), 6.44–6.47 (m, 2H), 6.58–6.62 (m, 1H). 13C-NMR (CDCl3, 100 MHz): δ = 20.0, 52.1, 53.3, 61.8, 91.0, 120.7, 121.3, 136.0, 140.4, 170.5, 178.3 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C11H13NO3Na 230.0793, found 230.0785.
(±)-(2R,3R,3aR)-Methyl 2-hydroxy-3a,4-dimethyl-3,3a-dihydro-2H-indole-3-carboxylate (10t) (150.6 mg, 34%). Off-white solid: 1H-NMR (CDCl3, 500 MHz): δ = 1.13 (s, 3H), 1.91 (s, 3H), 2.91 (d, 1H, J = 7.4 Hz), 3.77 (s, 3H), 5.4 (bs, 1H), 5.80 (d, 1H, J = 6.0 Hz), 5.85 (d, 1H, J = 7.4 Hz), 6.27 (d, 1H, J = 9.7 Hz), 6.50 (dd, 1H, J = 9.7 and 6.0 Hz). 13C-NMR (CDCl3, 125 MHz): δ = 15.4, 20.1, 52.0, 57.9, 60.5, 94.1, 118.4, 118.6, 136.5, 150.3, 171.6, 179.3 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C12H15NO3Na 244.0950, found 244.0952.
(±)-(2R,3R,3aS)-Methyl 4-chloro-2-hydroxy-3a-methyl-3,3a-dihydro-2H-indole-3-carboxylate (10u) (128.8 mg, 27%). Off-white solid: 1H-NMR (CDCl3, 500 MHz): δ = 1.30 (s, 3H), 3.02 (d, 1H, J = 7.5 Hz), 3.77 (s, 3H), 5.7 (bs, 1H), 5.75 (d, 1H, J = 7.5 Hz), 6.16 (d, 1H, J = 6.3 Hz), 6.42 (d, 1H, J = 9.6 Hz), 6.55 (dd, 1H, J = 9.6 and 6.3 Hz). 13C-NMR (CDCl3, 125 MHz): δ = 19.8, 52.2, 59.9, 61.0, 93.8, 119.5, 120.4, 135.5, 144.5, 170.8, 177.5 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C11H12ClNO3 264.0403, found 264.0404.
The relative stereochemistry was assigned based on the 1HNMR, COSY, HSQC and NOESY experiments. The syn relationship between H(2) and H(3) was assigned based on the observed 3J(H,H) = 7.5 Hz and their NOE interactions. The syn relationship between Me(3a) and H(3) was assigned based on their NOE interactions.
(±)-Methyl (2R,3R,3aR)-2-hydroxy-3a-methoxy-3,3a-dihydro-2H-indole-3-carboxylate (10z) (195.8 mg, 22%). Off-white solid: 1H-NMR (CDCl3, 500 MHz): δ = 2.87 (d, 1H, J = 6.1 Hz), 3.03 (s, 3H), 3.79 (s, 3H), 3.9 (bs, 1H), 6.21 (d, 1H, J = 6.1 Hz), 6.43 (d, 1H, J = 9.6 Hz), 6.51 (ddd, 1H, J = 9.6, 5.5 and 1.0 Hz), 6.58–6.62 (m, 2H), 6.67 (d, 1H, J = 9.6 Hz). 13C-NMR (CDCl3, 125 MHz): δ = 52.2, 52.4, 65.4, 81.4, 91.8, 123.3, 127.4, 131.4, 134.4, 168.6, 172.3 ppm. HRMS (ESI+) m/z [M + Na]+ calculated for C11H13NO4Na 246.0742, found 246.0735.
4. Conclusions
In summary, we reported a practical (scalable) and instrumentally simple methodology to synthesize N-unprotected polysubstituted indoles endowed with an electron-withdrawing group at C-3 position. The importance of these N-unprotected heterocycles in organic synthesis (molecular platforms), asymmetric catalysis (chiral catalysts), medicinal chemistry (therapeutics) and materials sciences (molecular devices) ensures the practical use of this methodology in different fields of organic chemistry. Studies aimed at the use of these 3-EWG-indoles as molecular platforms for catalytic asymmetric dearomatization and their therapeutic/biological annotations are in progress in our lab.
Supplementary Materials
1H and 13C-NMR spectra for compounds 6 and 10 are available online.
Author Contributions
R.D.-R. and D.T. conducted experimental work; D.T. and F.G.-T. initiated the project, designed experiments and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Spanish Ministry of Science, Innovation and Universities (MICINN), State Research Agency (AEI) and the European Regional Development Funds (ERDF) (PGC2018-094503-B-C21).
Acknowledgments
Authors thank La Laguna University for the use of SEGAI.
Conflicts of Interest
The authors declare no conflict of interest.
References and Note
- Barluenga, J.; Valdés, C. Five-membered heterocycles: Indole and related systems. In Modern Heterocyclic Chemistry; Alvarez-Builla, J., Vaquero, J.J., Barluenga, J., Eds.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2011; pp. 377–531. [Google Scholar]
- Dhuguru, J.; Skouta, R. Role of indole scafolds as pharmacophores in the development of anti-lung cancer agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Indole: A privileged scaffold for the design of anti-cancer agents. Eur. J. Med. Chem. 2019, 183, 111691–111709. [Google Scholar] [CrossRef] [PubMed]
- Goyal, D.; Kaur, A.; Goyal, B. Benzofuran and indole: Promising scaffolds for drug development in Alzheimer’s disease. ChemMedChem 2018, 13, 1275–1299. [Google Scholar] [CrossRef] [PubMed]
- Chadha, N.; Silakari, O. Indoles as therapeutics of interest in medicinal chemistry: Bird’s eye view. Eur. J. Med. Chem. 2017, 134, 159–184. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. Indole Ring Synthesis: From Natural Products to Drug Discovery; John Wiley & Sons: Chichester, UK, 2016. [Google Scholar]
- Owczarczyk, Z.R.; Braunecker, W.A.; Garcia, A.; Larsen, R.; Nardes, A.M.; Kopidakis, N.; Ginley, D.S.; Olson, D.C. 5,10-Dihydroindolo[3,2-b]indole-based copolymers with alternating donor and acceptor moieties for organic photovoltaics. Macromolecules 2013, 46, 1350–1360. [Google Scholar] [CrossRef]
- Nie, G.M.; Bai, Z.M.; Yu, W.Y.; Zhang, L. Electrochemiluminescence biosensor for ramos cells based on a nanostructured conducting polymer composite material (PICA-MWNTs). J. Polym. Sci. Part A Polym. Chem. 2013, 51, 2385–2392. [Google Scholar] [CrossRef]
- Manickam, M.; Iqbal, P.; Belloni, M.; Kumar, S.; Preece, J.A. A brief review of carbazole-based photorefractive liquid crystalline materials. Isr. J. Chem. 2012, 52, 917–934. [Google Scholar] [CrossRef]
- Li, T.Z.; Liu, S.J.; Tan, W.; Shi, F. For a recent review on the current state of the field, see: Catalytic asymmetric construction of axially chiral indole-based frameworks: An emerging area. Chem. Eur. J. 2020, 26. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Jiang, F.; Shi, F. Organocatalytic asymmetric synthesis of indole-based chiral heterocycles: Strategies, reactions, and outreach. Acc. Chem. Res. 2020, 53, 425–446. [Google Scholar] [CrossRef]
- Ma, C.; Sheng, F.T.; Wang, H.Q.; Deng, S.; Zhang, Y.C.; Jiao, Y.; Tan, W.; Shi, F. Atroposelective access to oxindole-based axially chiral styrenes via the strategy of catalytic kinetic resolution. J. Am. Chem. Soc. 2020, 142, 15686–15696. [Google Scholar] [CrossRef]
- Lai, Y.T.; Wang, T.; O’Dell, S.; Louder, M.K.; Schön, A.; Cheung, C.S.; Chuang, G.Y.; Druz, A.; Lin, B.; McKee, K.; et al. Lattice engineering enables definition of molecular features allowing for potent small-molecule inhibition of HIV-1 entry. Nat. Commun. 2019, 10, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Huang, B.; Zhan, P.; De Clercq, E.; Liu, X. Discovery of small molecular inhibitors targeting HIV-1 gp120eCD4 interaction drived from BMS-378806. J. Eur. Med. Chem. 2014, 86, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Guo, M.; Li, X.; Jia, F.; Li, C.; Yang, Y.; Cao, M.; Jiang, N.; Ma, E.; Zhai, X.; et al. Discovery of novel indole-based allosteric highly potent ATX inhibitors with great in vivo efficacy in a mouse lung fibrosis model. J. Med. Chem. 2020, 63, 7326–7346. [Google Scholar] [CrossRef] [PubMed]
- Apirakkan, O.; Gavrilović, I.; Cowan, D.A.; Abbate, V. In vitro phase i metabolic profiling of the synthetic cannabinoids AM-694, 5F-NNEI, FUB-APINACA, MFUBINAC, and AMB-FUBINACA. Chem. Res. Toxicol. 2020, 33, 1653–1664. [Google Scholar] [CrossRef]
- Liou, J.P. 3-Aroylindoles display antitumor activity in vitro and in vivo: Effects of N1-substituents on biological activity. J. Med. Chem. 2017, 125, 1268–1278. [Google Scholar]
- Smith, A.C.; Kung, D.W.; Shavnya, A.; Brandt, T.A.; Dent, P.D.; Genung, N.E.; Cabral, S.; Panteleev, J.; Herr, M.; Yip, K.N.; et al. Evolution of the synthesis of AMPK activators for the treatment of diabetic nephropathy: From three preclinical candidates to the investigational new drug PF-06409577. Org. Process. Res. Dev. 2018, 22, 681–696. [Google Scholar] [CrossRef]
- Wu, Y.S.; Coumar, M.S.; Chang, J.Y.; Sun, H.Y.; Kuo, F.M.; Kuo, C.C.; Chen, Y.J.; Chang, C.Y.; Hsiao, C.L.; Liou, J.P.; et al. Synthesis and evaluation of 3-aroylindoles as anticancer agents: Metabolite approach. J. Med. Chem. 2009, 52, 4941–4945. [Google Scholar] [CrossRef]
- Liou, J.P.; Chang, Y.L.; Kuo, F.M.; Chang, C.W.; Tseng, H.Y.; Wang, C.C.; Yang, Y.N.; Chang, J.Y.; Lee, S.J.; Hsieh, H.P.; et al. Concise synthesis and structure-activity relationships of combretastatin A-4 analogues, 1-aroylindoles and 3-aroylindoles, as novel classes of potent antitubulin agents. J. Med. Chem. 2004, 47, 4247–4257. [Google Scholar] [CrossRef]
- Kuo, C.C.; Hsieh, H.P.; Pan, W.Y.; Chen, C.P.; Liou, J.P.; Lee, S.J.; Chang, Y.L.; Chen, L.T.; Chen, C.T.; Chang, J.Y.; et al. BPR0L075, a novel synthetic indole compound with antimitotic activity in human cancer cells, exerts effective antitumoral activity in vivo. Cancer Res. 2004, 64, 4621–4628. [Google Scholar] [CrossRef] [Green Version]
- Huck, C.J.; Sarlah, D. Shaping Molecular Landscapes: Recent Advances, Opportunities, and Challenges in Dearomatization. Chem 2020, 6, 1589–1603. [Google Scholar] [CrossRef]
- Li, K.; Gonçalves, T.P.; Huang, K.W.; Lu, Y. Dearomatization of 3-nitroindoles by a phosphine-catalyzed enantioselective [3 + 2] annulation reaction. Angew. Chem. Int. Ed. 2019, 58, 5427–5431. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, J.; Tu, Y.; Zhang, J. Phosphine-catalyzed enantioselective dearomative [3 + 2]-cycloaddition of 3-nitroindoles and 2-nitrobenzofurans. Angew. Chem. Int. Ed. 2019, 58, 5422–5426. [Google Scholar] [CrossRef] [PubMed]
- Suo, J.J.; Liu, W.; Du, J.; Ding, C.H.; Hou, X.L. Diastereo-and enantioselective palladium-catalyzed dearomative [3 + 2] cycloaddition of 3-nitroindoles. Chem. Asian J. 2018, 13, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Zhang, F.; Cai, Y.; Guo, Y.L.; You, S.L. Stereodivergent synthesis of tetrahydrofuroindoles through Pd-catalyzed asymmetric dearomative formal [3 + 2] cycloaddition. Angew. Chem. Int. Ed. 2018, 57, 2134–2138. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhu, Z.Q.; Gu, L.; Wan, X.; Mei, G.J.; Shi, F. Catalytic asymmetric dearomative [3 + 2] cycloaddition of electron-deficient indoles with all-carbon 1,3-dipoles. J. Org. Chem. 2018, 83, 2341–2348. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Tong, F.; Sun, B.B.; Fan, W.T.; Chen, J.B.; Hu, D.; Wang, X.W. Pd-catalyzed asymmetric dearomative cycloaddition for construction of optically active pyrroloindoline and cyclopentaindoline derivatives: Access to 3a-aminopyrroloindolines. J. Org. Chem. 2018, 83, 2882–2891. [Google Scholar] [CrossRef]
- Yue, D.F.; Zhao, J.Q.; Chen, X.Z.; Zhou, Y.; Zhang, X.M.; Xu, X.Y.; Yuan, W.C. Multiple hydrogen-bonding bifunctional thiourea-catalyzed asymmetric dearomative [4 + 2] annulation of 3-nitroindoles: Highly enantioselective access to hydrocarbazole skeletons. Org. Lett. 2017, 19, 4508–4511. [Google Scholar] [CrossRef]
- Gerten, L.; Stanley, L.M. Enantioselective dearomative [3 + 2] cycloadditions of indoles with azomethine ylides derived from alanine imino esters. Org. Chem. Front. 2016, 3, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.Q.; Wu, Z.J.; Zhou, M.Q.; Xu, X.Y.; Zhang, X.M.; Yuan, W.C. Zn-catalyzed diastereo-and enantioselective cascade reaction of 3-isothiocyanato oxindoles and 3-nitroindoles: Stereocontrolled syntheses of polycyclic spirooxindoles. Org. Lett. 2015, 17, 5020–5023. [Google Scholar] [CrossRef]
- Awata, A.; Arai, T. PyBidine/Copper catalyst: Asymmetric exo’-selective [3 + 2] cycloaddition using imino ester and electrophilic indole. Angew. Chem. Int. Ed. 2014, 53, 10462–10465. [Google Scholar] [CrossRef]
- Li, Y.; Tur, F.; Nielsen, R.P.; Jiang, H.; Jensen, F.; Jørgensen, K.A. Enantioselective formal [4 + 2] cycloadditions to 3-nitroindoles by trienamine catalysis: Synthesis of chiral dihydrocarbazoles. Angew. Chem. Int. Ed. 2016, 55, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.P.; Tendoung, J.J.Y.; Treguier, B. Advances in dearomatization strategies of indoles. Tetrahedron 2015, 71, 3549–3591. [Google Scholar] [CrossRef]
- Kumar, N.; Parle, A. 3-Substituted indole: A review. Int. J. Chem. Stud. 2019, 72, 848–856. [Google Scholar]
- Norwood, V.M.; Huigens, R.W. Harnessing the chemistry of the indole heterocycle to drive discoveries in biology and medicine. ChemBioChem 2019, 20, 2273–2297. [Google Scholar] [CrossRef]
- Joule, J.A. Indole and its derivatives. Chapter 10.13. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations; Thomas, E.J., Ed.; George Thieme Verlag: Stuttgart, Germany, 2000; Volume 10, pp. 361–652. [Google Scholar]
- Li, L.H.; Niu, Z.J.; Liang, Y.M. New Friedel–Crafts strategy for preparing 3-acylindoles. Org. Biomol. Chem. 2018, 16, 7792–7796. [Google Scholar] [CrossRef]
- Wu, W.; Su, W. Mild and selective Ru-catalyzed formylation and Fe-catalyzed acylation of free (NH) indoles using anilines as the carbonyl source. J. Am. Chem. Soc. 2011, 133, 11924–11927. [Google Scholar] [CrossRef]
- Guchhait, S.K.; Kashyap, M.; Kamble, H. ZrCl4-Mediated regio-and chemoselective friedel–crafts acylation of indole. J. Org. Chem. 2011, 76, 4753–4758. [Google Scholar] [CrossRef]
- Katritzky, R.; Suzuki, K.; Singh, S.K.; He, H.Y. Regiospecific C-acylation of pyrroles and indoles using N-acylbenzotriazoles. J. Org. Chem. 2003, 58, 5720–5723. [Google Scholar] [CrossRef]
- Okauchi, T.; Itonaga, M.; Minami, T.; Owa, T.; Kitoh, K.; Yoshino, H. A general method for acylation of indoles at the 3-position with acyl chlorides in the presence of dialkylaluminum chloride. Org. Lett. 2000, 2, 1485–1487. [Google Scholar] [CrossRef]
- Zhang, W.; Tang, J.; Yu, W.; Huang, Q.; Fu, Y.; Kuang, G.; Pan, C.; Yu, G. Visible light-driven C-3 functionalization of indoles over conjugated microporous polymers. ACS Catal. 2018, 8, 8084–8091. [Google Scholar] [CrossRef]
- Wu, C.J.; Meng, Q.Y.; Lei, T.; Zhong, J.J.; Liu, W.Q.; Zhao, L.M.; Li, Z.J.; Chen, B.; Tung, C.H.; Wu, L.Z.; et al. An oxidant-free strategy for indole synthesis via intramolecular C−C bond construction under visible light irradiation: Cross-coupling hydrogen evolution reaction. ACS Catal. 2016, 6, 4635–4639. [Google Scholar] [CrossRef]
- Li, X.; Gu, X.; Li, Y.; Li, P. Aerobic transition-metal-free visible-light photoredox indole C-3 formylation reaction. ACS Catal. 2014, 4, 1897–1900. [Google Scholar] [CrossRef]
- Zhang, P.; Xiao, T.; Xiong, S.; Dong, X.; Zhou, L. Synthesis of 3-acylindoles by visible-light induced intramolecular oxidative cyclization of o-alkynylated N,N-dialkylamines. Org. Lett. 2014, 16, 3264–3267. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, G.R.; Kuethe, J.T. Practical methodologies for the synthesis of indoles. Chem. Rev. 2006, 106, 2875–2911. [Google Scholar] [CrossRef]
- Neto, J.S.S.; Zeni, G. Recent advances in the synthesis of indoles from alkynes and nitrogen sources. Org. Chem. Front. 2020, 7, 155–210. [Google Scholar] [CrossRef]
- Inman, M.; Moody, C.J. Indole synthesis-something old, something new. Chem. Sci. 2013, 4, 29–41. [Google Scholar] [CrossRef]
- Tabolin, A.A.; Ioffe, S.L. Rearrangement of N-oxyenamines and related reactions. Chem. Rev. 2014, 114, 5426–5476. [Google Scholar] [CrossRef]
- Heravi, M.M.; Rohani, S.; Zadsirjan, V.; Zahedi, N. Fischer indole synthesis applied to the total synthesis of natural products. RSC Adv. 2017, 7, 52852–52887. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.L. Progress in the Fischer indole reaction. A review. Org. Prep. Proced. Int. 1993, 25, 607–632. [Google Scholar] [CrossRef]
- Bartoli, G.; Dalpozzo, R.; Nardib, M. Applications of Bartoli indole synthesis. Chem. Soc. Rev. 2014, 43, 4728–4750. [Google Scholar] [CrossRef]
- Dalpozzo, R.; Bartoli, G. Bartoli indole synthesis. Curr. Org. Chem. 2005, 9, 163–178. [Google Scholar] [CrossRef]
- Fischer, E.E.; Jourdan, F. Ueber die hydrazine der brenztraubensäure. Eur. J. Inorg. Chem. 1883, 16, 2241–2245. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, L.; Zhang, L. Combining Zn ion catalysis with homogeneous gold catalysis: An efficient annulation approach to N-protected indoles. Chem. Sci. 2013, 4, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Guo, L.; Liu, F.; Miao, Z.; Feng, L.; Gao, H. A copper-catalyzed synthesis of polysubstituted indoles using N-arylhydroxylamines and vinyliodonium salts using the same disconnection pattern has been recently described, although its application to 3-EWG-indoles was not reported. Copper-catalyzed tandem o-vinylation of arylhydroxylamines/[3,3]-rearrangement/cyclization: Synthesis of highly substituted indoles and benzoindoles. ACS Catal. 2019, 9, 3906–3912. [Google Scholar]
- Tejedor, D.; Delgado-Hernández, S.; Colella, L.; García-Tellado, F. Catalytic hydrocyanation of activated terminal alkynes. Chem. Eur. J. 2019, 25, 15046–15049. [Google Scholar] [CrossRef]
- Tejedor, D.; Álvarez-Méndez, S.J.; López-Soria, J.M.; Martín, V.S.; García-Tellado, F. A robust and general protocol for the Lewis-base-catalysed reaction of alcohols and alkyl propiolates. Eur. J. Org. Chem. 2014, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Mola, L.; Font, J.; Bosch, L.; Caner, J.; Costa, A.M.; Etxebarría-Jardí, G.; Pineda, O.; De Vicente, D.; Vilarrasa, J. Nucleophile-catalyzed additions to activated triple bonds. Protection of lactams, imides, and nucleosides with MocVinyl and related groups. J. Org. Chem. 2013, 78, 5832–5842. [Google Scholar] [CrossRef] [Green Version]
- Tejedor, D.; Santos-Expósito, A.; Méndez-Abt, G.; Ruiz-Pérez, C.; García-Tellado, F. Trialkylamine versus trialkylphosphine: Catalytic conjugate addition of alcohols to alkyl propiolates. Synlett 2009, 1223–1226. [Google Scholar] [CrossRef] [Green Version]
- Feierfeil, J.; Magauer, T. De novo synthesis of benzannelated heterocycles. Chem. Eur. J. 2018, 24, 1455–1458. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.M.A.; Prabhakar, S.; Lobo, A.M. A Synthesis of the amaryllidaceae alkaloid pratosine. J. Nat. Prod. 1996, 59, 744–747. [Google Scholar] [CrossRef]
- Hwu, J.R.; Patel, H.V.; Lin, R.J.; Gray, M.O. Novel methods for the synthesis of functionalized indoles from arylhydroxylamines and activated acetylenes. J. Org. Chem. 1994, 59, 1577–1582. [Google Scholar] [CrossRef]
- Toyota, M.; Fukumoto, K. Tandem Michael addition-[3,3]sigmatropic rearrangement processes. Part 2. Construction of cyclopropa[3,4]pyrrolo [3,2-e]indol-4-one (CPI) unit of antitumour antibiotic CC-1065. J. Chem. Soc. Perkin Trans. 1992, 1, 547–552. [Google Scholar] [CrossRef]
- Blechert, S. Hetero-Cope-rearrangements, region-controlled synthesis of indoles. Helv. Chim. Acta 1985, 68, 1835–1843. [Google Scholar] [CrossRef]
- Chehardoli, G.; Bahmani, A. Synthetic strategies, SAR studies, and computer modeling of indole 2 and 3-carboxamides as the strong enzyme inhibitors: A review. Mol. Divers. 2020, 1–16. [Google Scholar] [CrossRef]
- Grandberg, I.; Belyaeva, L.D.; Dmitriev, L.B. Ratio of indole isomers formed during the fischer cyclization of meta-substituted diethyl ketone phenylhydrazones. Chem. Het. Compd. 1971, 7, 1131. [Google Scholar] [CrossRef]
- Krenske, E.H.; Burns, J.M.; McGeary, R.P. Claisen rearrangements of benzyl vinyl ethers: Theoretical investigation of mechanism, substituent effects, and regioselectivity. Org. Biomol. Chem. 2017, 15, 7887–7893. [Google Scholar] [CrossRef]
- An ortho-methyl group dropped the yield of the reaction to 25%.
- Lee, K.N.; Lei, Z.; Morales-Rivera, C.A.; Liu, P.; Ngai, M.Y. Mechanistic studies on intramolecular C–H trifluoromethoxylation of (hetero)arenes via OCF3-migration. Org. Biomol. Chem. 2016, 14, 5599. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.Q.; Marchini, M.; Xiao, W.J.; Ceroni, P.; Bandini, M. Visible-light-induced direct photocatalytic carboxylation of indoles with CBr4/MeOH. Chem. Eur. J. 2015, 50, 18052–18056. [Google Scholar] [CrossRef]
- Abe, T.; Yamada, K. Amination/cyclization cascade by acid-catalyzed activation of indolenine for the one-pot synthesis of phaitanthrin E. Org. Lett. 2016, 18, 6504–6507. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, C.; Yang, X.; Su, W. Decarboxylative acylation of N-free indoles enabled by a catalytic amount of copper catalyst and liquid-assisted grinding. Org. Biomol. Chem. 2019, 17, 4446–4451. [Google Scholar] [CrossRef]
- Wynne, J.H.; Lloyd, C.T.; Jensen, S.D.; Boson, S.; Stalick, W.M. 3-Acylindoles via a one-pot, regioselective Friedel-Crafts reaction. Synthesis 2004, 2277–2282. [Google Scholar] [CrossRef]
- Peng, J.; Liu, L.; Hu, Z.; Huang, J.; Zhu, Q. Direct carboxamidation of indoles by palladium-catalyzed C–H activation and isocyanide insertion. Chem. Commun. 2012, 48, 3772–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velavan, A.; Sumathi, S.; Balasubramanian, K.K. AlMe3-mediated region- and chemoselective reactions of indole with carbamoyl electrophiles. Eur. J. Org. Chem. 2013, 3148–3157. [Google Scholar] [CrossRef]
- Yamazaki, K.; Nakamura, Y.; Kondo, Y. Solid-phase synthesis of indolecarboxylates using palladium-catalyzed reactions. J. Org. Chem. 2003, 68, 6011–6019. [Google Scholar] [CrossRef]
- Linton, E.C.; Kozlowski, M.C. Catalytic enantioselective Meerwein−Eschenmoser Claisen rearrangement: Asymmetric synthesis of allyl oxindoles. J. Am. Chem. Soc. 2008, 130, 16162–16163. [Google Scholar] [CrossRef]
- Fisher, M.J.; McMurray, L.; Lu, S.; Morse, C.L.; Liow, J.S.; Zoghbi, S.S.; Kowalski, A.; Tye, G.L.; Innis, R.B.; Aigbirhio, F.I.; et al. [Carboxyl-11 C]labelling of four high-affinity cPLA2α inhibitors and their evaluation as radioligands in mice by positron emission tomography. ChemMedChem 2018, 13, 138–146. [Google Scholar] [CrossRef]
- Natarajan, R.; Rappa, J.P.; Unnikrishnan, P.A.; Radhaman, S.; Prathapan, S. A new method for the synthesis of 3-substituted indoles. Synlett 2015, 26, 2467–2471. [Google Scholar]
- Zhang, Z.W.; Xue, H.; Li, H.; Kang, H.; Feng, J.; Lin, A.; Liu, S. Collective synthesis of 3-acylindoles, indole-3-carboxylic esters, indole-3-sulfinic acids, and 3-(methylsulfonyl)indoles from free (N-H) indoles via common N-indolyl triethylborate. Org. Lett. 2016, 18, 3918–3921. [Google Scholar] [CrossRef]
- Ji, X.; Huang, H.; Wu, W.; Li, X.; Jiang, H. Palladium-catalyzed oxidative coupling of aromatic primary amines and alkenes under molecular oxygen: Stereoselective assembly of (Z)-enamines. J. Org. Chem. 2013, 38, 11155–11162. [Google Scholar] [CrossRef]
- Rashad, M.; La Vecchia, L.; Prasad, K.; Repic, O. A convenient synthesis of 3-substituted 1H-indoles. Synth. Commun. 1995, 25, 95–100. [Google Scholar] [CrossRef]
- Yoo, W.J.; Capdevila, M.G.; Du, X.; Kobayashi, S. Base-mediated carboxylation of unprotected indole derivatives with carbon dioxide. Org. Lett. 2012, 14, 5326–5329. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 6a–ad are available from the authors. |
Scheme 1.
Strategy for the synthesis of indoles 6 with electron-withdrawing group at the C-3 position (3-EWG indoles). (a) General [3,3] sigmatropic disconnection. (b) Zn/Au- catalyzed synthesis of indoles. (c) This work: consecutive two-step reaction manifold.
Scheme 1.
Strategy for the synthesis of indoles 6 with electron-withdrawing group at the C-3 position (3-EWG indoles). (a) General [3,3] sigmatropic disconnection. (b) Zn/Au- catalyzed synthesis of indoles. (c) This work: consecutive two-step reaction manifold.

Scheme 2.
DABCO-catalyzed reaction of N-Benzoyl-N-phenylhydroxylamine (4a-N-Bz) and methyl propiolate (5a).
Scheme 2.
DABCO-catalyzed reaction of N-Benzoyl-N-phenylhydroxylamine (4a-N-Bz) and methyl propiolate (5a).

Scheme 3.
Coupling reduction and cascade condensation/rearrangement/elimination reactions in a consecutive 2-step protocol.
Scheme 3.
Coupling reduction and cascade condensation/rearrangement/elimination reactions in a consecutive 2-step protocol.

Scheme 4.
Alkyne scope of the protocol. Reaction conditions: N-phenylhydroxylamine (4a) (4.0 mmol), alkyne 5 (4.2 mmol), DABCO (5 mol%), CH2Cl2 (20 mL), 0 °C to RT, 1 h. a Overnight.
Scheme 4.
Alkyne scope of the protocol. Reaction conditions: N-phenylhydroxylamine (4a) (4.0 mmol), alkyne 5 (4.2 mmol), DABCO (5 mol%), CH2Cl2 (20 mL), 0 °C to RT, 1 h. a Overnight.

Scheme 5.
Nitroarene scope of the protocol. Reaction conditions: 1) nitroarene 3 (4 mmol), NH2NH2.H2O (4.4 mmol), Rh/C (5%) (20 mg), THF (20 mL), 0 °C to RT, 3 h, filtration on celite plug and concentration; 2) methyl propiolate (5a) (4.2 mmol), DABCO (5 mol%), CH2Cl2 (20 mL), 0 °C to RT, 1 h. a50 mmol scale. b10s (21%). c10t (34%). d10u (27%). e10z (22%) (See Scheme 6 for structures).
Scheme 5.
Nitroarene scope of the protocol. Reaction conditions: 1) nitroarene 3 (4 mmol), NH2NH2.H2O (4.4 mmol), Rh/C (5%) (20 mg), THF (20 mL), 0 °C to RT, 3 h, filtration on celite plug and concentration; 2) methyl propiolate (5a) (4.2 mmol), DABCO (5 mol%), CH2Cl2 (20 mL), 0 °C to RT, 1 h. a50 mmol scale. b10s (21%). c10t (34%). d10u (27%). e10z (22%) (See Scheme 6 for structures).

Scheme 6.
Alternative [3,3]-sigmatropic rearrangement outcome of ortho-substituted N-oxyenamines.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of the reaction conditions.

| entry | DBACO | Temperature | Time | Yield (6a) |
|---|---|---|---|---|
| 1 | 10 mol% | RT | 1 h | 68% |
| 2 | 10 mol% | 0 °C to RT | 1 h | 83% |
| 3 | 1 mol% | 0 °C to RT | Overnight | 75% |
| 4 | 5 mol% | 0 °C to RT | 1 h | 82% |
| 5 | 5 mol% | −25 °C to RT | 2 h | 86% |
Red: To highlight that these are the best conditions.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tejedor, D.; Diana-Rivero, R.; García-Tellado, F. A General and Scalable Synthesis of Polysubstituted Indoles. Molecules 2020, 25, 5595. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235595
AMA Style
Tejedor D, Diana-Rivero R, García-Tellado F. A General and Scalable Synthesis of Polysubstituted Indoles. Molecules. 2020; 25(23):5595. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235595
Chicago/Turabian StyleTejedor, David, Raquel Diana-Rivero, and Fernando García-Tellado. 2020. "A General and Scalable Synthesis of Polysubstituted Indoles" Molecules 25, no. 23: 5595. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235595