
Plasmodial Kinase Inhibitors Targeting Malaria: Recent Developments


Aix Marseille Univ, CNRS, ICR UMR 7273, Equipe Pharmaco-Chimie Radicalaire, Faculté de Pharmacie, 13385 Marseille CEDEX 05, France
*
Authors to whom correspondence should be addressed.
Molecules 2020, 25(24), 5949; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245949
Submission received: 10 November 2020
/
Revised: 11 December 2020
/
Accepted: 11 December 2020
/
Published: 15 December 2020
(This article belongs to the Special Issue Kinase Inhibitors 2021)
Abstract
:Recent progress in reducing malaria cases and ensuing deaths is threatened by factors like mutations that induce resistance to artemisinin derivatives. Multiple drugs are currently in clinical trials for malaria treatment, including some with novel mechanisms of action. One of these, MMV390048, is a plasmodial kinase inhibitor. This review lists the recently developed molecules which target plasmodial kinases. A systematic review of the literature was performed using CAPLUS and MEDLINE databases from 2005 to 2020. It covers a total of 60 articles and describes about one hundred compounds targeting 22 plasmodial kinases. This work highlights the strong potential of compounds targeting plasmodial kinases for future drug therapies. However, the majority of the Plasmodium kinome remains to be explored.
1. Introduction
Malaria is the deadliest parasitic disease with an estimated 405,000 deaths and 213 million cases in 2018 [1], mainly in Africa. Of the estimated victims, 67% are children below five years old. The disease is caused by five Plasmodium species: P. falciparum (responsible for most of the deaths), P. vivax and three less important species, P. ovale, P. malariae and P. knowlesi. With the use of artemisinin-based combined therapies (ACT), combining an artemisinin derivate and another antimalarial drug, and vector control measures, the Word Health Organization hoped that 2020 would see a 40% reduction in malaria cases and deaths compared to 2015 [2]. However, this objective is not going to be achieved.
The vector, mosquitoes from the Anopheles genus, is becoming resistant to pyrethroid insecticides (used in long-lasting insecticide nets) in Africa [3]. Moreover, mutations on the Pfkelch13 protein, plasmodial protein recently described to be involved in hemoglobin endocytosis mechanisms [4], mediate resistance to artemisinin and its derivates. Pfkelch13 mutations are linked [5]:
- In vitro to a decreased susceptibility during a ring stage assay,
- In vivo to a delayed parasitic clearance.
Increased prevalence of these mutations in South-East Asia is leading to clinical failures of ACT treatments [6]. Millions of people in Africa are threatened: recent researches indicate the appearance of in vitro artemisinin resistance in Rwanda [7], and the spread of city-dwelling A. stephensi in Africa [8]. Thus, research and development aimed at finding new drug therapies with novel mechanisms of action is a priority. Medicines for Malaria Venture, a non-profit organization, aims to develop new antimalarial drug therapies. To guide drug discovery, target candidate profiles (TCP) have been defined for antimalarial molecules setting clear goals for new therapy [9]. There are five TCP: blood-stage killer, hypnozoites killer, hepatic schizonts killer (chemoprotection), gametocytes killer, and transmission blocker. The best compounds are the one following multiple or every TCP. One of these drug candidates, MMV390048, currently in phase II clinical trial, is the first plasmodial kinase inhibitor to reach this stage, opening the way for antimarial drugs targeting this type of protein.
Kinases are proteins catalyzing the addition of a phosphate group on a substrate, like a simple molecule or a protein. In the latter, the kinase, designed under the term of “protein kinase”, will modified the activity of the targeted protein after addition of the phosphate group. The complete set of encoded protein kinases in the genome of an organism is defined as a kinome. The human kinome consists of 518 encoded kinases [10] that play a role in the regulation of cellular processes, and whose dysregulation is involved in numerous cancers [11]. The two first human kinase inhibitors approved by the Food and Drug Administration (FDA) were rapamycin in 1999 (indicated for immunosuppression) and imatinib in 2001. Importantly, the small molecule imatinib inhibits the BCR-Abl chimeric protein, found mainly in chronic myeloid leukemia. This finding accelerated research on human kinase inhibitors and as of 1 March 2019, 48 small molecules designed as human kinase inhibitors were approved by the FDA [12], the majority of them used in oncology. According to PKIDB (a website tracking human kinase inhibitors in clinical trials), as of July 2020, 253 human kinase inhibitors were currently in clinical trials [13].
Mapping of the P. falciparum kinome, depending on the literature source, indicates 85 (65 for eukaryotic protein kinases (ePKs) and 20 for ePKs-related proteins) [14] or 99 [15] encoding genes, and a significant number of these proteins do not possess a human ortholog [16]. These kinases are highly conserved among the different Plasmodium species. Moreover, 36 plasmodial kinases (in P. falciparum) are identified as likely to be essential for the asexual blood-stage [17], and 15 kinases are known to be involved in P. berghei development in mosquitoes [18]. Thus, developing selective plasmodial kinase inhibitors could lead to multiple new antimalarial drugs with new mechanisms of action.
This review of the literature lists molecules proven to be inhibitors of plasmodial kinases and describes their properties.
2. Methods
This systematic review complies with the PRISMA statement defining the PRISMA checklist and PRISMA flowchart [19].
2.1. Data Sources and Search Parameters
Two databases were used: Scifinder (http://scifinder.cas.org, CAPLUS and MEDLINE database) and Pubmed (https://pubmed.ncbi.nlm.nih.gov/, MEDLINE database). In both databases, the keywords and Boolean operator “kinase inhibitor AND malaria” were used. The advanced search options were set to “journal” for document type and English or French for language. The research was conducted on articles published from 1 January 2005 to 1 August 2020.
2.2. Article Selection
Articles were selected by one reviewer, with two rounds of selection. During the first round, for each database set, articles’ titles and abstracts were assessed to eliminate irrelevant material (outside medicinal chemistry or biological fields, non-malaria related, non-filtered reviews, duplicates). To be discussed in the following review, articles had to follow the inclusion and exclusion criteria listed in Table 1.
3. Results
Four hundred and fifty-six articles from Scifinder (after applying the “remove duplicates” tool) and 366 articles from Pubmed were obtained. During the first round of selection, 348 Scifinder articles and 286 Pubmed articles were excluded.
The two sets were combined, and duplicates removed, leaving 121 articles. During the second round of selection, 64 articles were excluded leading to 57 selected articles; three additional articles were added in the dataset while searching for further protein information.
4. Discussion
4.1. Molecules Targeting P. falciparum Calcium-Dependant Protein Kinases (PfCDPKs)
CDPKs can be found in plants and alveolate protists including Apicomplexan parasites like T. gondii and Plasmodium spp [20]. They belong to the calmodulin-dependent kinases (CaMK) family [14]. The CDPK proteins, possessing seven members (CDPK1 to CDPK7), are involved in multiple parasitic stages [21], and constitute interesting targets as they are not found in humans. Only inhibitors for PfCDPK1 and PfCDPK4 have been described in the literature.
4.1.1. Molecules Targeting P. falciparum Calcium-Dependent Protein Kinase 1 (PfCDPK1)
As discovered by Zhao et al. [22], PfCDPK1 is involved in the invasion of red blood cells (RBC) by merozoites and their egress through activation of a protein motor complex [23,24]. More recent studies indicate the role of PfCDPK1 in gametogenesis, mosquito infection [25], and regulation of PfPKA (P. falciparum (cAMP)-dependent protein kinase) [26]. PfCDPK1 was assumed to be an essential protein for Plasmodium since knocking out the gene during blood-stages was impossible [24]. Bansal et al., starting from a P. falciparum strain with a mutant PfCDPK1 having reduced activity, recently managed to knock out completely PfCDPK1 [25].
In 2008, Green et al. [23] used compound K252a (Figure 1, compound 1) to study the motor complex regulated by PfCDPK1. This indolopyrrolocarbazole displayed an IC50 of 45 nM toward PfCDPK1 and was able to reduce RBC invasion by merozoites with an EC50 of 348 nM.
Kato et al. screened a library of 20,000 compounds on PfCDPK1 [24]. The most active class of compounds was a series of 2,6,9-trisubstitued purines and the most potent molecule, called purfalcamine (Figure 1, compound 2), showed an IC50 value of 17 nM on PfCDPK1. EC50 in a parasitic proliferation assay on multiple P. falciparum strains including a multi-resistant W2 strain was determined, as well as cytotoxic values on multiple cell lines. However, the compound lacked activity in vivo in the four-day Peter’s test [27]: only a delay in onset of parasitemia was observed. Other compounds from the 2,6,9-trisubstituted purine series lacked PfCDPK1 affinity or P. falciparum in vitro activity when the fluorine atom was replaced by a carboxylic acid or a tert-butoxy group. P. falciparum in vitro activity was also suppressed when the 4-aminocyclohexyl was replaced by a 4-hydroxycyclohexyl group.
In 2009, Lemercier et al. identified two PfCDPK1 inhibitors after a screening of 54,000 compounds [28]: the indolizine 3 and the imidazopyridazine 4 (Figure 2). 3 displayed a Ki of 262 nM on PfCDPK1, while 4 possessed a Ki of 37 nM on PfCDPK1 and an IC50 on P. falciparum of 5.7 µM. Out of 46 human kinases tested, compound 3 inhibited two human kinases and compound 4, five (at 10 µM concentration). Modulation work focusing on the phenolic hydroxyl group of 4 showed a tolerance on the Ki value when this 3-hydroxyl was replaced with H-bond acceptors like 3,4-methylenedioxyl or 3-methoxyl, but in the latter accompanied with a 4-hydroxyl substitution.
Screening of 35,000 compounds by Chapman et al. [29] displayed imidazopyridazine as a promising series, in line with the discovery of compound 4. An extensive Structure-Activity Relationship (SAR) work started on this scaffold (Table 2). Chapman et al. [30] explored the side chains at positions 3 and 6 of the imidazopyridazine core. Work on the R1 substituent largely targeted the introduction of cyclic amines, while work on R2 addressed para- or meta-functionalized phenyls, pyridines, or pyrimidines. Compound 5 (Table 2) displayed interesting results with an IC50 on PfCDPK1 of 13 nM, an IC50 on Pf3D7 of 400 nM, and the ability to reduce by 46% the parasitemia, in a P. berghei mouse model per os during a four-day Peter’s test. Finally, 5 showed inhibition toward 12 human kinases (out of 73 tested) ranging between 50% and 80% at 1 µM.
Large et al. continued this work with the introduction of N-substituted imidazoles at position 3 of the imidazopyridazine core while modulating position 6 with substituents like aminocyclohexyl or N-methylpiperidine [31]. This resulted in the synthesis of compound 6 (Table 2) showing an improved activity on Pf3D7 compared to compound 5 and improved ligand-lipophilicity efficiency (LLE) with a score of 6, compared to 4.5 for compound 5. Inhibition assays on human kinases were not performed on compound 6, but other compounds from this series showed lower selectivity than compound 5.
The last work on this series was done by Chapman et al. [32] using docking studies on the T. gondii CDPK1 ortholog structure to guide them. Starting from 5, they started by modulating R2 with N-substituted 2-aminopyrimidines, hoping for improved in vivo results. Compound 7 (Table 2) emerged as the best compound with improved in vitro parameters. However, low PAMPA (parallel artificial membrane permeability assay) permeability (4 Papp/nms−1) led to poor results in vivo, with only 4% reduction of parasitemia in the four-day Peter’s test [27]. Out of 66 human kinases tested at 1 µM concentration, 7 inhibited nine human kinases by at least 80%. Additional work was then done: the side chain at position 6 was modulated with different cyclic amines, and at position 3, the fluorine atom position on both cycles was explored along with the nature of the two cycles (phenyls or pyridines). This led to compound 8 (Table 2) with slightly lower activity in vitro than 7. The reduction of parasitaemia in P. berghei mouse model was slightly better at 51% than that of 5 at 46% (Table 2).
In 2016, Crowther et al. screened 14,000 compounds on multiple plasmodial kinases including PfCDPK1 [33]. One hundred and eighty-one molecules showed a sub-micromolar IC50 on PfCDPK1 and 12 compounds grouped in four chemical series displayed an IC50 equal to or below 20 nM. The best compounds from each series are represented in Figure 3.
4.1.2. Molecules Targeting Plasmodium falciparum Calcium-Dependent Protein Kinase 4 (PfCDPK4)
PfCDPK4 is a key enzyme for the exflagellation of gametocytes, a mechanism leading to the formation of microgametocytes, the male gametocytes, in the mosquito midgut [34,35,36].
In 2012, Ojo et al. described the properties of BKI-1 (compound 13), a disubstituted pyrazolo[3,4-d]pyrimidin-4-amine [36] (Figure 4). 13 displayed an IC50 on PfCDPK4 of 4.1 nM, an IC50 on PfCDPK1 of 136 nM, an EC50 toward asexual stages of 2 µM (unsurprisingly since CDPK4 is important in the sexual stage) and was able to inhibit P. falciparum exflagellation with an EC50 of 35 nM. 13′s selectivity was assessed with IC50 measures on HsABL and HsSRC, two human kinases, who were superior to 50 and 20 µM, respectively. Lastly, on a P. berghei-infected mouse model, 13 was able to completely inhibit the formation of oocysts in mosquitoes at the intraperitoneal dose of 10 mg/kg.
Ojo et al. then carried out minor pharmacomodulations on 13 [37] including the replacement of the piperidine with a N-methylpiperidine, morpholine or pyrane heterocycle, replacement of the naphthalene by a quinoline and replacement of the methoxy group by an ethoxy group. Activities on the enzyme and the parasite were maintained for all molecules except the quinoline analog without a methoxy group. Compound 14, the N-methypiperidine analog of 13 (Figure 4) showed similar in vitro activity parameters to 13 but clear improvements in in vitro and in vivo absorption-distribution-metabolism-elimination-toxicology (ADMET) parameters (Table 3). Selectivity against human kinases was conserved as only one protein out of the 80 assessed was found to be inhibited. The only downside to 14 was its hERG channel activity, which was reduced, from 0.767 µM to more than 10 µM, by replacing the N-methylpiperidine with a pyrane.
This work was followed by a massive medicinal chemistry study by Vidadala et al. focused on the modulation of the lateral chains of the pyrazolo[3,4-d]pyrimidin-4-amine core [38]. ATP binding sites of both PfCDPK1 and PfCDPK4 are very similar to the one found in T. gondii CDPK1. Thus TgCDPK1 was used for docking studies with previously described PfCDPK4 inhibitors to guide pharmacomodulations. The C3 side-chain was the first to be modulated while keeping an isopropyl or tert-butyl group as N-substituent. The best C3 group was then used during the modulation of the N-substituent. A scaffold hopping strategy was also carried out, changing the pyrazolopyrimidine to an imidazopyrazine core. This led to compound 15 with an interesting dual activity on PfCDPK1 and PfCDPK4 (Figure 4) and a conserved selectivity versus HsSRC (IC50 > 10 µM). The scaffold hopping strategy was successful, as illustrated by compound 16. The authors stated that they are now seeking to enhance the metabolic stability of their compounds in vivo, since they need to remain in the bloodstream for a period of three to four weeks to have their effect on gametocytes.
In 2016, Huang et al. used on 5-aminopyrazole-4-carboxamides, known to work on Toxoplasma gondii and Cryptosporidium parvum CDPK1 (orthologs of PfCDPK4), to obtain new PfCDPK4 inhibitors [39]. They synthesized 28 compounds; most of which have IC50 toward PfCDPK4 below 100 nM. The best compound regarding PfCDPK4 inhibition was 17, while 18 was interesting with a dual PfCDPK1/4 inhibition (Figure 5). These two compounds did not possess any activity on hERG channels. 17 was selective as it possessed an IC50 superior to 30 µM against HsSRC. Work on this series is currently focused on C. parvum therapy [40].
In the Crowther et al. screening [33], 55 compounds showed an activity below the micromolar range against PfCDPK4. The best compound (19) displayed an IC50 of 21 nM (Figure 5).
The only thing that prevents PfCDPK4 inhibitors from entering more advanced pre-clinical studies is the long-term bioavailability they would need to be active against gametocytes. Moreover, finding from Vidadala et al. [38] and data from screening by Crowther et al. [33], identified compounds providing dual inhibition of PfCDPK1 and PfCPDK4, offering the prospect of compounds with action on two stages out of three of the Plasmodium cycle.
4.2. Molecules Targeting Plasmodium falciparum Choline Kinase (PfCK)
Choline kinase is an enzyme involved in the synthesis of phosphatidylcholine; phospholipid most frequently found in P. falciparum. It transforms choline into phosphocholine, and is also involved in the synthesis of phosphatidylethanolamine, the second most common phospholipid [41]. Inhibition of PfCK has been shown to reduce parasite growth in vitro [42,43]. The active sites of PfCK have 69% similarity with the human enzyme HsCKα1 [44].
In 2007, Choubey et al. [45] studied hexadecyltrimethylammonium bromide (compound 20, Figure 6). It displayed in vitro, at 10 µM and 20 µM, parasitic growth inhibition of 62% and 81%, and reduced phosphocholine synthesis by 57% at 10 µM. In a P. yoelii mouse model, it was able to reduce parasitemia by almost 50% after four days of intravenous doses of 5 mg/kg.
A screening by Crowther et al. on around 5000 commercially available small molecules led to the discovery of three hits against PfCK [46] (compounds 21, 22 and 23, Figure 7).
While studying PfCK metabolic activities, Serrán-Aguilera et al. used two compounds (24 and 25, Table 4) to inhibit the enzyme [41], which possessed a Ki between 30 and 40 µM (when assessed for choline conversion to phosphocholine). To explore the activity of these pyridinium salts on PfCK, Schiafino-Ortega et al. assessed 1,2-diphenoxyethane salts with a symmetrical structure [47] close to 24 and 25. Bisquinolinium bromide salts derivatives were the most potent with submicromolar activity on P. falciparum. However, their activity on the PfCK enzyme was found to be above 2 µM for all this series, raising doubts about their real mechanism of action. The best compound of this series was compound 26, with an improved PfCKCho IC50 (formation of phosphocholine is measured) reduced from 103 to 2.4 µM, but it no longer showed P. falciparum in vitro activity (Table 4). Compound 26 was not selective as it also inhibited the human HsCKα1 enzyme.
Most of the molecules presented above (from 21 to 25) probably target more than just PfCK, as enzyme IC50 values not in line IC50 on the parasite: 24 is the best example of such behavior. Furthermore, the design of these compounds needs to take into account the human choline kinase. Thus, PfCK inhibitors are potentially valuable compounds, they are still in the early stages of development and promising hits have yet to be discovered.
4.3. Molecules Targeting Plasmodium falciparum Casein Kinase 2 (PfCK2)
Casein kinases are serine/threonine protein kinases found in eukaryotic organisms. Two casein kinases can be found in P. falciparum: CK1 and CK2. PfCK2 is thought to be a key enzyme during the asexual blood-stage of the parasite. It has been shown that many proteins are possibly phosphorylated, and thus have their activity regulated, via PfCK2 during the asexual blood-stage [48]. Both PfCK1 and PfCK2 have been demonstrated to be essential for the asexual blood-stage [17,49].
4.4. Molecules Targeting Plasmodium falciparum Cyclin-Dependent-Like Kinase (PfCLK)
The CLK family includes four enzymes (PfCLK1 to PfCLK 4) involved in the phosphorylation (and the activity modulation) mainly of serine-arginine-rich proteins found in spliceosomes [14,52,53]. Spliceosomes are complexes of proteins involved in the removal of introns in pre-messenger RNA. All PfCLKs are considered essential for the asexual blood-stage [17]. Inhibition of CLKs was first linked to a schizonticide and gametocytocidal effect [53]. A more recent study on PfCLK3 showed that inhibition of this protein affected all three stages of the malaria cycle [54].
In 2014, Kern et al. studied the effect of inhibiting PfCLKs [53]. The authors did not manage to disrupt the genes directly and therefore turned to chemical compounds. They screened 63 HsCLK inhibitors against PfCLKs. Two compounds, 28 and chlorhexidine (29) showed interesting results regarding PfCLK inhibition and Pf3D7 growth inhibition; results are summarized in Table 5.
In 2017, Bendjeddou et al. tested an imidazopyridazine series on multiple human and parasitic kinases, including PfCLK1 [55]. Two compounds (30 and 31, Figure 9) showed activity on PfCLK1 below 100 nM but lacked selectivity as they also targeted human kinases, in some cases with IC50 below 100 nM.
More recently, Alam et al. screened 30,000 compounds on PfCLK1 and PfCLK3 [54]. TCMDC-135793 (compound 32) and TCMDC-135051 (compound 33) were among the most potent and selective compounds on PfCLK1 and PfCLK3, respectively (Figure 10). Target was confirmed for 33 with PfDd2 strain possessing a mutant PfCLK3 that showed an increased IC50 value. Compund 33 was active at all stages, inhibiting:
- Schizont development in blood-stage,
- Sporozoite invasion and development in the liver stage,
- Gametocyte development,
- Exflagellation in the mosquito midgut.
SAR work associated with 33 was carried out by Mahindra et al. [56]. Modulations targeting the substituents of the phenyl ring at position 2 of the pyrrolo[2,3-b]pyridine core led to analogs possessing an IC50 against PfCLK3 below 100 nM. However, micromolar activity was found against Pf3D7 when the diethylamine was changed to dimethylamine, primary amine or morpholine substituents. The position of the methoxy substituent appeared to be important for activity. Similar modifications were carried out on the other phenyl ring: removing the isopropyl substituent or replacing it by methyl led to compounds with micromolar activities against Pf3D7, and removing the carboxylic acid or changing it to an ethyl ester resulted in coumpounds with micromolar activity on PfCLK3. Replacing the carboxylic acid to a tetrazole, a known bioisoster, led to compound 34 showing a slight improvement in PfCLK3 activity, with an IC50 of 19 nM but with an increased Pf3D7 IC50 to 270 nM. 34 stability was conserved in vitro in mouse liver microsomes with an intrinsic clearance at 2.32 mL/min/g of liver, compared to 33 with 1.33.
4.5. Molecules Targeting Coenzyme A Synthesis Pathway Kinases: Plasmodium falciparum Pantothenate Kinase and Dephospho-Coenzyme A Kinase (PfPanK & PfDPCK)
Coenzyme A is a molecule found in many metabolic pathways in eukaryotic life forms. In P. falciparum, coenzyme A is synthesized in five steps starting from pantothenate; two of these steps involve kinases:
- the first step, transforming pantothenate into 4′-phosphopantothenate, is catalyzed by PfPanK,
- the last step, transforming dephospho-coenzyme A into coenzyme A, is catalyzed by PfDPCK.
While the synthesis and the function of coenzyme A are well preserved around all organisms that possess it [57], the enzymes’ coding sequences can differ.
Studies on the inhibition of PfPanK focused on the synthesis of pantothenate (Table 6, compound 35) analogs able to competitively inhibit 4′-phosphopantothenate synthesis. Spry et al. synthesized ten analogs with various chains on the nitrogen atom of the amide group [58]. Compound 36 was the best of the series, with an IC50 of 76 µM on Pf3D7 (at 1 µM pantothenate concentration). Increasing the pantothenate concentration increased this IC50 suggesting a mechanism of action related to the pantothenate metabolism. Changing the terminal group of the chain or its length did not affect the activity. Spry et al. recently investigated the synthesis of analogs of CJ-15,801 (37) [59], a trans enamide analog of pantothenate. Modulations were focused on the ester and modifying the diol side chain. Compound 38 showed an improved IC50 of 13 µM on Pf3D7 compared to 36 µM for 37 but most importantly a 100-fold decreased of the PfPanK IC50 value. In 2017, Chiu et al. described compound 39 as a pantothenate kinase inhibitor with an IC50 of 30 nM on PfW2 [60]. Adding more pantothenate to the growing medium increased this value. The authors also demonstrated that 39 was able to inhibit the PfPanK homolog of S. cervisiae, Cab1.
Concerning PfDPCK, work by Fletcher et al. identified some potent compounds [61]. The activity of assessed compounds on P. falciparum was counterbalanced by adding coenzyme A to the culture medium to identified compounds active on coenzyme A synthesis. Similar rescue tests were then done with PfPanK or PfDPCK substrates. Four compounds (40–43, Figure 11) showed possible activity on PfDPCK, affecting the blood-stage and gametocytes.
4.6. Molecules Targeting Plasmodium falciparum FIKK Kinases (PfFKks)
FIKK kinases are named after a shared sequence of four amino acids (Phe-Ile-Lys-Lys or FIKK) and are an important group of kinases in malaria parasites [14]. While P. vivax and P. berghei possess one FIKK kinase, P. falciparum differs completely, possessing 20 FIKK kinases. The FIKK kinase gene may be essential for the parasite [18], yet the role of these proteins remains unclear. PfFKks are exported into the RBC cytosol (except PfFKk8), where they possibly interact with RBC’s cytoskeletal proteins and membrane [62,63].
The only studies on inhibitors of FKks were performed by Lin et al. [64,65], who screened compounds on PfFKk8 and P. vivax FKk (catalytic domain only for the latter). In both articles, emodin (44, Figure 12), an anthraquinone, showed an IC50 of 1.9 µM and 2 µM against these tested proteins, respectively.
4.7. Molecules Targeting Plasmodium falciparum Guanylate Kinase (PfGK)
Guanylate kinase catalyzes the transformation of (deoxy)guanosine-monophosphate into (deoxy)guanosine-diphosphate. However, this transformation can potentially be bypassed by P. falciparum thymidylate kinase, which can also catalyze the PfGK reaction [66]. In their screening [46], Crowther et al. included guanylate kinase as one of their screened proteins but used PvGK (possibly because guanylate kinase expression is greater at liver stage [67]). Three compounds emerged as potential hits (45 to 47, Figure 13), but as the authors stated, structural features do not currently make them good candidates for further medicinal chemistry studies.
4.8. Molecules Targeting Plasmodium falciparum Glycogen Synthase Kinase 3 (PfGSK3)
PfGSK3 is possibly an essential enzyme for the parasite asexual blood-stage [17] but its implication in biological pathways is not clear. One of its phosphorylation targets, the apical membrane antigen 1 (AMA1) [68], is required for RBC invasion by merozoites. PfGSK-3 possesses a human ortholog, HsGSK3. HsGSK3 hyperactivation is linked to Alzheimer’s disease, diabetes, or cancers and many compounds inhibiting HsGSK3 have been described [69].
In 2013, Fugel et al. realized a high-throughput screening of 10,000 molecules on PfGSK3 [70]. Multiple molecules with a thieno[2,3-b]pyridine core were found to be hits against PfGSK3. Forty-three analogs from four different thieno[2,3-b]pyridines scaffolds were synthesized. The 3,6-diamino-4-(2-halophenyl)-2-benzoylthieno[2,3-b]pyridine-5-nitriles showed the best results on PfGSK3 and Sus scrofa GSK3 (used for selectivity comparison). HsGSK3 IC50 and PfNF54LUC EC50 tests were done on the best compound, 48 (Table 7).
Based on a docking study of 48 on an analogy model of PfGSK3, Masch el al. synthesized 23 analogs of 48 with different substituents (halogens, ethers, alkylated amines, cyclic amines) at position 4 of the 2-chlorophenyl cycle [71] (Table 7). Compound 49 showed lower activity on PfGSK3 but its potency on in vitro parasites was improved compared to 48. Aqueous solubility was improved, going from 1.5 µM for 48 to 4.8 µM for 49. The authors explored the possible axial chirality created by the 2-chlorophenyl cycle. They isolated the two isomers of 50 and compared their activity: (+)-50 was active but not (−)-50. According to the authors, this difference arose from the chlorine atom position in (−)-50, which prevented it from fitting into the protein binding pocket.
4.9. Molecules Targeting Plasmodium falciparum Hexokinase (PfHK)
Hexokinase is the first enzyme implicated in glycolysis, catalyzing the transformation of glucose into glucose 6-phosphate. Studies showed that PfHK inhibition (directly or indirectly by inhibiting glucose transporters) is linked to parasite death during the asexual blood-stage [72,73]. PfHK only possesses 24% similarity with human glucokinase, suggesting the possibility of designing selective PfHK inhibitors [74].
In 2013, Harris et al. screened a small library of compounds on PfHK [74]: three simple benzoisothiazolinones (51–53, Table 8) and the closely-related selenium compound ebselen (54) showed activity against PfHK below 300 nM. These values were not completely correlated regarding EC50 against Pf3D7, as stated by the authors, who suggested that cell permeability and off-target interaction problems might explain the difference.
4.10. Molecules Targeting Plasmodium falciparum Mitogen-Activated Protein Kinase 2 (PfMAP2)
Mitogen-activated protein kinases (MAPK) are proteins involved in signal transduction and are the key to many cellular processes in eukaryotic organisms. Two MAPKs are found in P. falciparum: PfMAP1 and PfMAP2. Very recently, Hitz et al. defined the role of these two proteins: neither is essential for asexual development, but PfMAP2 is essential for exflagellation, a mechanism that appears to be directly linked to this protein without the involvement of PfMAP1 [76]. Brumlik et al. screened some Hsp38α (a human MAPK) inhibitors to assess their activity on P. falciparum [77]. One compound (57) displayed sub-micromolar activity on PfW2 (Figure 14).
4.11. Molecules Targeting Plasmodium falciparum MO15-Related Protein Kinase (PfMRK)
PfMRK is a cyclin-dependent kinase located in the nucleus. It interacts with two other proteins [78]: CDK-activating kinase assembly factor (PfMAT1) and cyclin 1 (Pfcyc-1). These three proteins are considered likely to be important for gene expression and DNA replication, and PfMRK is believed to be essential for the asexual blood-stage [17]. PfMRK shares the most properties, regarding human proteins, with HsCDK7.
Woodard et al. screened 12 isoquinoline and naphthalene sulfonamides, known for being kinase inhibitors, on PfMRK [79]. Only 58 displayed a sub-micromolar activity on PfMRK but was not active on P. falciparum in vitro. The same team later designed some chalcones based on an in-silico model of HsCDK7 [80]. One of these chalcones, 59, displayed a 1.3 µM activity against PfMRK associated with an in vitro activity of 4.6 µM on PfW2. Caridha et al. tested 27 sulfonamide compounds substituted by phenyl or thiophene cycles [81]. Ten molecules, including compound 60 (Figure 15), possessed a sub-micromolar activity on PfMRK, but none was active on PfW2. Cytotoxicity of compound 60 on five different cell lines was assessed, and all the values were around or above 25 µM.
4.12. Molecules Targeting Plasmodium falciparum NIMA-Related Kinase 1 (PfNEK1)
Never-in-mitosis/Aspergillus (NIMA) related protein kinases are found in multiple eukaryotic organisms and are involved in the cell division process. Four NEK proteins have been discovered in Plasmodium [82]:
- PfNEK2 and PfNEK4 are related to sexual stages of the parasite,
- PfNEK1 and PfNEK3 can interact with PfMAP2 and,
- PfNEK1 is also expressed during schizogony (it is the only NEK essential in the blood-stage) and in male gametocytes.
Laurent et al. and Desoubzdanne et al. found two natural compounds targeting this protein (Figure 16): xestoquinone 61 and alisiaquinone 62 [83,84] displayed interesting in vitro parameters and an in vivo activity on a mouse model at 5 mg/kg during a four-day Peter’s test [27]. Xestoquinone 61 was found to be selective of PfNEK1 versus other plasmodial and human kinases.
4.13. Molecules Targeting Plasmodium falciparum Phosphatidyl-Inositol 4-Kinase (PfPI4K)
Phosphatidyl-inositol 4-kinase catalyzes the transformation of phosphatidyl-inositol into phosphatidyl-inositol 4 phosphate (PI4P). PI4P is a key secondary messenger involved in multiple cellular pathways. Inhibition of PfPI4K led to activity on every stage of the Plasmodium cycle [85] with inhibition of:
- Development of schizonts and hypnozoites in the liver stage,
- Asexual growth and gametocytogenesis in blood-stage,
- Reduction of oocysts in mosquito midgut in the sexual stage.
PfPI4K inhibition phenotypic consequences were found by McNamara et al. using four compounds as tools (three are showed in Table 10) [85]. The most assessed compound, KDU691 (63) an imidazopyrazine, showed promising in vivo activity on luciferase-expressing P. berghei-infected mice: with a single dose of 7.5 mg/kg, 63 was able to protect mice from what would be otherwise a fatal infection. 63 is selective of PfPI4K versus other human kinases including HsPI4K (IC50 = 7.9 µM). Other tested compounds included the imidazopyridazine KAI715 (64) and the quinoxaline BQR695 (65). Their in vitro results are summed up in Table 10. PfPI4K was confirmed as the target of these compounds by analysis of resistant clones and artificial modifications of PfPI4K gene were also associated with drug resistance.
PfPI4K inhibitors are the most advanced plasmodial kinase inhibitors in terms of drug development. One compound, MMV390048 (66), is currently in phase II of clinical trials and is the only plasmodial kinase inhibitor currently in clinical trial (Figure 17) [86]. MMV390048 PI4K inhibitory effects, with the consequence of a multistage activity, were demonstrated by Paquet et al. using chemoproteomics studies [87]. This inhibition is selective, as HsPI4Kα and HsPI4Kβ were not targeted by 66. Numerous in vivo experiments were performed (on mice, rats, dogs, and monkeys): multistage potency was conserved in in vivo models and 66 displayed very good pharmacokinetic parameters including bioavailability of 74% and a half-time of 66 h at an oral dose of 5 mg/kg on monkeys. All these parameters pointed 66 as a strong lead compound candidate for clinical studies for single exposure radical cure; it could also be promising for chemoprevention. Compound 66 originally emerged from a SAR study by Younis et al. which, starting from compound 67, focused on modulation of the methoxyphenol group [88].
Initial human clinical studies on 66 “showed high variability in exposure, which was attributed to low aqueous solubility” [89]. To solve, this problem, Brunshwig et al., synthesized UCT943 (68), an analog of 66 where the methylsulfone was replaced by a piperazinyl carboxamide and the aminopyridine by an aminopyrazine to improve aqueous solubility [89]. This second generation of PfPI4K inhibitor showed improved parameters on every stage of the malaria cycle. ADMET properties were impacted by the modification but the in vivo activity was conserved.
Continuing on this new aminopyrazine scaffold, Gibhard et al., synthesized two aminopyrazines (Figure 18) to explore a potential prodrug strategy by conversion of a sulfoxide into a sulfone [90]. Compounds 69 (pyrazine analog of 66) and 70 showed nanomolar activities in vitro. Solubility was better at physiological pH for the sulfoxide derivative compared to the sulfone. In vivo, the 90% effective dose (ED90) per os was at 0.12 mg.kg−1 for both compounds compared to 0.57 mg.kg−1 for 66 and 0.25 mg.kg-1 for 68 (on Pf3D7 infected mouse model). As expected by the authors, 70 was found to be rapidly converted in vivo into 69, validating the sulfoxide prodrug approach for this series of compounds.
Other compounds with original scaffolds have been discovered since the development of 66. Kandepedu et al. described a series of 1,5-naphthyridines, most of them having submicromolar activities on PfNF54 [91]. Starting from MMV024101 (71), a SAR study was performed on the two substituents of the naphthyridine core (Figure 19) and 48 analogs were synthesized. Compound 73 showed good activity on PfNF54 with an IC50 of 63 nM and good metabolic stability (t1/2 = 33 h at 5 mg/kg per os) but limited oral bioavailability (39%) on a mouse model.
Liang et al. described a bipyridine series targeting PfPI4K from screening in their compound library [92]. Compound 74 displayed an activity around 4 µM on Pf3D7 but was able to inhibit PfPI4K with an IC50 of 7.7 nM (Figure 20). An in-silico homology model of PfPI4K was then used to rationally guide modulations on 74: the 2-chloro-3-sulfonamide pyridine core was kept and modulations were performed on the two side chains. Compound 75 displayed the best activity on Pf3D7 of 25.1 nM together with an IC50 on PfPI4K of 0.9 nM. This activity was shown to be selective of PfPI4K versus human kinases including HsPI3K or HsPI4K. With oral bioavailability of 80% and a t1/2 of 3.6 h on a mouse model (at 10 mg/kg per os), 75 was able in vivo to cure blood-stage P. yoelii at 80 mg/kg/7 days and to stop liver-stage infection by P. berghei sporozoites at a single dose of 1 mg/kg in a mouse model.
4.14. Molecules Targeting Plasmodium falciparum Protein Kinase 5, 6, 7 and 9 (PfPK5, PfPK6, PfPK7 and PfPK9)
PfPK5 is a cyclin-dependent-like kinase [93], essential for the parasite blood-stage [17], with functions still unclear. Because of its homology with HsCDK2 [94], PfPK5 could be linked to cell division regulation [95]. PfPK6 is another cyclin-dependent like kinase most likely essential for the asexual blood-stage [17,18,96]. PfPK7 is an orphan kinase (it does not cluster with defined eukaryotic kinase groups) distantly related to the MAPKK proteins (mitogen-activated protein kinase) [97]. Disrupting PfPK7 showed that it was not essential for the asexual blood-stage and was linked to a reduced growth rate during the asexual blood-stage and a lower number of oocysts in the mosquito midgut [98]. PfPK9 is another orphan kinase, essential for the blood-stage [17], with currently only one known phosphorylation target: the E2 ubiquitin-conjugating enzyme 13 (PfUBC13) [99]. The human homolog, HsUBC13, is involved in DNA repair and immune responses [100].
4.14.1. Molecules Targeting Plasmodium falciparum Protein Kinase 5 (PfPK5)
Compound 76 is a known HsCDK2 and PfPK5 inhibitor, but is 1000 times more potent toward the human protein (Figure 21). Eubanks et al. synthesized analogs of 76 to try to obtain selective compounds against PfPK5 but their modifications failed [95]. Since PfPK5 crystal structure was available, authors also realized an in-silico screening of 35,000 compounds against PfPK5 followed by an in vitro high throughput binding screening. A chromen-2-one-based compound was found interesting and analogs of this compound were purchased and tested. Compound 77 was the most interesting analog, with a Kd(app) of 3.8 µM and 5 µM against PfPK5 and HsCDK2 respectively but lacked activity against PfDd2 in vitro (Figure 21).
4.14.2. Molecules Targeting Plasmodium falciparum Protein Kinase 6 (PfPK6)
PfPK6 inhibitors have only been identified as hit compounds in the screening by Crowther et al. Figure 22 displays two compounds (78 and 79) having an IC50 against PfPK6 of around 60 nM but lacked selectivity toward PfPK6.
4.14.3. Molecules Targeting Plasmodium falciparum Protein Kinase 7 (PfPK7)
Starting from hit compound 80 identified by a high-throughput screening campaign, Bouloc et al. synthesized a series of 3-amino-6-phenyl-imidazopyridazines (Figure 23) [101]. Thirty-five analogs were synthesized with modulations targeting the substitution of the amine at position 3 and the nature of the para-substituent on the phenyl at position 6. Compound 81 was the best compound of this series, with an IC50 of 0.13 µM and 1.09 µM against PfPK7 and Pf3D7 respectively.
Merckx et al. realized a screening of compounds against PfPK7, where two molecules (82 and 83, Figure 24) with an imidazopyridazine or pyrazolopyrimidine core displayed moderate activity on both kinase and Plasmodium inhibition assays [102]. Klein et al. tested their series of pyrazolopyrimidines against PfPK7 [103]. Compound 84 was shown to inhibit PfPK7 at 10 µM by approximately 60% (Figure 24).
4.14.4. Molecules Targeting Plasmodium falciparum Protein Kinase 9 (PfPK9)
PfPK9 inhibitors have only been explored by Raphemot et al. [100]. Starting from a screening of 3,200 molecules, authors discovered that taketinib (85), an inhibitor of HsTAK1, was an inhibitor of PfPK9 (Table 11). 85 used at its EC50 on P. berghei infected HuH7 cells (7.3 µM) was able to increase the size of intracellular parasites but not their numbers, suggesting a deregulation of growth pathways by inhibition of PbPK9. Analogs of taketinib were synthesized to obtain selective compounds against PfPK9: analog 86 displayed a decreased affinity toward PfPK9 at 4.1 µM but was unable to interact with HsTAK1 (Table 11).
4.15. Molecules Targeting cGMP Cyclin-Dependent Protein Kinase (PfPKG)
PfPKG is a serine/threonine kinase involved in mechanisms at all the stages of parasite life: parasite motility, hepatocyte invasion, asexual blood-stage development, and gametocytogenesis [104,105,106,107,108]. Compared to HsPKG, PfPKG possesses a smaller gatekeeper (the amino acid at the entrance of the catalytic domain), making it easier to develop molecules selective toward PfPKG. Inhibition of PfPKG led to multistage activity [109,110] with inhibition of:
- sporozoite invasion during the liver-stage,
- development of asexual blood-stage,
- development of gametocytes,
- exflagellation in the mosquito midgut.
Two different scaffolds were explored for PfPKG inhibitors: imidazopyridines and trisubstituted five-membered aromatic cycles which could be viewed as ring-simplified analogs of the first series.
4.15.1. Imidazopyridines Targeting PfPKG
Starting from compound 87 (Table 12), designed originally as a PKG inhibitor against Eimeria tenella in chickens [111], Baker et al. realized a SAR study and synthesized nine analogs [110]. This work led to compound 88 with an IC50 against PfPKG reduced from 3.1 to 0.16 nM and IC50 against Pf3D7 strain reduced from 395 to 2.1 nM. 88 was also able to reduce blood-stage growth of P. berghei in a mouse model during a four-day Peter’s test by around 60% (25 mg/kg/twice per day per os) [27]. Additional work to better understand the properties of the imidazopyridine series was carried out by Large et al., leading to compound 89 [112] which showed lower potency on P. falciparum than 87 but better selectivity and metabolic stability.
Another study by Large et al. involved modifying the imidazopyridine core using a scaffold hopping strategy [113]. Compound 90, a pyrazolopyridine analog of 87, displayed activities on both PfPKG and Pf3D7 similar to 87 but with lower metabolic stability (22% of compound remaining after 30 min incubation with mouse liver microsome) due to a more lipophilic molecule (Figure 25).
4.15.2. Trisubstitued Five-Membered Aromatic Cycles Targeting PfPKG
Work on trisubstituted five-membered aromatic cycles started with Diaz et al., who tested compound 91 on P. falciparum (Figure 26) [114]. 91 was designed initially as an anticoccidial compound targeting Eimeria tenella PKG [115]. 91 displayed nanomolar activity against multiple recombinant PfPKG and an IC50 of 0.49 µM against P. falciparum NF54 strain. Activity in vivo on a mouse model infected by P. berghei was also assessed. At 50 mg/kg/7 days by intraperitoneal injections, 91 was able to delay the death of all the mouse groups by 12 days.
Tsagris et al. decided to change the pyrrole core in 91 into a thiazole while keeping key substituents from both compounds 87 and 91 [116]. The authors started from compound 92 which displayed nanomolar inhibition of PfPKG but lacked good potency in vitro against Pf3D7 (Figure 27). 15 analogs were synthesized, with modifications targeting all three substituents, leading to compound 93. Using the same substituents as 87 led to improved potency but 93 displayed an hERG IC50 of 1.3 µM (Figure 27). 93 was assessed against human kinases and inhibited all the tested kinases below 70% at 100 nM. Starting from 93, Matralis et al. continued pharmacomodulations, mainly on the N-substituent of aminopyrimidine and the replacement of N-methypiperidine [117]. Multiple compounds showed improved potency, including compound 94 (Figure 27). 94 was able to kill parasites rapidly, like artemisinin. Thus, the authors decided to assess 94 in a binding assay against other plasmodial kinases and found that it was also able to bind PfCLK2. This dual activity was linked to the fast-killing profile, since compounds only targeting PfPKG possessed a slow killing profile. Moreover, 94 also showed an affinity toward PfCDPK1 and PfCDPK4.
From three isoxazole-based hits on PfPKG of their own compounds’ library, Ul Mahmood et al. explored the three positions on the isoxazole cycle while keeping key features like aminopyrimidine from previously described PfPKG inhibitors [118]. 29 analogs were synthesized and tested on PfPKG: six of them displayed an IC50 below 50 nM, including compound 95, the most potent one (Figure 28). Finally, MMV030084 (compound 96, Figure 28), a trisubstituted imidazole, was described by Vanaershot et al. as a multistage active compound (no gametocytocidal effect) targeting PfPKG [109]. Using knockout studies, PfPKG was confirmed to be the main target of 96.
4.16. Molecules Targeting Plasmodium falciparum Thymidylate Kinase (PfTMK)
PfTMK is the enzyme catalyzing the phosphorylation of thymidine monophosphate to thymidine diphosphate. PfTMK is involved in the de novo synthesis of purine bases and is the only way for the parasite to create these bases [119]. This pathway also includes major drug targets such as dihydrofolate reductase (PfDHFR, targeted by sulfadoxine) and more recently, dihydroorotate dehydrogenase (PfDHODH, targeted by DSM265 [120]). PfTMK is also able to catalyze the phosphorylation of deoxyguanosine monophosphate into deoxyguanosine diphosphate.
Research on PfTMK inhibitors has been centered on thymidine analogs. Cui et al. synthesized a large number of thymidine analogs bearing a urea side chain [121]. Compound 97 was used as a starting point for pharmacomodulations which led to compound 98 (Figure 29). Modulations showed several structural features important for potency: α-anomers were more potent than β, thiourea decreased potency whereas hydrophobic para-substituent on the phenyl urea increased potency. 98 displayed nanomolar activity on Pf3D7 but with low microsomal stability. Surprisingly, no inhibitory assays were performed on the most potent compounds of the series, meaning no clear conclusion can be drawn on the target of these compounds.
Simpler structures were explored by Kato et al., who replaced the tetrahydrofuran by a cyclopentene [122]. This led to compound 99, showing a weak activity with a Ki of 20 µM on PfTMK (Figure 30). 99 was then used by Noguchi et al. as a starting point for pharmacomodulations on the side chain of the cyclopentene core [123]. Compound 100 with a fluoroethanol substituent displayed better potency than 99, with a Ki of 14 µM (Figure 30).
5. Conclusions
Plasmodial kinases appear to be promising targets for new drug therapies against malaria. In the best cases, their inhibition can impact every stage of the parasitic cycle. This is due to the involvement of kinases in most of the cellular mechanisms. Zhang et al. created a mutagenesis index score (MIS) for each gene of the P. falciparum asexual blood-stage, illustrating the essentialness of each of these genes (Figure 31) [124]: only five of the kinases described in this review are considered dispensable (MIS close to 1) but two of them (PfCDPK4 and PfPK7) are known to be key proteins during exflagellation.
MMV390048 (66), targeting PfPI4K is currently the only plasmodial kinase inhibitor in clinical trial. However, this could quickly change as the way is opened to other PfPI4K inhibitors. This makes PfPI4K inhibitors the most advanced plasmodial kinase inhibitors in the drug development process (Table 13). PfCDPK4 inhibitors are getting close to a pre-clinical stage while PfCDPK1, PfCLK3, PfGSK3 and PfPKG inhibitors are promising compounds awaiting further studies. This represents only six out of the 22 kinases described in this review. Many are still in a hit discovery stage, sometimes without progress for more than ten years.
The development of plasmodial kinase inhibitors is slowed by a lack of X-ray structural data, challenges regarding selectivity against human kinases, and loss of potency going from protein to parasite activity. Moreover, new antiplasmodial hit molecules without a defined target usually have their mechanism of action explored based on commercial antiplasmodial drugs. If these compounds possess multistage activity, they should be tested on plasmodial kinases.
There is still plenty of room for new kinase inhibitors: only 22 out of 86 (or 99) estimated plasmodial kinases were found to be targeted by the compounds in this review. In addition to plasmodial kinases inhibitors, other new promising drugs in development targeting plasmodial proteins such as PfDHODH or PfATP4 will be important for the creation of new powerful drug combinations to reduce numbers of malaria cases and deaths in years to come.
Author Contributions
Conceptualization, N.P.; methodology, R.M. and N.P.; formal analysis, R.M.; data curation, R.M.; writing—original draft preparation, R.M.; writing—review and editing, N.P. and P.V.; supervision, N.P. and P.V.; funding acquisition, N.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by “Fondation pour la Recherche Médicale (FRM)”, project code DCM20181039565.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- World Health Organization World Malaria Report 2019; World Health Organization: Geneva, Switzerland, 2019; ISBN 978-92-4-156572-1.
- World Health Organization; Global Malaria Programme. Global Technical Strategy for Malaria, 2016–2030; World Health Organization: Geneva, Switzerland, 2015; ISBN 978-92-4-156499-1. [Google Scholar]
- Ranson, H.; Lissenden, N. Insecticide Resistance in African Anopheles Mosquitoes: A Worsening Situation that Needs Urgent Action to Maintain Malaria Control. Trends Parasitol. 2016, 32, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.; Scharf, S.; Schmidt, S.; Jonscher, E.; Hoeijmakers, W.A.M.; Flemming, S.; Toenhake, C.G.; Schmitt, M.; Sabitzki, R.; Bergmann, B.; et al. A Kelch13-defined endocytosis pathway mediates artemisinin resistance in malaria parasites. Science 2020, 367, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Ouji, M.; Augereau, J.-M.; Paloque, L.; Benoit-Vical, F. Plasmodium falciparum resistance to artemisinin-based combination therapies: A sword of Damocles in the path toward malaria elimination. Parasite 2018, 25, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phyo, A.P.; Win, K.K.; Thu, A.M.; Swe, L.L.; Htike, H.; Beau, C.; Sriprawat, K.; Winterberg, M.; Proux, S.; Imwong, M.; et al. Poor response to artesunate treatment in two patients with severe malaria on the Thai–Myanmar border. Malaria J. 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Uwimana, A.; Legrand, E.; Stokes, B.H.; Ndikumana, J.-L.M.; Warsame, M.; Umulisa, N.; Ngamije, D.; Munyaneza, T.; Mazarati, J.-B.; Munguti, K.; et al. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat. Med. 2020, 26, 1602–1608. [Google Scholar] [CrossRef]
- Sinka, M.E.; Pironon, S.; Massey, N.C.; Longbottom, J.; Hemingway, J.; Moyes, C.L.; Willis, K.J. A new malaria vector in Africa: Predicting the expansion range of Anopheles stephensi and identifying the urban populations at risk. Proc. Natl. Acad. Sci. USA 2020, 117, 24900–24908. [Google Scholar] [CrossRef]
- Burrows, J.N.; Duparc, S.; Gutteridge, W.E.; Hooft van Huijsduijnen, R.; Kaszubska, W.; Macintyre, F.; Mazzuri, S.; Möhrle, J.J.; Wells, T.N.C. New developments in anti-malarial target candidate and product profiles. Malaria J. 2017, 16. [Google Scholar] [CrossRef] [Green Version]
- Manning, G. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug. Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Carles, F.; Bourg, S.; Meyer, C.; Bonnet, P. PKIDB: A Curated, Annotated and Updated Database of Protein Kinase Inhibitors in Clinical Trials. Molecules 2018, 23, 908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.; Equinet, L.; Packer, J.; Doerig, C. Protein kinases of the human malaria parasite Plasmodium falciparum: The kinome of a divergent eukaryote. BMC Genom. 2004, 5, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anamika; Srinivasan, N.; Krupa, A. A genomic perspective of protein kinases in Plasmodium falciparum. Proteins 2004, 58, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucet, I.S.; Tobin, A.; Drewry, D.; Wilks, A.F.; Doerig, C. Plasmodium kinases as targets for new-generation antimalarials. Future Med. Chem. 2012, 4, 2295–2310. [Google Scholar] [CrossRef] [PubMed]
- Solyakov, L.; Halbert, J.; Alam, M.M.; Semblat, J.-P.; Dorin-Semblat, D.; Reininger, L.; Bottrill, A.R.; Mistry, S.; Abdi, A.; Fennell, C.; et al. Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat. Commun. 2011, 2, 565. [Google Scholar] [CrossRef] [PubMed]
- Tewari, R.; Straschil, U.; Bateman, A.; Böhme, U.; Cherevach, I.; Gong, P.; Pain, A.; Billker, O. The Systematic Functional Analysis of Plasmodium Protein Kinases Identifies Essential Regulators of Mosquito Transmission. Cell Host Microbe 2010, 8, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Group, T.P. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.F.; Harmon, A. Plants, symbiosis and parasites: A calcium signalling connection. Nat. Rev. Mol. Cell. Bio. 2005, 6, 555–566. [Google Scholar] [CrossRef]
- Billker, O.; Lourido, S.; Sibley, L.D. Calcium-Dependent Signaling and Kinases in Apicomplexan Parasites. Cell Host Microbe 2009, 5, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Kappes, B.; Franklin, R. Gene Structure and Expression of an Unusual Protein Kinase from Plasmodium falciparum Homologous at Its Carboxyl Terminus with the EF Hand Calcium-binding Proteins. J. Biol. Chem. 1993, 268, 4347–4354. [Google Scholar]
- Green, J.L.; Rees-Channer, R.R.; Howell, S.A.; Martin, S.R.; Knuepfer, E.; Taylor, H.M.; Grainger, M.; Holder, A.A. The motor complex of Plasmodium falciparum: Phosphorylation by a calcium-dependent protein kinase. J. Biol. Chem. 2008, 283, 30980–30989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, N.; Sakata, T.; Breton, G.; Le Roch, K.G.; Nagle, A.; Andersen, C.; Bursulaya, B.; Henson, K.; Johnson, J.; Kumar, K.A.; et al. Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat. Chem. Biol. 2008, 4, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Molina-Cruz, A.; Brzostowski, J.; Liu, P.; Luo, Y.; Gunalan, K.; Li, Y.; Ribeiro, J.M.C.; Miller, L.H. PfCDPK1 is critical for malaria parasite gametogenesis and mosquito infection. Proc. Natl. Acad. Sci. USA 2018, 115, 774–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Kumar, M.; Ekka, R.; Dvorin, J.D.; Paul, A.S.; Madugundu, A.K.; Gilberger, T.; Gowda, H.; Duraisingh, M.T.; Keshava Prasad, T.S.; et al. PfCDPK1 mediated signaling in erythrocytic stages of Plasmodium falciparum. Nat. Commun. 2017, 8, 63. [Google Scholar] [CrossRef]
- Peters, W.; Robinson, B.L. Chapter 92—Malaria. In Handbook of Animal Models of Infection; Zak, O., Sande, M.A., Eds.; Academic Press: London, UK, 1999; pp. 757–773. ISBN 978-0-12-775390-4. [Google Scholar]
- Lemercier, G.; Fernandez-Montalvan, A.; Shaw, J.P.; Kugelstadt, D.; Bomke, J.; Domostoj, M.; Schwarz, M.K.; Scheer, A.; Kappes, B.; Leroy, D. Identification and characterization of novel small molecules as potent inhibitors of the plasmodial calcium-dependent protein kinase 1. Biochemistry 2009, 48, 6379–6389. [Google Scholar] [CrossRef]
- Ansell, K.H.; Jones, H.M.; Whalley, D.; Hearn, A.; Taylor, D.L.; Patin, E.C.; Chapman, T.M.; Osborne, S.A.; Wallace, C.; Birchall, K.; et al. Biochemical and antiparasitic properties of inhibitors of the Plasmodium falciparum calcium-dependent protein kinase PfCDPK1. Antimicrob. Agents Chemother. 2014, 58, 6032–6043. [Google Scholar] [CrossRef] [Green Version]
- Chapman, T.M.; Osborne, S.A.; Bouloc, N.; Large, J.M.; Wallace, C.; Birchall, K.; Ansell, K.H.; Jones, H.M.; Taylor, D.; Clough, B.; et al. Substituted imidazopyridazines are potent and selective inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1). Bioorg. Med. Chem. Lett. 2013, 23, 3064–3069. [Google Scholar] [CrossRef] [Green Version]
- Large, J.M.; Osborne, S.A.; Smiljanic-Hurley, E.; Ansell, K.H.; Jones, H.M.; Taylor, D.L.; Clough, B.; Green, J.L.; Holder, A.A. Imidazopyridazines as potent inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1): Preparation and evaluation of pyrazole linked analogues. Bioorg. Med. Chem. Lett. 2013, 23, 6019–6024. [Google Scholar] [CrossRef] [Green Version]
- Chapman, T.M.; Osborne, S.A.; Wallace, C.; Birchall, K.; Bouloc, N.; Jones, H.M.; Ansell, K.H.; Taylor, D.L.; Clough, B.; Green, J.L.; et al. Optimization of an imidazopyridazine series of inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1). J. Med. Chem. 2014, 57, 3570–3587. [Google Scholar] [CrossRef]
- Crowther, G.J.; Hillesland, H.K.; Keyloun, K.R.; Reid, M.C.; Lafuente-Monasterio, M.J.; Ghidelli-Disse, S.; Leonard, S.E.; He, P.; Jones, J.C.; Krahn, M.M.; et al. Biochemical Screening of Five Protein Kinases from Plasmodium falciparum against 14,000 Cell-Active Compounds. PLoS ONE 2016, 11, e0149996. [Google Scholar] [CrossRef] [Green Version]
- Billker, O.; Dechamps, S.; Tewari, R.; Wenig, G.; Franke-Fayard, B.; Brinkmann, V. Calcium and a Calcium-Dependent Protein Kinase Regulate Gamete Formation and Mosquito Transmission in a Malaria Parasite. Cell 2004, 117, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Sudo, A.; Kobayashi, K.; Sugi, T.; Tohya, Y.; Akashi, H. Characterization of Plasmodium falciparum calcium-dependent protein kinase 4. Parasitol. Int. 2009, 58, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Ojo, K.K.; Pfander, C.; Mueller, N.R.; Burstroem, C.; Larson, E.T.; Bryan, C.M.; Fox, A.M.W.; Reid, M.C.; Johnson, S.M.; Murphy, R.C.; et al. Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J. Clin. Invest. 2012, 122, 2301–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojo, K.K.; Eastman, R.T.; Vidadala, R.; Zhang, Z.; Rivas, K.L.; Choi, R.; Lutz, J.D.; Reid, M.C.; Fox, A.M.W.; Hulverson, M.A.; et al. A specific inhibitor of PfCDPK4 blocks malaria transmission: Chemical-genetic validation. J. Infect. Dis. 2014, 209, 275–284. [Google Scholar] [CrossRef]
- Vidadala, R.S.R.; Ojo, K.K.; Johnson, S.M.; Zhang, Z.; Leonard, S.E.; Mitra, A.; Choi, R.; Reid, M.C.; Keyloun, K.R.; Fox, A.M.W.; et al. Development of potent and selective Plasmodium falciparum calcium-dependent protein kinase 4 (PfCDPK4) inhibitors that block the transmission of malaria to mosquitoes. Eur. J. Med. Chem. 2014, 74, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Hulverson, M.A.; Zhang, Z.; Choi, R.; Hart, K.J.; Kennedy, M.; Vidadala, R.S.R.; Maly, D.J.; Van Voorhis, W.C.; Lindner, S.E.; et al. Aminopyrazole-4-carboxamide analogues are selective inhibitors of Plasmodium falciparum microgametocyte exflagellation and potential malaria transmission blocking agents. Bioorg. Med. Chem. Lett. 2016, 26, 5487–5491. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Hulverson, M.A.; Choi, R.; Arnold, S.L.M.; Zhang, Z.; McCloskey, M.C.; Whitman, G.R.; Hackman, R.C.; Rivas, K.L.; Barrett, L.K.; et al. Development of 5-Aminopyrazole-4-carboxamide-based Bumped-Kinase Inhibitors for Cryptosporidiosis Therapy. J. Med. Chem. 2019, 62, 3135–3146. [Google Scholar] [CrossRef]
- Serran-Aguilera, L.; Denton, H.; Rubio-Ruiz, B.; Lopez-Gutierrez, B.; Entrena, A.; Izquierdo, L.; Smith, T.K.; Conejo-Garcia, A.; Hurtado-Guerrero, R. Plasmodium falciparum Choline Kinase Inhibition Leads to a Major Decrease in Phosphatidylethanolamine Causing Parasite Death. Sci. Rep. 2016, 6, 33189. [Google Scholar] [CrossRef] [Green Version]
- Ancelin, M.L.; Calas, M.; Bompart, J.; Cordina, G.; Martin, D.; Ben Bari, M.; Jei, T.; Druilhe, P.; Vial, H.J. Antimalarial Activity of 77 Phospholipid Polar Head Analogs: Close Correlation Between Inhibition of Phospholipid Metabolism and In Vitro Plasmodium Falciparum Growth. Blood 1998, 91, 1426–1437. [Google Scholar] [CrossRef]
- Ancelin, M.L.; Calas, M.; Vidal-Sailhan, V.; Herbuté, S.; Ringwald, P.; Vial, H.J. Potent Inhibitors of Plasmodium Phospholipid Metabolism with a Broad Spectrum of In Vitro Antimalarial Activities. Antimicrob. Agents Chemoter. 2003, 47, 2590–2597. [Google Scholar] [CrossRef] [Green Version]
- Serran-Aguilera, L.; Nuti, R.; Lopez-Cara, L.C.; Rios-Marco, P.; Carrasco, M.P.; Marco, C.; Entrena, A.; Macchiarulo, A.; Hurtado-Guerrero, R. Choline Kinase Active Site Provides Features for Designing Versatile Inhibitors. Curr. Top. Med. Chem. 2014, 14, 2684–2693. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Maity, P.; Guha, M.; Kumar, S.; Srivastava, K.; Puri, S.K.; Bandyopadhyay, U. Inhibition of Plasmodium falciparum choline kinase by hexadecyltrimethylammonium bromide: A possible antimalarial mechanism. Antimicrob. Agents Chemoter. 2007, 51, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowther, G.J.; Napuli, A.J.; Gilligan, J.H.; Gagaring, K.; Borboa, R.; Francek, C.; Chen, Z.; Dagostino, E.F.; Stockmyer, J.B.; Wang, Y.; et al. Identification of inhibitors for putative malaria drug targets among novel antimalarial compounds. Mol. Biochem. Parasit. 2011, 175, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiafino-Ortega, S.; Baglioni, E.; Perez-Moreno, G.; Marco, P.R.; Marco, C.; Gonzalez-Pacanowska, D.; Ruiz-Perez, L.M.; Carrasco-Jimenez, M.P.; Lopez-Cara, L.C. 1,2-Diphenoxiethane salts as potent antiplasmodial agents. Bioorg. Med. Chem. Lett. 2018, 28, 2485–2489. [Google Scholar] [CrossRef]
- Pease, B.N.; Huttlin, E.L.; Jedrychowski, M.P.; Talevich, E.; Harmon, J.; Dillman, T.; Kannan, N.; Doerig, C.; Chakrabarti, R.; Gygi, S.P.; et al. Global Analysis of Protein Expression and Phosphorylation of Three Stages of Plasmodium falciparum Intraerythrocytic Development. J. Proteome Res. 2013, 12, 4028–4045. [Google Scholar] [CrossRef] [Green Version]
- Holland, Z.; Prudent, R.; Reiser, J.-B.; Cochet, C.; Doerig, C. Functional Analysis of Protein Kinase CK2 of the Human Malaria Parasite Plasmodium falciparum. Eukaryot. Cell 2009, 8, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Carrillo, D.; Lin, J.; El Sahili, A.; Wei, M.; Sze, S.K.; Cheung, P.C.F.; Doerig, C.; Lescar, J. The protein kinase CK2 catalytic domain from Plasmodium falciparum: Crystal structure, tyrosine kinase activity and inhibition. Sci. Rep. 2018, 8, 7365. [Google Scholar] [CrossRef] [Green Version]
- Testing the Safety and Tolerability of CX-4945 in Patients with Recurrent Medulloblastoma Who May or May Not Have Surgery—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03904862 (accessed on 4 September 2020).
- Talevich, E.; Mirza, A.; Kannan, N. Structural and evolutionary divergence of eukaryotic protein kinases in Apicomplexa. BMC Evol. Biol. 2011, 11, 321. [Google Scholar] [CrossRef] [Green Version]
- Kern, S.; Agarwal, S.; Huber, K.; Gehring, A.P.; Strödke, B.; Wirth, C.C.; Brügl, T.; Abodo, L.O.; Dandekar, T.; Doerig, C.; et al. Inhibition of the SR protein-phosphorylating CLK kinases of Plasmodium falciparum impairs blood stage replication and malaria transmission. PLoS ONE 2014, 9, e105732. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.M.; Sanchez-Azqueta, A.; Janha, O.; Flannery, E.L.; Mahindra, A.; Mapesa, K.; Char, A.B.; Sriranganadane, D.; Brancucci, N.M.B.; Antonova-Koch, Y.; et al. Validation of the protein kinase PfCLK3 as a multistage cross-species malarial drug target. Science 2019, 365. [Google Scholar] [CrossRef] [Green Version]
- Bendjeddou, L.Z.; Loaëc, N.; Villiers, B.; Prina, E.; Späth, G.F.; Galons, H.; Meijer, L.; Oumata, N. Exploration of the imidazo[1,2-b]pyridazine scaffold as a protein kinase inhibitor. Eur. J. Med. Chem. 2017, 125, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Mahindra, A.; Janha, O.; Mapesa, K.; Sanchez-Azqueta, A.; Alam, M.M.; Amambua-Ngwa, A.; Nwakanma, D.C.; Tobin, A.B.; Jamieson, A.G. Development of Potent PfCLK3 Inhibitors Based on TCMDC-135051 as a New Class of Antimalarials. J. Med. Chem. 2020, 63, 9300–9315. [Google Scholar] [CrossRef] [PubMed]
- Genschel, U. Coenzyme A Biosynthesis: Reconstruction of the Pathway in Archaea and an Evolutionary Scenario Based on Comparative Genomics. Mol. Biol. Evol. 2004, 21, 1242–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spry, C.; Chai, C.L.L.; Kirk, K.; Saliba, K.J. A class of pantothenic acid analogs inhibits Plasmodium falciparum pantothenate kinase and represses the proliferation of malaria parasites. Antimicrob. Agents Chemother. 2005, 49, 4649–4657. [Google Scholar] [CrossRef] [Green Version]
- Spry, C.; Sewell, A.L.; Hering, Y.; Villa, M.V.J.; Weber, J.; Hobson, S.J.; Harnor, S.J.; Gul, S.; Marquez, R.; Saliba, K.J. Structure-activity analysis of CJ-15,801 analogues that interact with Plasmodium falciparum pantothenate kinase and inhibit parasite proliferation. Eur. J. Med. Chem. 2018, 143, 1139–1147. [Google Scholar] [CrossRef] [Green Version]
- Chiu, J.E.; Thekkiniath, J.; Choi, J.-Y.; Perrin, B.A.; Lawres, L.; Plummer, M.; Virji, A.Z.; Abraham, A.; Toh, J.Y.; Zandt, M.V.; et al. The antimalarial activity of the pantothenamide α-PanAm is via inhibition of pantothenate phosphorylation. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, S.; Avery, V.M. A novel approach for the discovery of chemically diverse anti-malarial compounds targeting the Plasmodium falciparum Coenzyme A synthesis pathway. Malaria J. 2014, 13, 343. [Google Scholar] [CrossRef]
- Brandt, G.S.; Bailey, S. Dematin, a human erythrocyte cytoskeletal protein, is a substrate for a recombinant FIKK kinase from Plasmodium falciparum. Mol. Biochem. Parasit. 2013, 191, 20–23. [Google Scholar] [CrossRef]
- Kats, L.M.; Fernandez, K.M.; Glenister, F.K.; Herrmann, S.; Buckingham, D.W.; Siddiqui, G.; Sharma, L.; Bamert, R.; Lucet, I.; Guillotte, M.; et al. An exported kinase (FIKK4.2) that mediates virulence-associated changes in Plasmodium falciparum-infected red blood cells. Int. J. Parasitol. 2014, 44, 319–328. [Google Scholar] [CrossRef]
- Lin, B.C.; Harris, D.R.; Kirkman, L.M.D.; Perez, A.M.; Qian, Y.; Schermerhorn, J.T.; Hong, M.Y.; Winston, D.S.; Xu, L.; Lieber, A.M.; et al. The anthraquinone emodin inhibits the non-exported FIKK kinase from Plasmodium falciparum. Bioorg. Chem. 2017, 75, 217–223. [Google Scholar] [CrossRef]
- Lin, B.C.; Harris, D.R.; Kirkman, L.M.D.; Perez, A.M.; Qian, Y.; Schermerhorn, J.T.; Hong, M.Y.; Winston, D.S.; Xu, L.; Brandt, G.S. FIKK Kinase, a Ser/Thr Kinase Important to Malaria Parasites, Is Inhibited by Tyrosine Kinase Inhibitors. ACS Omega 2017, 2, 6605–6612. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Kitade, Y. Molecular Characterization, Heterologous Expression and Kinetic Analysis of Recombinant Plasmodium falciparum Thymidylate Kinase. J. Biochem. 2008, 144, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Tarun, A.S.; Peng, X.; Dumpit, R.F.; Ogata, Y.; Silva-Rivera, H.; Camargo, N.; Daly, T.M.; Bergman, L.W.; Kappe, S.H.I. A combined transcriptome and proteome survey of malaria parasite liver stages. Proc. Natl. Acad. Sci. USA 2008, 105, 305–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, B.; Harvey, K.L.; Wilcke, L.; Ruch, U.; Engelberg, K.; Biller, L.; Lucet, I.; Erkelenz, S.; Heincke, D.; Spielmann, T.; et al. Hierarchical phosphorylation of apical membrane antigen 1 is required for efficient red blood cell invasion by malaria parasites. Sci. Rep. 2016, 6, 34479. [Google Scholar] [CrossRef] [Green Version]
- Saraswati, A.P.; Ali Hussaini, S.M.; Krishna, N.H.; Babu, B.N.; Kamal, A. Glycogen synthase kinase-3 and its inhibitors: Potential target for various therapeutic conditions. Eur. J. Med. Chem. 2018, 144, 843–858. [Google Scholar] [CrossRef]
- Fugel, W.; Oberholzer, A.E.; Gschloessl, B.; Dzikowski, R.; Pressburger, N.; Preu, L.; Pearl, L.H.; Baratte, B.; Ratin, M.; Okun, I.; et al. 3,6-Diamino-4-(2-halophenyl)-2-benzoylthieno[2,3-b]pyridine-5-carbonitriles are selective inhibitors of Plasmodium falciparum glycogen synthase kinase-3. J. Med. Chem. 2013, 56, 264–275. [Google Scholar] [CrossRef]
- Masch, A.; Nasereddin, A.; Alder, A.; Bird, M.J.; Schweda, S.I.; Preu, L.; Doerig, C.; Dzikowski, R.; Gilberger, T.W.; Kunick, C. Structure-activity relationships in a series of antiplasmodial thieno[2,3-b]pyridines. Malaria J. 2019, 18, 89. [Google Scholar] [CrossRef]
- Slavic, K.; Straschil, U.; Reininger, L.; Doerig, C.; Morin, C.; Tewari, R.; Krishna, S. Life cycle studies of the hexose transporter of Plasmodium species and genetic validation of their essentiality. Mol. Microbiol. 2010, 75, 1402–1413. [Google Scholar] [CrossRef] [Green Version]
- van Schalkwyk, D.A.; Priebe, W.; Saliba, K.J. The Inhibitory Effect of 2-Halo Derivatives of d-Glucose on Glycolysis and on the Proliferation of the Human Malaria Parasite Plasmodium falciparum. J. Pharmacol. Exp. Ther. 2008, 327, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Harris, M.T.; Walker, D.M.; Drew, M.E.; Mitchell, W.G.; Dao, K.; Schroeder, C.E.; Flaherty, D.P.; Weiner, W.S.; Golden, J.E.; Morris, J.C. Interrogating a Hexokinase-Selected Small-Molecule Library for Inhibitors of Plasmodium falciparum Hexokinase. Antimicrob. Agents Chemother. 2013, 57, 3731–3737. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.I.; Patrick, S.L.; Blanding, W.M.; Dwivedi, V.; Suryadi, J.; Golden, J.E.; Coussens, N.P.; Lee, O.W.; Shen, M.; Boxer, M.B.; et al. Identification of Novel Plasmodium falciparum Hexokinase Inhibitors with Antiparasitic Activity. Antimicrob. Agents Chemother. 2016, 60, 6023–6033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitz, E.; Balestra, A.C.; Brochet, M.; Voss, T.S. PfMAP-2 is essential for male gametogenesis in the malaria parasite Plasmodium falciparum. Sci. Rep. 2020, 10, 11930. [Google Scholar] [CrossRef] [PubMed]
- Brumlik, M.J.; Nkhoma, S.; Kious, M.J.; Thompson, G.R., 3rd; Patterson, T.F.; Siekierka, J.J.; Anderson, T.J.C.; Curiel, T.J. Human p38 mitogen-activated protein kinase inhibitor drugs inhibit Plasmodium falciparum replication. Exp. Parasitol. 2011, 128, 170–175. [Google Scholar] [CrossRef] [Green Version]
- Jirage, D.; Chen, Y.; Caridha, D.; O’Neil, M.T.; Eyase, F.; Witola, W.H.; Mamoun, C.B.; Waters, N.C. The malarial CDK Pfmrk and its effector PfMAT1 phosphorylate DNA replication proteins and co-localize in the nucleus. Mol. Biochem. Parasit. 2010, 172, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodard, C.L.; Keenan, S.M.; Gerena, L.; Welsh, W.J.; Geyer, J.A.; Waters, N.C. Evaluation of broad spectrum protein kinase inhibitors to probe the architecture of the malarial cyclin dependent protein kinase Pfmrk. Bioorg. Med. Chem. Lett. 2007, 17, 4961–4966. [Google Scholar] [CrossRef]
- Geyer, J.A.; Keenan, S.M.; Woodard, C.L.; Thompson, P.A.; Gerena, L.; Nichols, D.A.; Gutteridge, C.E.; Waters, N.C. Selective inhibition of Pfmrk, a Plasmodium falciparum CDK, by antimalarial 1,3-diaryl-2-propenones. Bioorg. Med. Chem. Lett. 2009, 19, 1982–1985. [Google Scholar] [CrossRef] [PubMed]
- Caridha, D.; Kathcart, A.K.; Jirage, D.; Waters, N.C. Activity of substituted thiophene sulfonamides against malarial and mammalian cyclin dependent protein kinases. Bioorg. Med. Chem. Lett. 2010, 20, 3863–3867. [Google Scholar] [CrossRef]
- Carvalho, T.G.; Doerig, C.; Reininger, L. Nima- and Aurora-related kinases of malaria parasites. Biochim. Biophys. Acta 2013, 1834, 1336–1345. [Google Scholar] [CrossRef]
- Laurent, D.; Jullian, V.; Parenty, A.; Knibiehler, M.; Dorin, D.; Schmitt, S.; Lozach, O.; Lebouvier, N.; Frostin, M.; Alby, F.; et al. Antimalarial potential of xestoquinone, a protein kinase inhibitor isolated from a Vanuatu marine sponge Xestospongia sp. Bioorg. Med. Chem. 2006, 14, 4477–4482. [Google Scholar] [CrossRef]
- Desoubzdanne, D.; Marcourt, L.; Raux, R.; Chevalley, S.; Dorin, D.; Doerig, C.; Valentin, A.; Ausseil, F.; Debitus, C. Alisiaquinones and Alisiaquinol, Dual Inhibitors of Plasmodium falciparum Enzyme Targets from a New Caledonian Deep Water Sponge. J. Nat. Prod. 2008, 71, 1189–1192. [Google Scholar] [CrossRef]
- McNamara, C.W.; Lee, M.C.S.; Lim, C.S.; Lim, S.H.; Roland, J.; Nagle, A.; Simon, O.; Yeung, B.K.S.; Chatterjee, A.K.; McCormack, S.L.; et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature 2013, 504, 248–253. [Google Scholar] [CrossRef] [Green Version]
- MMV390048 POC in Patients with P. vivax and P. falciparum Malaria—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02880241 (accessed on 15 September 2020).
- Paquet, T.; Le Manach, C.; Cabrera, D.G.; Younis, Y.; Henrich, P.P.; Abraham, T.S.; Lee, M.C.S.; Basak, R.; Ghidelli-Disse, S.; Lafuente-Monasterio, M.J.; et al. Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younis, Y.; Douelle, F.; Feng, T.-S.; Cabrera, D.G.; Manach, C.L.; Nchinda, A.T.; Duffy, S.; White, K.L.; Shackleford, D.M.; Morizzi, J.; et al. 3,5-Diaryl-2-aminopyridines as a Novel Class of Orally Active Antimalarials Demonstrating Single Dose Cure in Mice and Clinical Candidate Potential. J. Med. Chem. 2012, 55, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Brunschwig, C.; Lawrence, N.; Taylor, D.; Abay, E.; Njoroge, M.; Basarab, G.S.; Le Manach, C.; Paquet, T.; Cabrera, D.G.; Nchinda, A.T.; et al. UCT943, a Next-Generation Plasmodium falciparum PI4K Inhibitor Preclinical Candidate for the Treatment of Malaria. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Gibhard, L.; Njoroge, M.; Paquet, T.; Brunschwig, C.; Taylor, D.; Lawrence, N.; Abay, E.; Wittlin, S.; Wiesner, L.; Street, L.J.; et al. Investigating Sulfoxide-to-Sulfone Conversion as a Prodrug Strategy for a Phosphatidylinositol 4-Kinase Inhibitor in a Humanized Mouse Model of Malaria. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandepedu, N.; Gonzàlez Cabrera, D.; Eedubilli, S.; Taylor, D.; Brunschwig, C.; Gibhard, L.; Njoroge, M.; Lawrence, N.; Paquet, T.; Eyermann, C.J.; et al. Identification, Characterization, and Optimization of 2,8-Disubstituted-1,5-naphthyridines as Novel Plasmodium falciparum Phosphatidylinositol-4-kinase Inhibitors with in Vivo Efficacy in a Humanized Mouse Model of Malaria. J. Med. Chem. 2018, 61, 5692–5703. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Jiang, Z.; Huang, Z.; Li, F.; Chen, C.; Hu, C.; Wang, W.; Hu, Z.; Liu, Q.; Wang, B.; et al. Discovery of 6′-chloro-N-methyl-5′-(phenylsulfonamido)-[3,3′-bipyridine]-5-carboxamide (CHMFL-PI4K-127) as a novel Plasmodium falciparum PI(4)K inhibitor with potent antimalarial activity against both blood and liver stages of Plasmodium. Eur. J. Med. Chem. 2020, 188, 112012. [Google Scholar] [CrossRef]
- Le Roch, K.; Sestier, C.; Dorin, D.; Waters, N.; Kappes, B.; Chakrabarti, D.; Meijer, L.; Doerig, C. Activation of a Plasmodium falciparum cdc2-related Kinase by Heterologous p25 and Cyclin H: Functionnal characterization of a P. falciparum Cyclin homologue. J. Biol. Chem. 2000, 275, 8952–8958. [Google Scholar] [CrossRef] [Green Version]
- Ross-Macdonald, P.B.; Graeser, R.; Kappes, B.; Franklin, R.; Williamson, D.H. Isolation and expression of a gene specifying a cdc2-like protein kinase from the human malaria parasite Plasmodium falciparum. Eur. J. Biochem. 1994, 220, 693–701. [Google Scholar] [CrossRef]
- Eubanks, A.L.; Perkins, M.M.; Sylvester, K.; Ganley, J.G.; Posfai, D.; Sanschargrin, P.C.; Hong, J.; Sliz, P.; Derbyshire, E.R. In silico Screening and Evaluation of Plasmodium falciparum Protein Kinase 5 (PK5) Inhibitors. ChemMedChem 2018, 13, 2479–2483. [Google Scholar] [CrossRef]
- Bracchi-Ricard, V.; Barik, S.; Delvecchio, C.; Doerig, C.; Chakrabarti, R.; Chakrabarti, D. PfPK6, a novel cyclin-dependent kinase/mitogen-activated protein kinase-related protein kinase from Plasmodium falciparum. Biochem. J. 2000, 347, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Dorin, D.; Semblat, J.-P.; Poullet, P.; Alano, P.; Goldring, J.P.D.; Whittle, C.; Patterson, S.; Chakrabarti, D.; Doerig, C. PfPK7, an atypical MEK-related protein kinase, reflects the absence of classical three-component MAPK pathways in the human malaria parasite Plasmodium falciparum: An atypical MEK-like enzyme in malarial parasites. Mol. Microbiol. 2004, 55, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Dorin-Semblat, D.; Sicard, A.; Doerig, C.; Ranford-Cartwright, L.; Doerig, C. Disruption of the PfPK7 Gene Impairs Schizogony and Sporogony in the Human Malaria Parasite Plasmodium falciparum. Eukaryot. Cell 2008, 7, 279–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip, N.; Haystead, T.A. Characterization of a UBC13 kinase in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2007, 104, 7845–7850. [Google Scholar] [CrossRef] [Green Version]
- Raphemot, R.; Eubanks, A.L.; Toro-Moreno, M.; Geiger, R.A.; Hughes, P.F.; Lu, K.-Y.; Haystead, T.A.J.; Derbyshire, E.R. Plasmodium PK9 Inhibitors Promote Growth of Liver-Stage Parasites. Cell. Chem. Biol. 2019, 26, 411–419.e7. [Google Scholar] [CrossRef]
- Bouloc, N.; Large, J.M.; Smiljanic, E.; Whalley, D.; Ansell, K.H.; Edlin, C.D.; Bryans, J.S. Synthesis and in vitro evaluation of imidazopyridazines as novel inhibitors of the malarial kinase PfPK7. Bioorg. Med. Chem. Lett. 2008, 18, 5294–5298. [Google Scholar] [CrossRef]
- Merckx, A.; Echalier, A.; Langford, K.; Sicard, A.; Langsley, G.; Joore, J.; Doerig, C.; Noble, M.; Endicott, J. Structures of P. falciparum Protein Kinase 7 Identify an Activation Motif and Leads for Inhibitor Design. Structure 2008, 16, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.; Dinér, P.; Dorin-Semblat, D.; Doerig, C.; Grøtli, M. Synthesis of 3-(1,2,3-triazol-1-yl)- and 3-(1,2,3-triazol-4-yl)-substituted pyrazolo[3,4-d]pyrimidin-4-amines via click chemistry: Potential inhibitors of the Plasmodium falciparum PfPK7 protein kinase. Org. Biomol. Chem. 2009, 7, 3421–3429. [Google Scholar] [CrossRef] [Green Version]
- McRobert, L.; Taylor, C.J.; Deng, W.; Fivelman, Q.L.; Cummings, R.M.; Polley, S.D.; Billker, O.; Baker, D.A. Gametogenesis in Malaria Parasites Is Mediated by the cGMP-Dependent Protein Kinase. PLoS Biol. 2008, 6, e139. [Google Scholar] [CrossRef] [Green Version]
- Moon, R.W.; Taylor, C.J.; Bex, C.; Schepers, R.; Goulding, D.; Janse, C.J.; Waters, A.P.; Baker, D.A.; Billker, O. A cyclic GMP signalling module that regulates gliding motility in a malaria parasite. PLoS Pathog. 2009, 5, e1000599. [Google Scholar] [CrossRef] [Green Version]
- Taylor, H.M.; McRobert, L.; Grainger, M.; Sicard, A.; Dluzewski, A.R.; Hopp, C.S.; Holder, A.A.; Baker, D.A. The malaria parasite cyclic GMP-dependent protein kinase plays a central role in blood-stage schizogony. Eukaryot. Cell 2010, 9, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brochet, M.; Collins, M.O.; Smith, T.K.; Thompson, E.; Sebastian, S.; Volkmann, K.; Schwach, F.; Chappell, L.; Gomes, A.R.; Berriman, M.; et al. Phosphoinositide Metabolism Links cGMP-Dependent Protein Kinase G to Essential Ca2+ Signals at Key Decision Points in the Life Cycle of Malaria Parasites. PLoS Biol. 2014, 12, e1001806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindasamy, K.; Jebiwott, S.; Jaijyan, D.K.; Davidow, A.; Ojo, K.K.; Van Voorhis, W.C.; Brochet, M.; Billker, O.; Bhanot, P. Invasion of hepatocytes by Plasmodium sporozoites requires cGMP-dependent protein kinase and calcium dependent protein kinase 4. Mol. Microbiol. 2016, 102, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Vanaerschot, M.; Murithi, J.M.; Pasaje, C.F.A.; Ghidelli-Disse, S.; Dwomoh, L.; Bird, M.; Spottiswoode, N.; Mittal, N.; Arendse, L.B.; Owen, E.S.; et al. Inhibition of Resistance-Refractory P. falciparum Kinase PKG Delivers Prophylactic, Blood Stage, and Transmission-Blocking Antiplasmodial Activity. Cell. Chem. Biol. 2020, 27, 806–816.e8. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.A.; Stewart, L.B.; Large, J.M.; Bowyer, P.W.; Ansell, K.H.; Jiménez-Díaz, M.B.; El Bakkouri, M.; Birchall, K.; Dechering, K.J.; Bouloc, N.S.; et al. A potent series targeting the malarial cGMP-dependent protein kinase clears infection and blocks transmission. Nat. Commun. 2017, 8, 430. [Google Scholar] [CrossRef]
- Biftu, T.; Feng, D.; Fisher, M.; Liang, G.-B.; Qian, X.; Scribner, A.; Dennis, R.; Lee, S.; Liberator, P.A.; Brown, C.; et al. Synthesis and SAR studies of very potent imidazopyridine antiprotozoal agents. Bioorg. Med. Chem. Lett. 2006, 16, 2479–2483. [Google Scholar] [CrossRef]
- Large, J.M.; Birchall, K.; Bouloc, N.S.; Merritt, A.T.; Smiljanic-Hurley, E.; Tsagris, D.J.; Wheldon, M.C.; Ansell, K.H.; Coombs, P.J.; Kettleborough, C.A.; et al. Potent bicyclic inhibitors of malarial cGMP-dependent protein kinase: Approaches to combining improvements in cell potency, selectivity and structural novelty. Bioorg. Med. Chem. Lett. 2019, 29, 126610. [Google Scholar] [CrossRef]
- Large, J.M.; Birchall, K.; Bouloc, N.S.; Merritt, A.T.; Smiljanic-Hurley, E.; Tsagris, D.J.; Wheldon, M.C.; Ansell, K.H.; Coombs, P.J.; Kettleborough, C.A.; et al. Potent inhibitors of malarial P. Falciparum protein kinase G: Improving the cell activity of a series of imidazopyridines. Bioorg. Med. Chem. Lett. 2019, 29, 509–514. [Google Scholar] [CrossRef]
- Diaz, C.A.; Allocco, J.; Powles, M.A.; Yeung, L.; Donald, R.G.K.; Anderson, J.W.; Liberator, P.A. Characterization of Plasmodium falciparum cGMP-dependent protein kinase (PfPKG): Antiparasitic activity of a PKG inhibitor. Mol. Biochem. Parasitol. 2006, 146, 78–88. [Google Scholar] [CrossRef]
- Biftu, T.; Feng, D.; Ponpipom, M.; Girotra, N.; Liang, G.-B.; Qian, X.; Bugianesi, R.; Simeone, J.; Chang, L.; Gurnett, A.; et al. Synthesis and SAR of 2,3-diarylpyrrole inhibitors of parasite cGMP-dependent protein kinase as novel anticoccidial agents. Bioorg. Med. Chem. Lett. 2005, 15, 3296–3301. [Google Scholar] [CrossRef]
- Tsagris, D.J.; Birchall, K.; Bouloc, N.; Large, J.M.; Merritt, A.; Smiljanic-Hurley, E.; Wheldon, M.; Ansell, K.H.; Kettleborough, C.; Whalley, D.; et al. Trisubstituted thiazoles as potent and selective inhibitors of Plasmodium falciparum protein kinase G (PfPKG). Bioorg. Med. Chem. Lett. 2018, 28, 3168–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matralis, A.N.; Malik, A.; Penzo, M.; Moreno, I.; Almela, M.J.; Camino, I.; Crespo, B.; Saadeddin, A.; Ghidelli-Disse, S.; Rueda, L.; et al. Development of Chemical Entities Endowed with Potent Fast-Killing Properties against Plasmodium falciparum Malaria Parasites. J. Med. Chem. 2019, 62, 9217–9235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ul Mahmood, S.; Cheng, H.; Tummalapalli, S.R.; Chakrasali, R.; Yadav Bheemanaboina, R.R.; Kreiss, T.; Chojnowski, A.; Eck, T.; Siekierka, J.J.; Rotella, D.P. Discovery of isoxazolyl-based inhibitors of Plasmodium falciparum cGMP-dependent protein kinase. RSC Med. Chem. 2020, 11, 98–101. [Google Scholar] [CrossRef]
- Reyes, P.; Rathod, P.K.; Sanchez, D.J.; Mrema, J.E.K.; Rieckmann, K.H.; Heidrich, H.-G. Enzymes of purine and pyrimidine metabolism from the human malaria parasite, Plasmodium falciparum. Mol. Biochem. Parasitol. 1982, 5, 275–290. [Google Scholar] [CrossRef]
- Ashton, T.D.; Devine, S.M.; Möhrle, J.J.; Laleu, B.; Burrows, J.N.; Charman, S.A.; Creek, D.J.; Sleebs, B.E. The Development Process for Discovery and Clinical Advancement of Modern Antimalarials. J. Med. Chem. 2019, 62, 10526–10562. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Carrero-Lerida, J.; Silva, A.P.G.; Whittingham, J.L.; Brannigan, J.A.; Ruiz-Perez, L.M.; Read, K.D.; Wilson, K.S.; Gonzalez-Pacanowska, D.; Gilbert, I.H. Synthesis and Evaluation of α-Thymidine Analogs as Novel Antimalarials. J. Med. Chem. 2012, 55, 10948–10957. [Google Scholar] [CrossRef]
- Kato, A.; Yasuda, Y.; Kitamura, Y.; Kandeel, M.; Kitade, Y. Carbocyclic thymidine derivatives efficiently inhibit Plasmodium falciparum thymidylate kinase (PfTMK). Parasitol. Int. 2012, 61, 501–503. [Google Scholar] [CrossRef]
- Noguchi, Y.; Yasuda, Y.; Tashiro, M.; Kataoka, T.; Kitamura, Y.; Kandeel, M.; Kitade, Y. Synthesis of carbocyclic pyrimidine nucleosides and their inhibitory activities against Plasmodium falciparum thymidylate kinase. Parasitol. Int. 2013, 62, 368–371. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, C.; Otto, T.D.; Oberstaller, J.; Liao, X.; Adapa, S.R.; Udenze, K.; Bronner, I.F.; Casandra, D.; Mayho, M.; et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 2018, 360, eaap7847. [Google Scholar] [CrossRef] [Green Version]
Scheme 1.
Flow diagram of article selection (derived from PRISMA flow diagram [19]).
Scheme 1.
Flow diagram of article selection (derived from PRISMA flow diagram [19]).

Figure 1.
Inhibitors of PfCDPK1 K252a 1 and purfalcamine 2.

Figure 2.
Inhibitors of PfCDPK1 discovered by Lemercier et al. [28].
Figure 2.
Inhibitors of PfCDPK1 discovered by Lemercier et al. [28].

Figure 3.
Best compounds, from each chemical series having a molecule with an IC50 equal to or below 20 nM toward PfCDPK1 in the screening realized by Crowther et al. [33].
Figure 3.
Best compounds, from each chemical series having a molecule with an IC50 equal to or below 20 nM toward PfCDPK1 in the screening realized by Crowther et al. [33].

Figure 4.
4-Aminopyrazolopyrimidine and 4-aminoimidazopyrazine inhibitors of PfCDPK4. 1 On mammalian fibroblast cells. 2 On HepG2 cells.
Figure 4.
4-Aminopyrazolopyrimidine and 4-aminoimidazopyrazine inhibitors of PfCDPK4. 1 On mammalian fibroblast cells. 2 On HepG2 cells.

Figure 5.
Aminopyrazole-carboxamide and indolone inhibitors of PfCDPK4.

Figure 6.
Hexadecyltrimethylammonium bromide 20 structure.

Figure 7.
Hits from Crowther et al. against PfCK [46].
Figure 7.
Hits from Crowther et al. against PfCK [46].

Figure 8.
CX-4945, an HsCK2 inhibitor in clinical trials, also inhibits PfCK2.

Figure 9.
Imidazopyridazines inhibitors of PfCLK1.

Figure 10.
Molecules inhibiting PfCLK1 or PfCLK3.

Figure 11.
Inhibitors of PfDPCK from Fletcher et al. [61]. 1 At 40 µM. EG = Early-stage Gametocytes, LG = Late-stage Gametocytes.
Figure 11.
Inhibitors of PfDPCK from Fletcher et al. [61]. 1 At 40 µM. EG = Early-stage Gametocytes, LG = Late-stage Gametocytes.

Figure 12.
Chemical structure of emodin.

Figure 13.
Inhibitors of PvGK.

Figure 14.
Inhibitor of Pfmap-2 discovered by Brumlik et al. [77].
Figure 14.
Inhibitor of Pfmap-2 discovered by Brumlik et al. [77].

Figure 15.
Inhibitors of PfMRK.

Figure 16.
Natural quinone inhibitors of Pfnek-1. 1 4-day Peter’s test at 5 mg/kg/4 days intraperitoneal.
Figure 16.
Natural quinone inhibitors of Pfnek-1. 1 4-day Peter’s test at 5 mg/kg/4 days intraperitoneal.

Figure 17.
Starting from 67, medicinal chemistry work led to 66 which was further improved to 68. EG = Early-stage gametocytes, LG = Late-stage gametocytes. 1 mouse model, 20 mg/kg per os.
Figure 17.
Starting from 67, medicinal chemistry work led to 66 which was further improved to 68. EG = Early-stage gametocytes, LG = Late-stage gametocytes. 1 mouse model, 20 mg/kg per os.

Figure 18.
Aminopyrazines derivatives used to study a sulfoxide-to sulfone produg approach.

Figure 19.
1,5-naphthyridines as inhibitors of PfPI4K.

Figure 20.
2-chloro-3-sulfonamide pyridines as inhibitors of PfPI4K.

Figure 21.
Inhibitors of PfPK5 discovered by Eubanks et al. [95].
Figure 21.
Inhibitors of PfPK5 discovered by Eubanks et al. [95].

Figure 22.
Molecules inhibiting PfPK6 discovered by Crowther et al. [33].
Figure 22.
Molecules inhibiting PfPK6 discovered by Crowther et al. [33].

Figure 23.
Imidazopyridazine inhibitors of PfPK7.


Figure 25.
Pyrazolopyridine 90 as inhibitor of PfPKG.

Figure 26.
Trisubstituted pyrrole as inhibitor of PfPKG.

Figure 27.
Trisubstituted thiazoles as inhibitors of PfPKG.

Figure 28.
Trisubstituted isoxazole and imidazole as inhibitors of PfPKG.

Figure 29.
α-thymidine urea analogs as inhibitors of PfTMK.

Figure 30.
Cyclopentene analogs of thymidine as inhibitors of PfTMK.

Figure 31.
Mutagenesis Index Score graph with protein kinases described in the review highlighted. All P. falciparum genes were ordered from the lowest to the highest score and then attributed an arbitrary rank number to obtain this curve. Data from Zhang et al. [124].
Figure 31.
Mutagenesis Index Score graph with protein kinases described in the review highlighted. All P. falciparum genes were ordered from the lowest to the highest score and then attributed an arbitrary rank number to obtain this curve. Data from Zhang et al. [124].


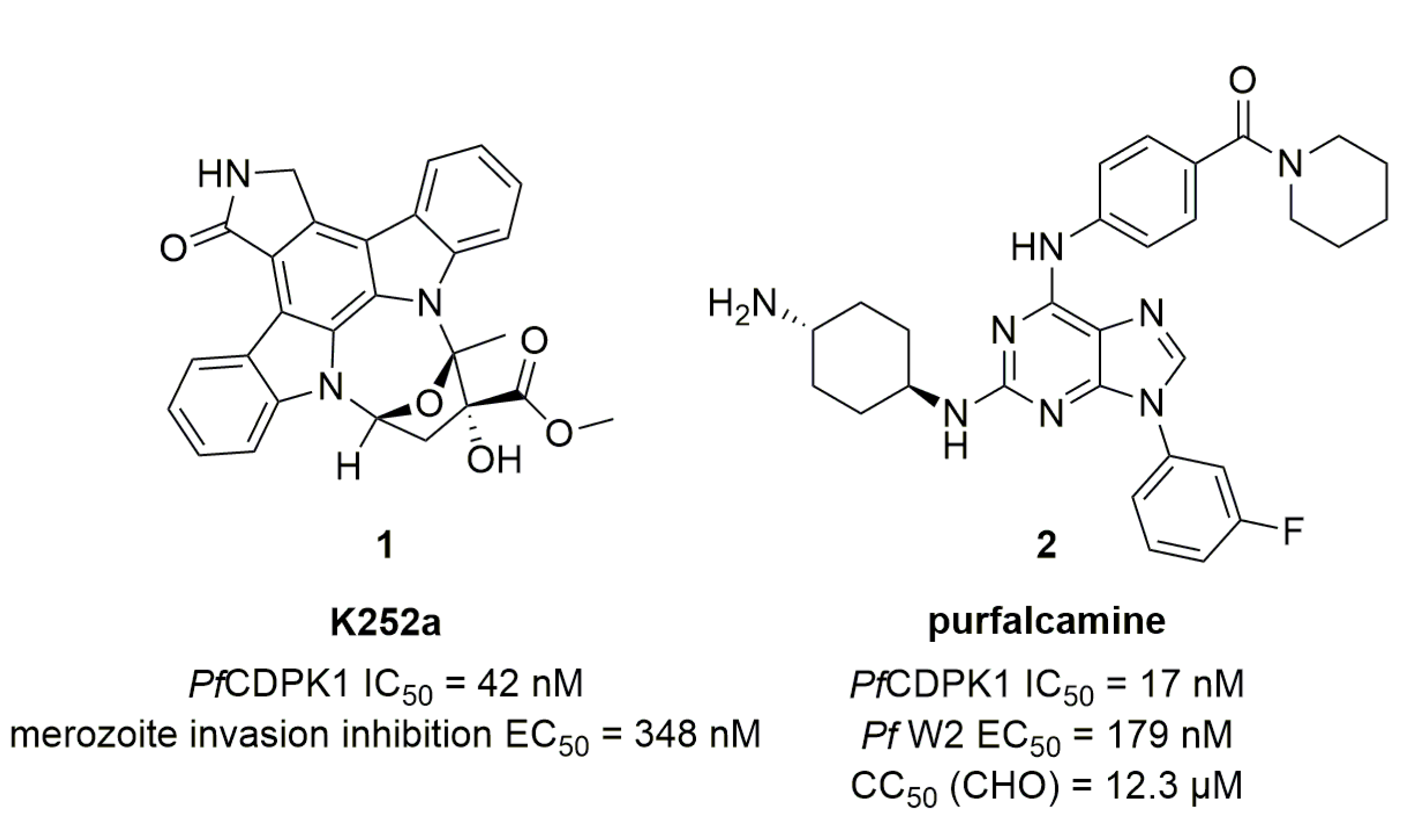{kind=link}
{kind=link}
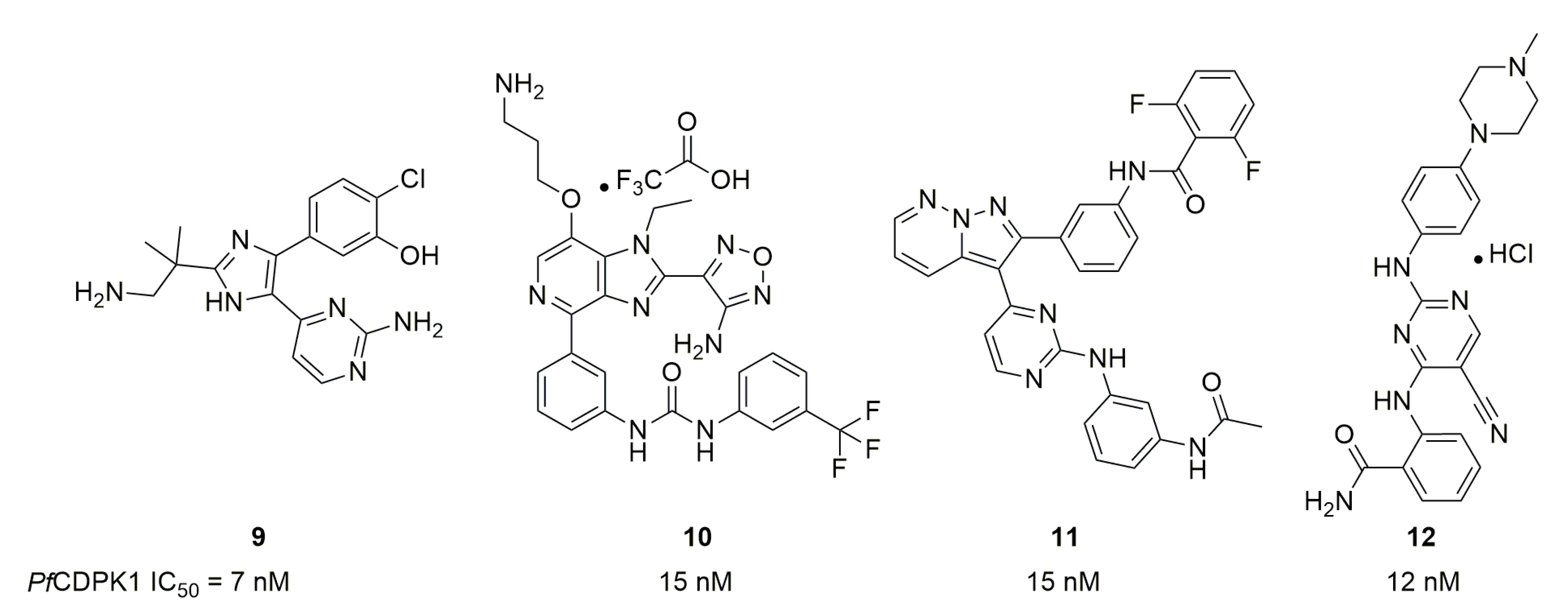{kind=link}
{kind=link}
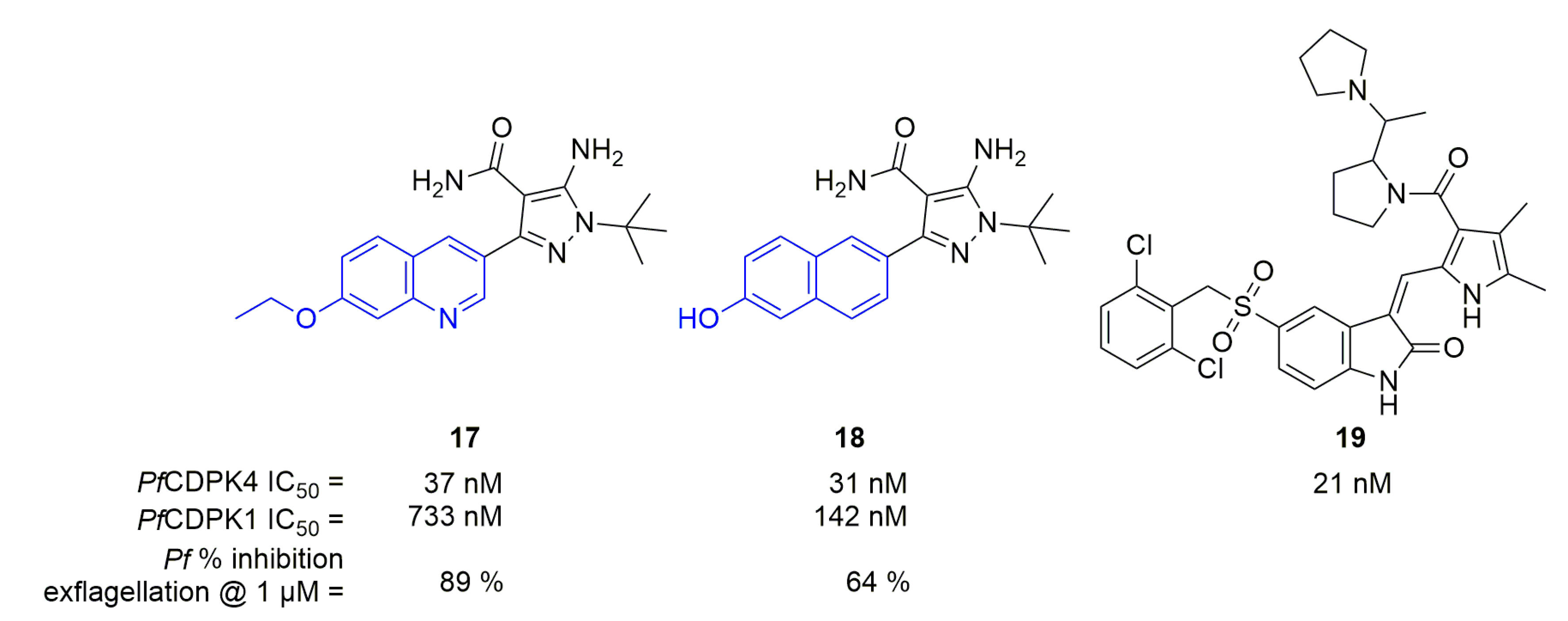{kind=link}
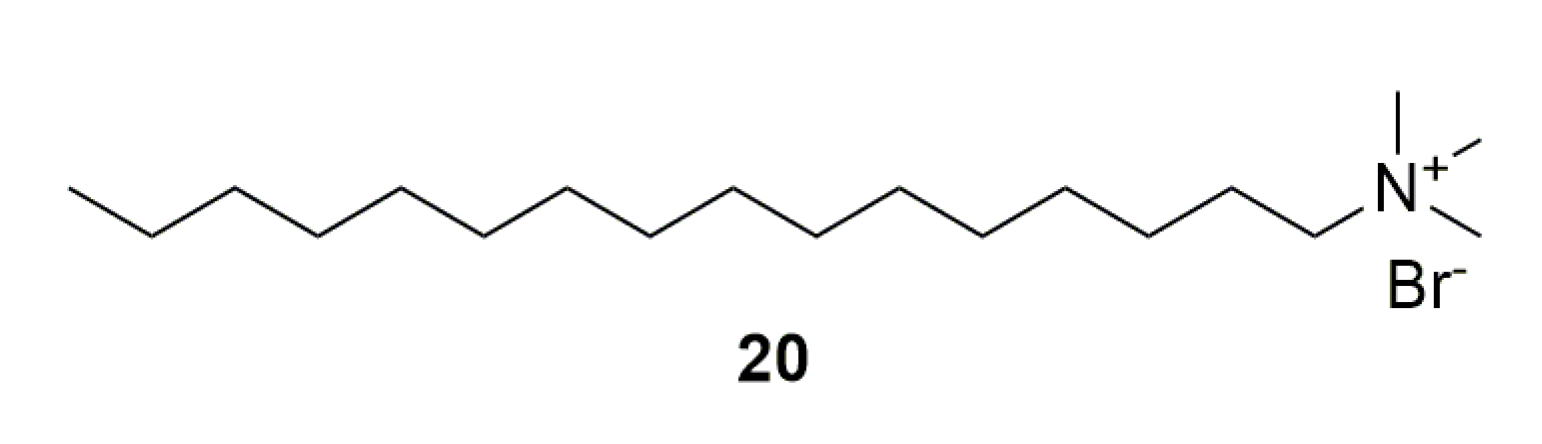{kind=link}
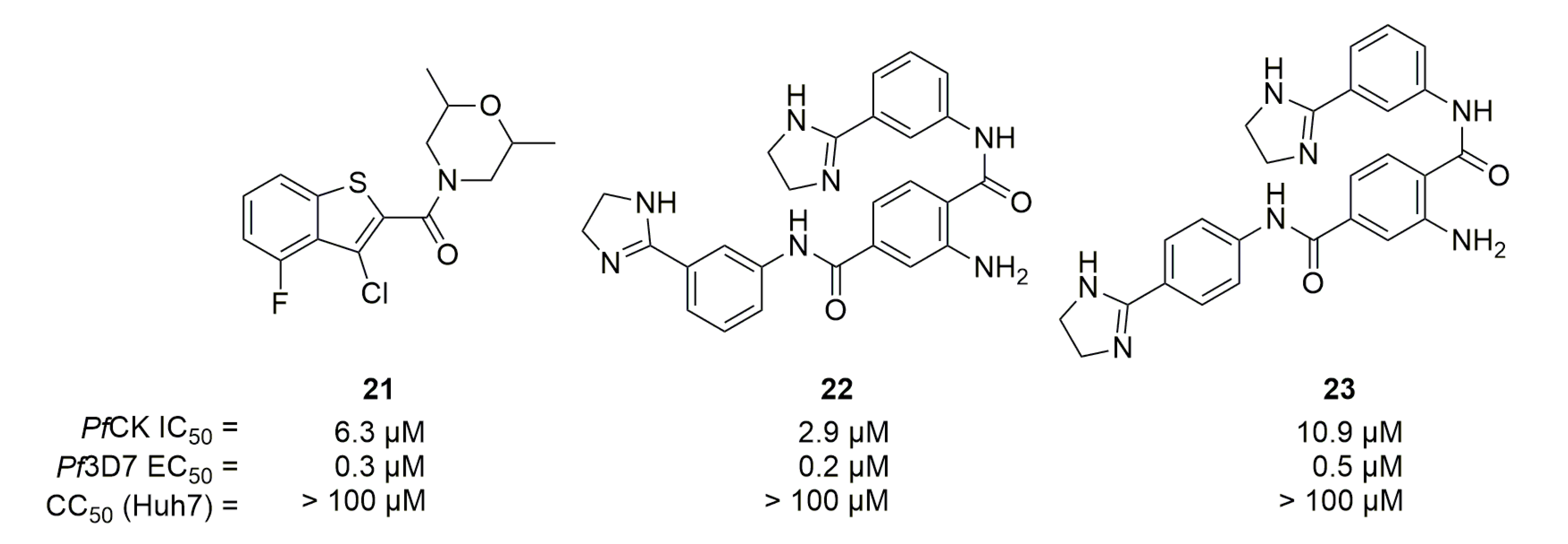{kind=link}
{kind=link}
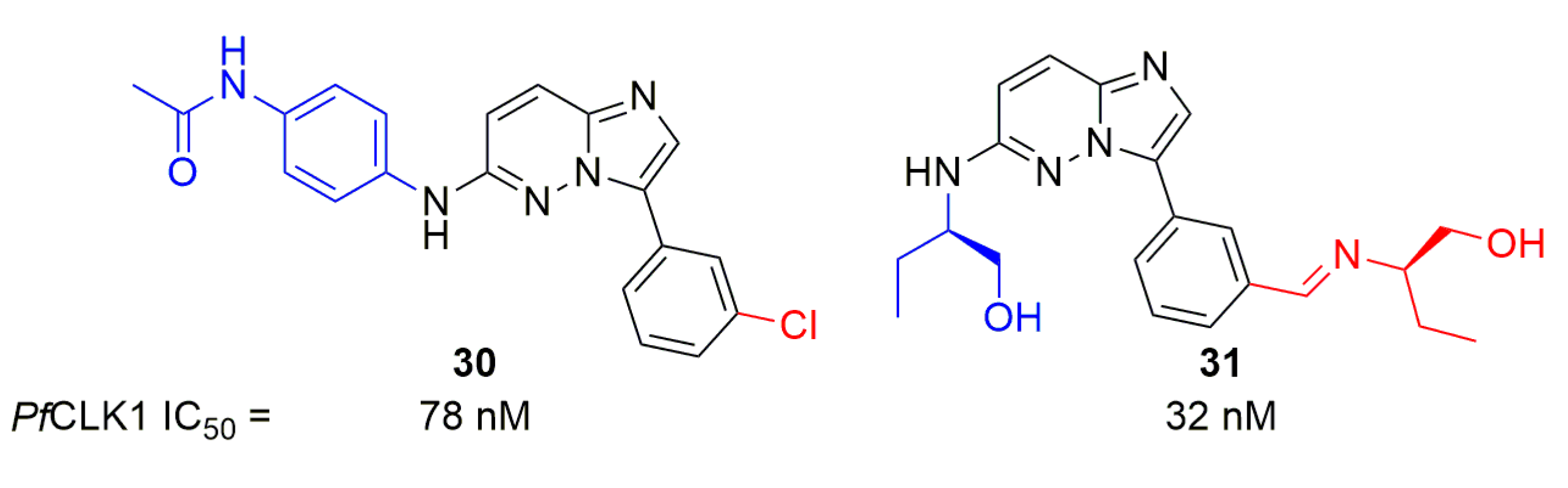{kind=link}
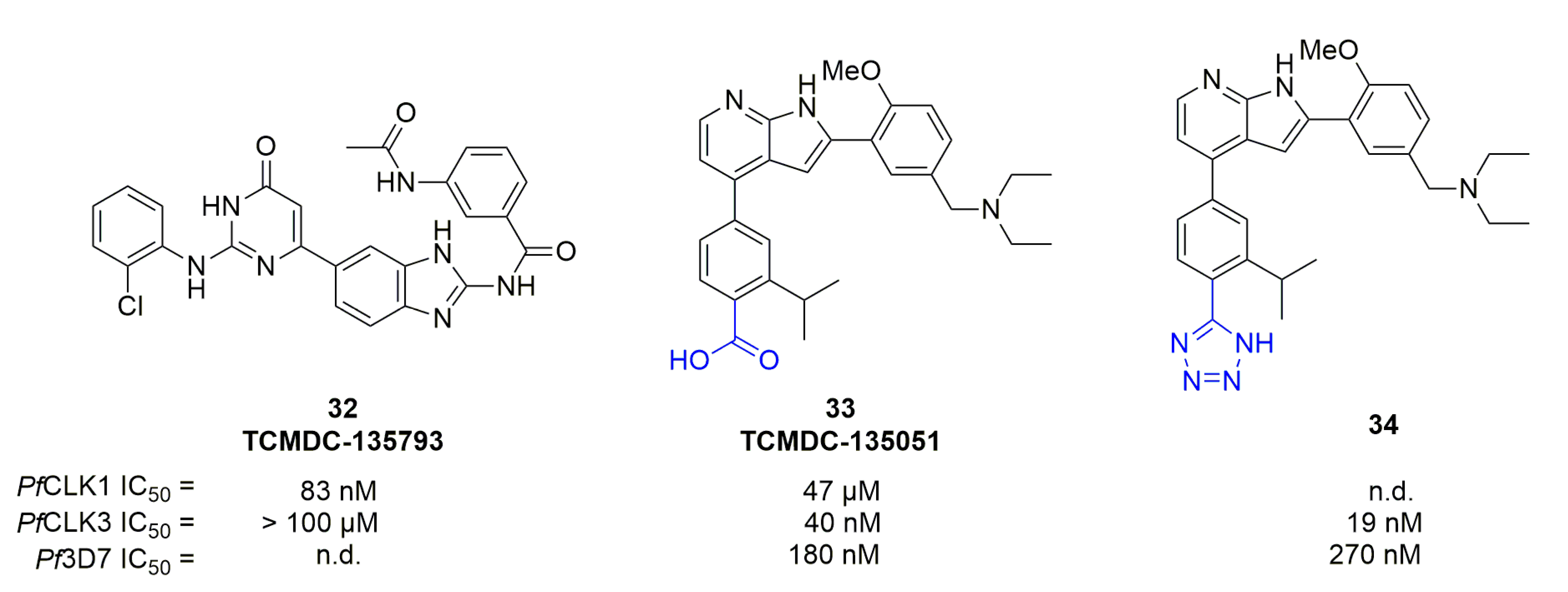{kind=link}
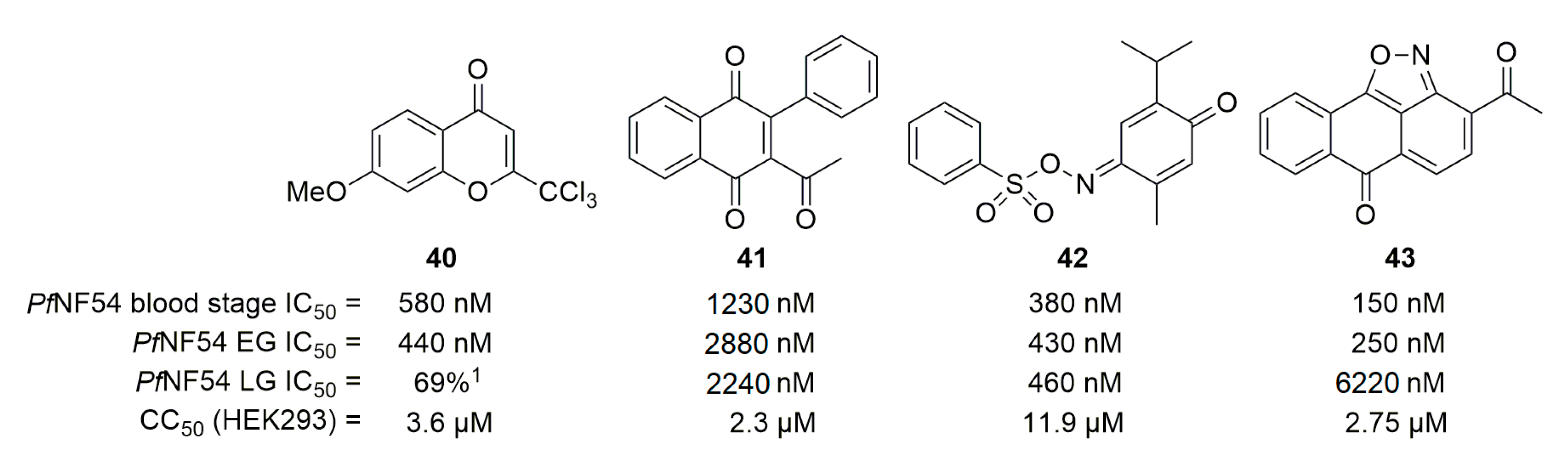{kind=link}
{kind=link}
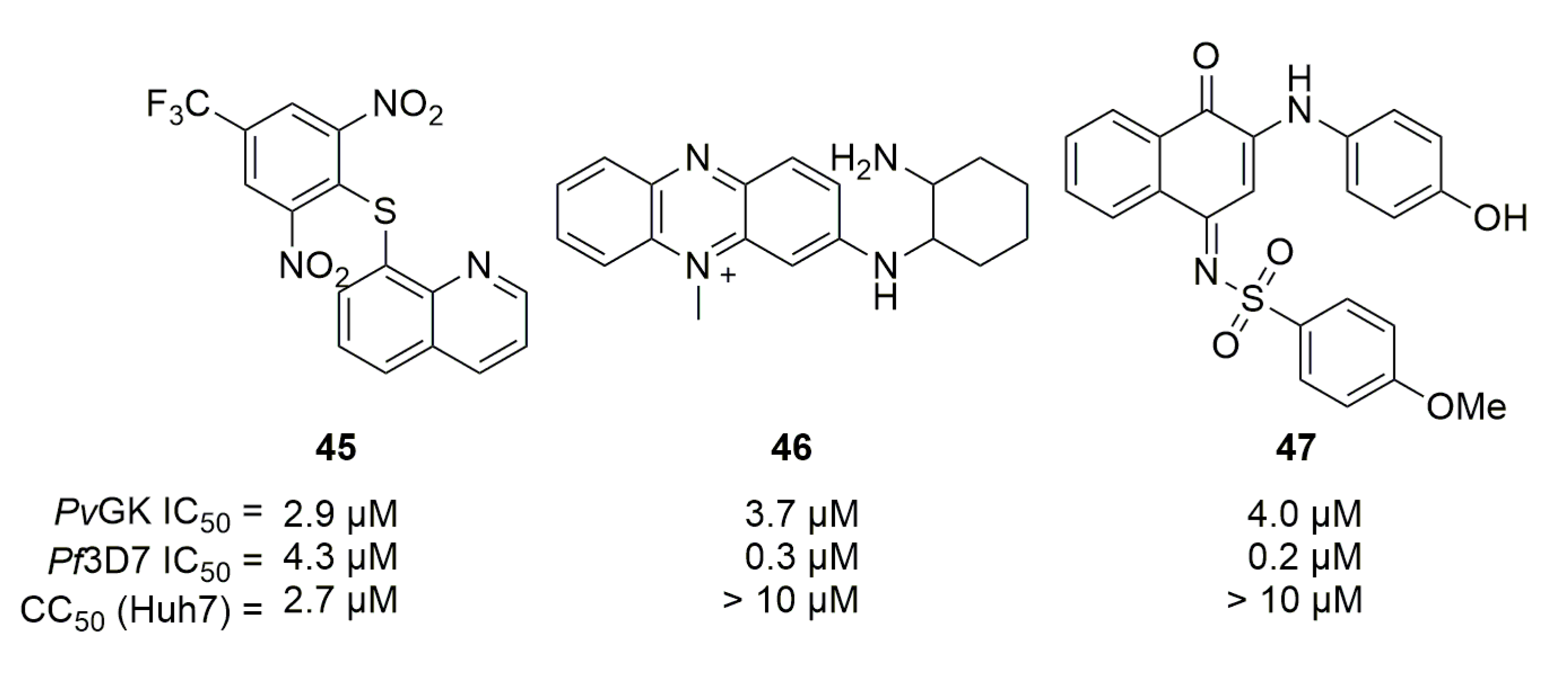{kind=link}
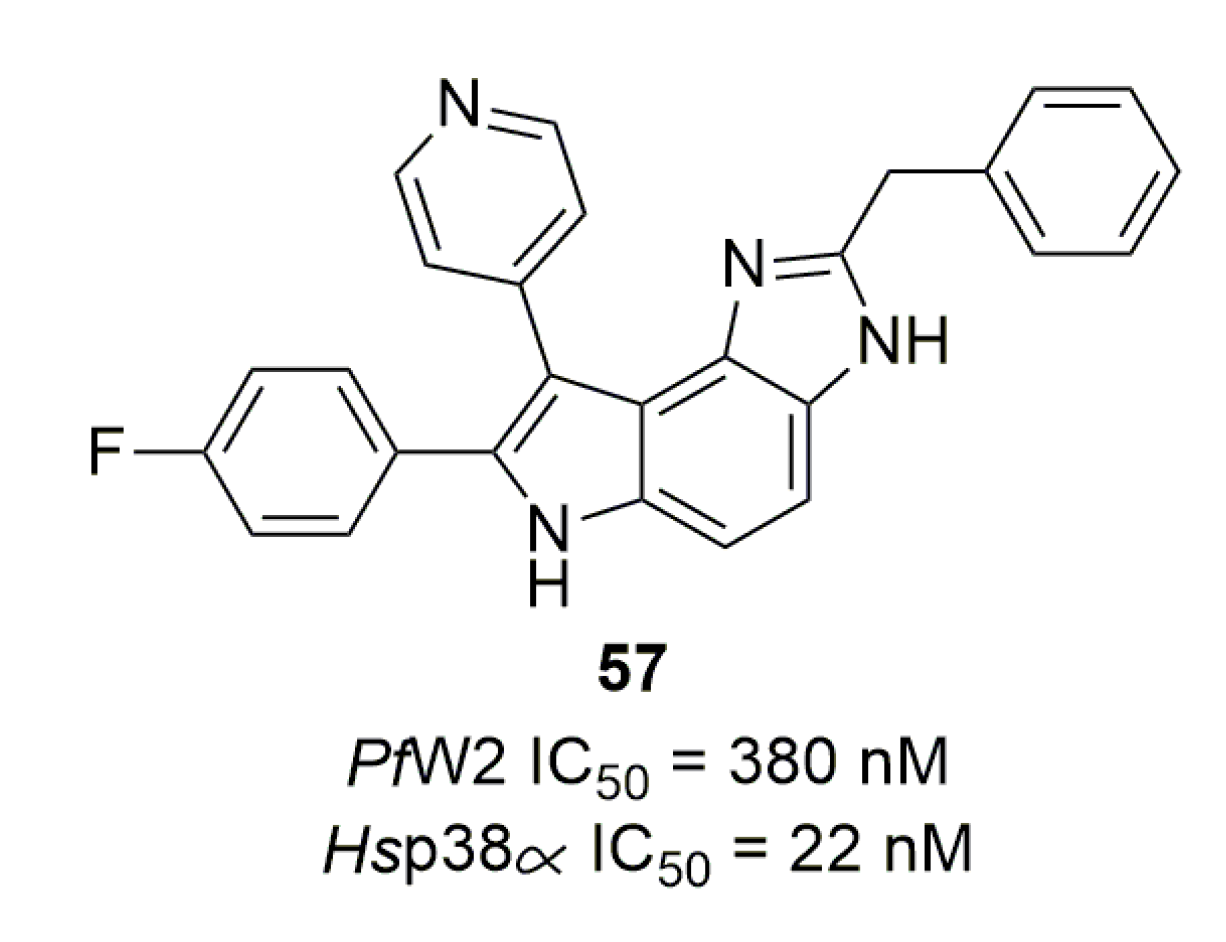{kind=link}
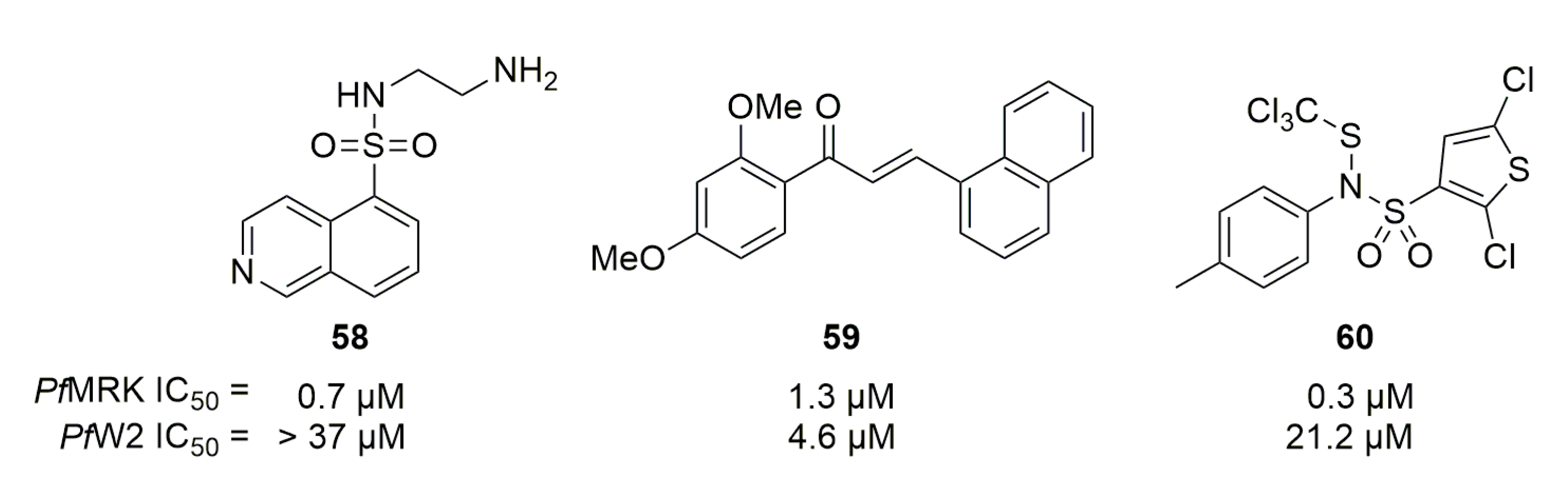{kind=link}
{kind=link}
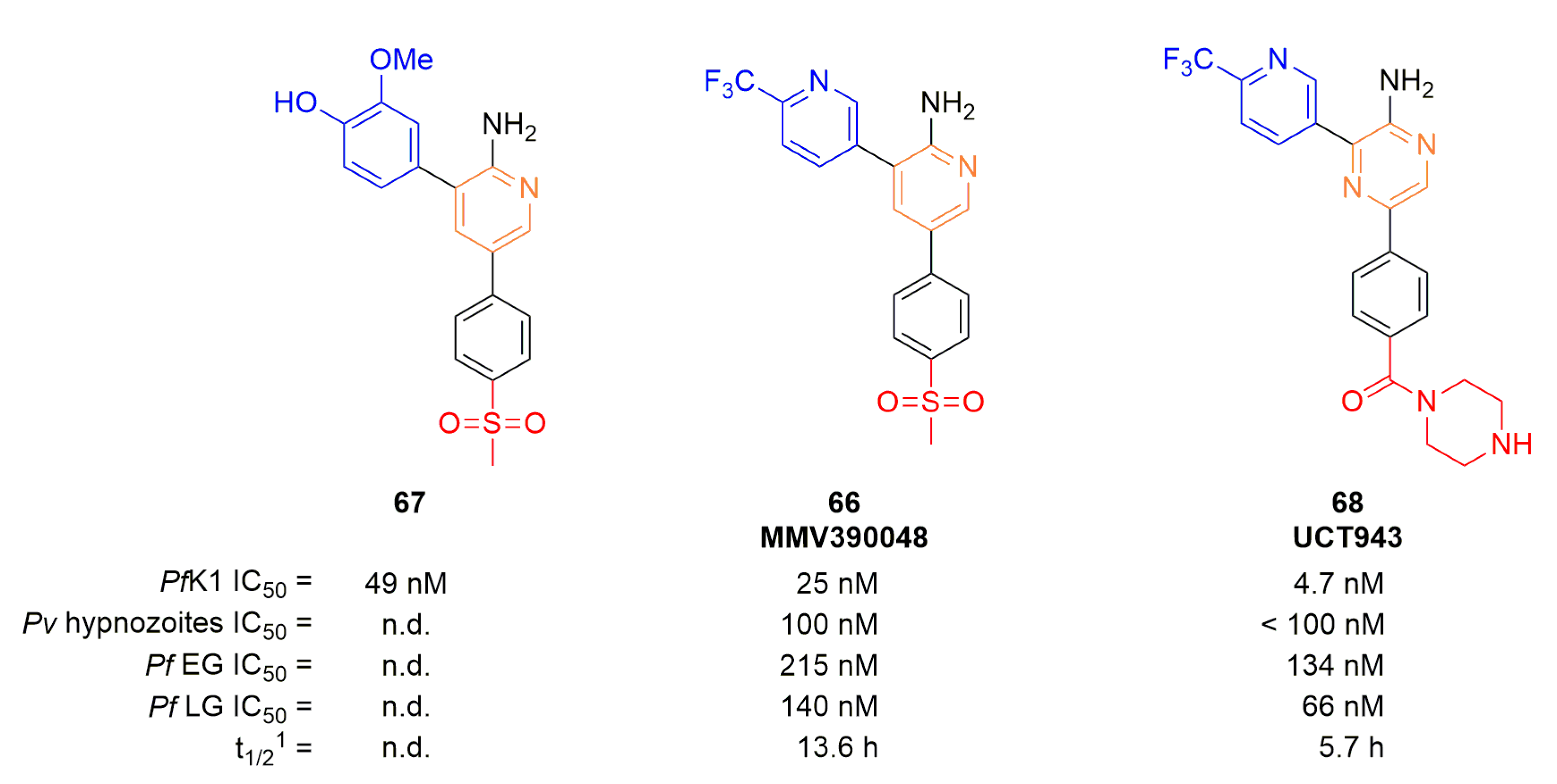{kind=link}
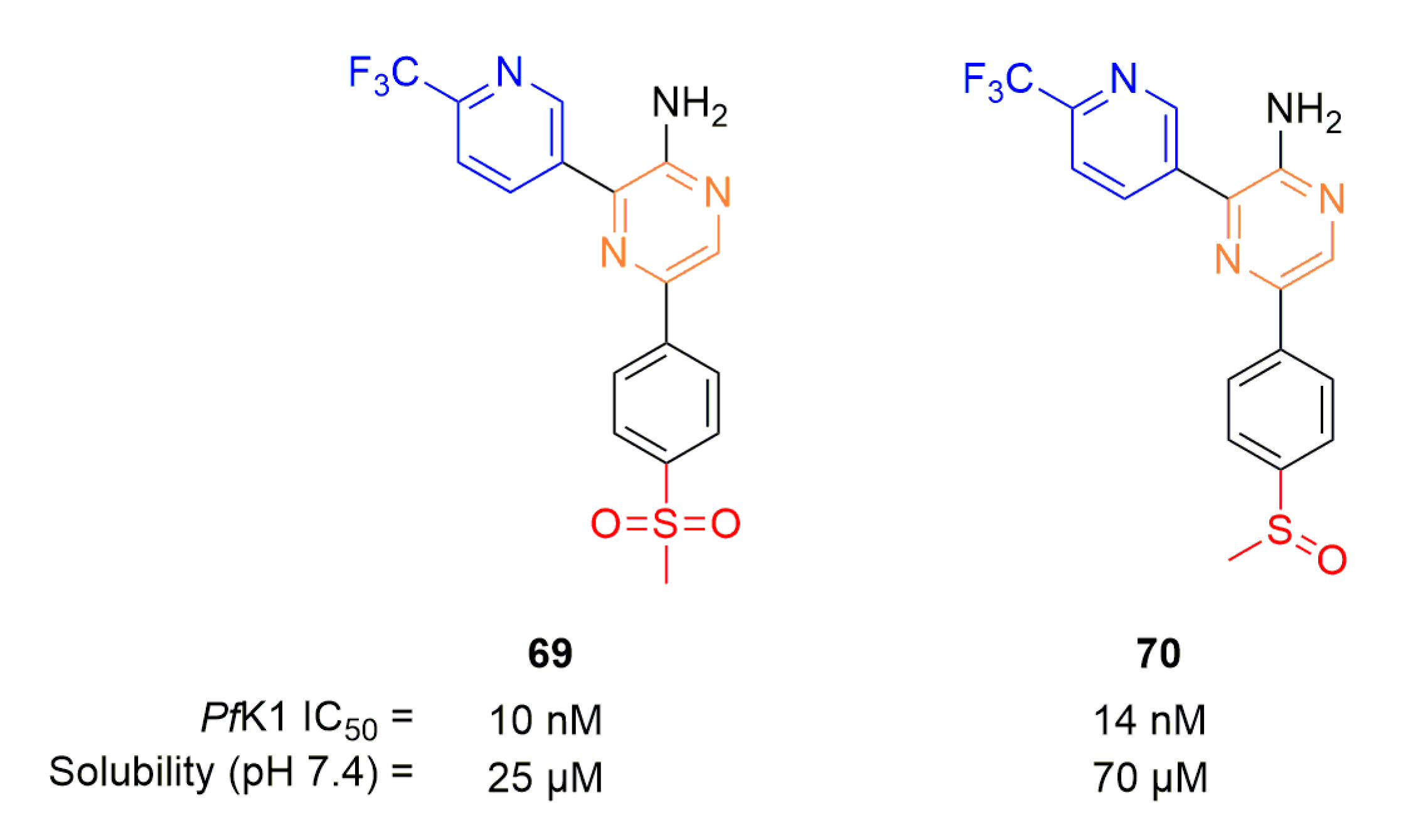{kind=link}
{kind=link}
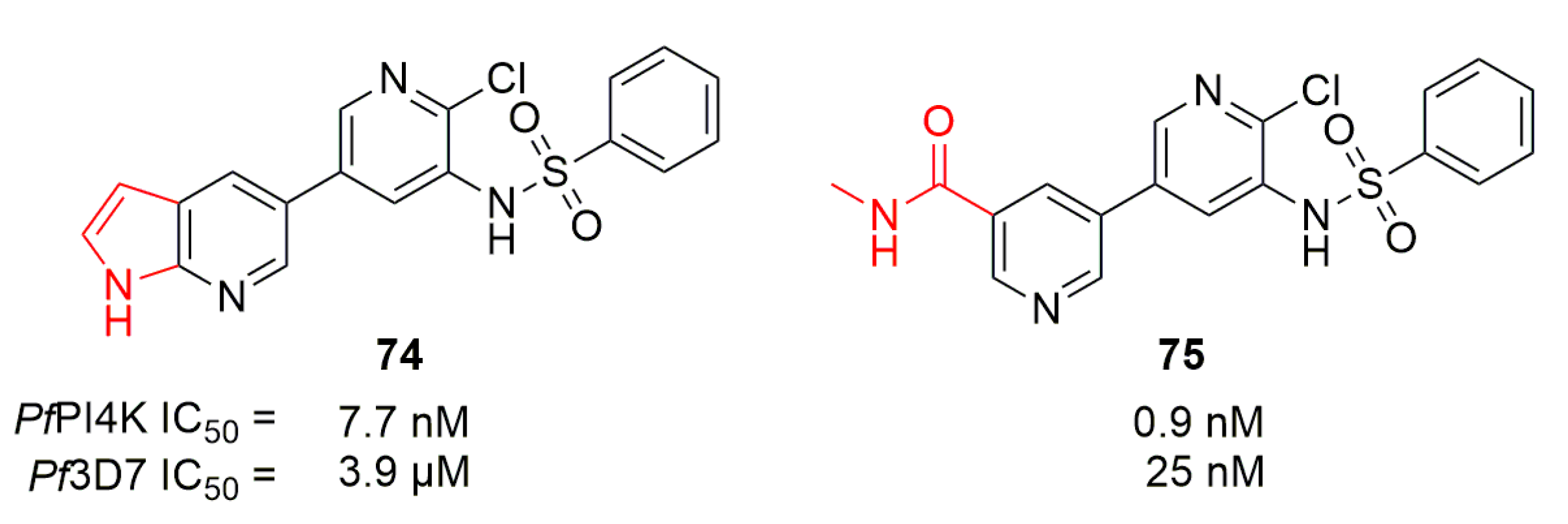{kind=link}
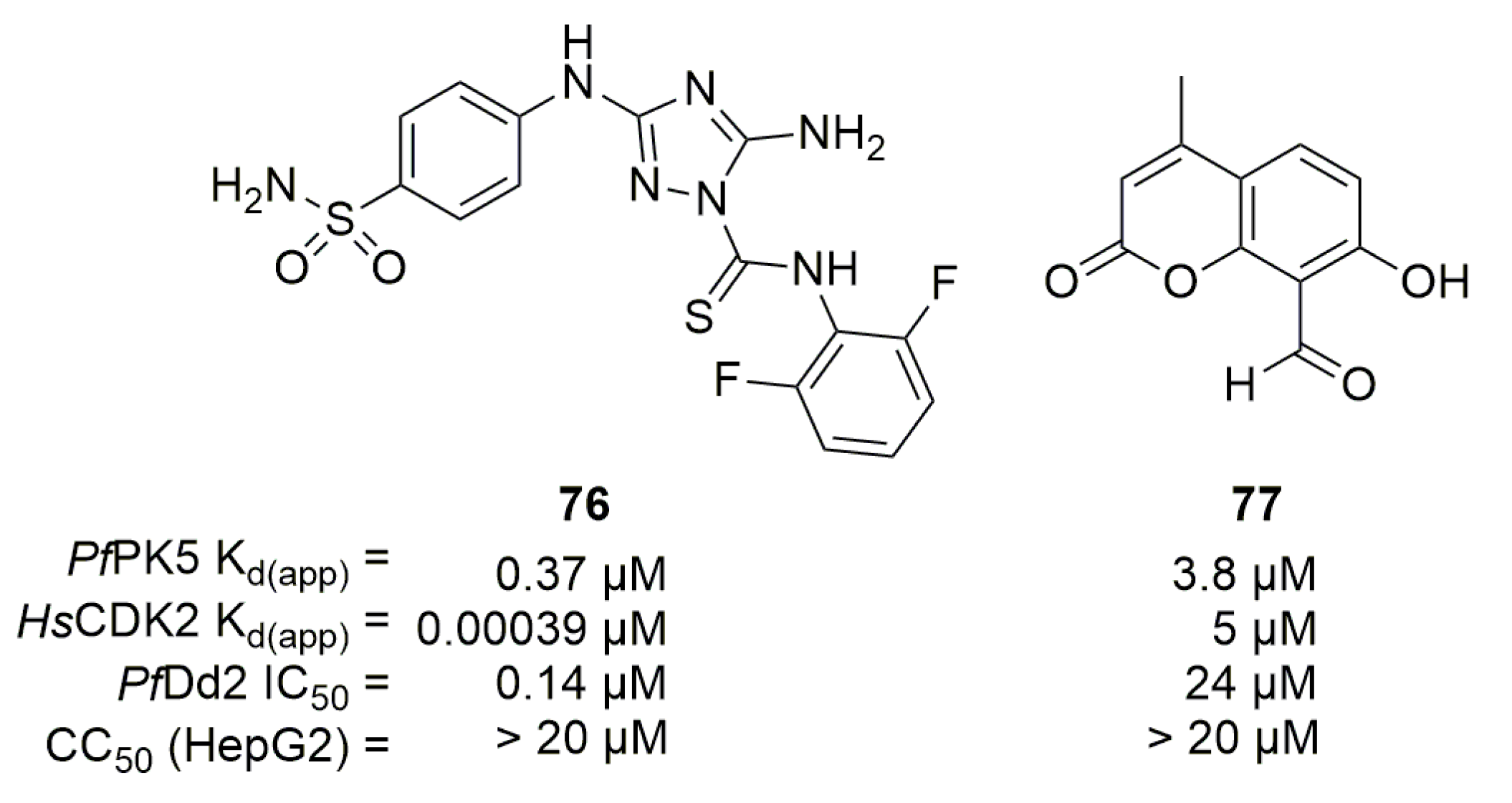{kind=link}
{kind=link}
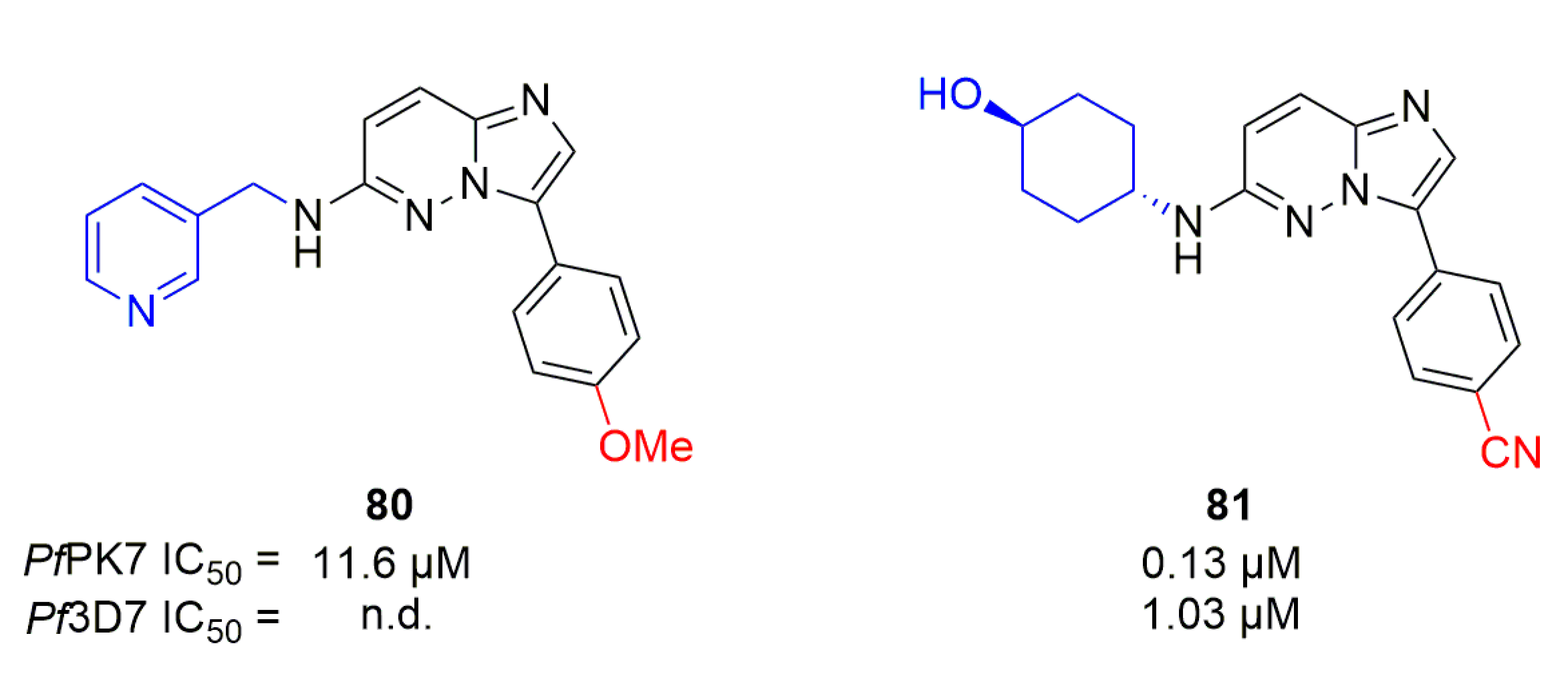{kind=link}
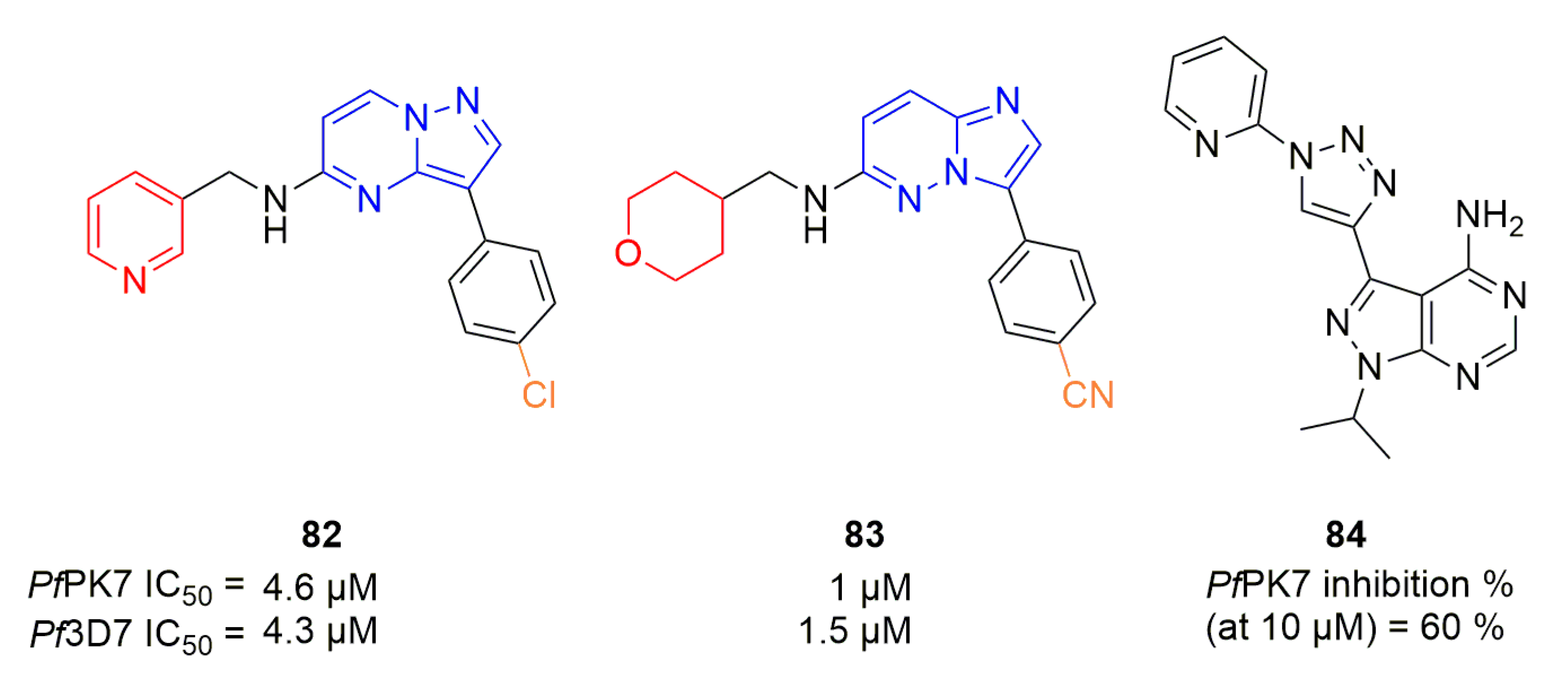{kind=link}
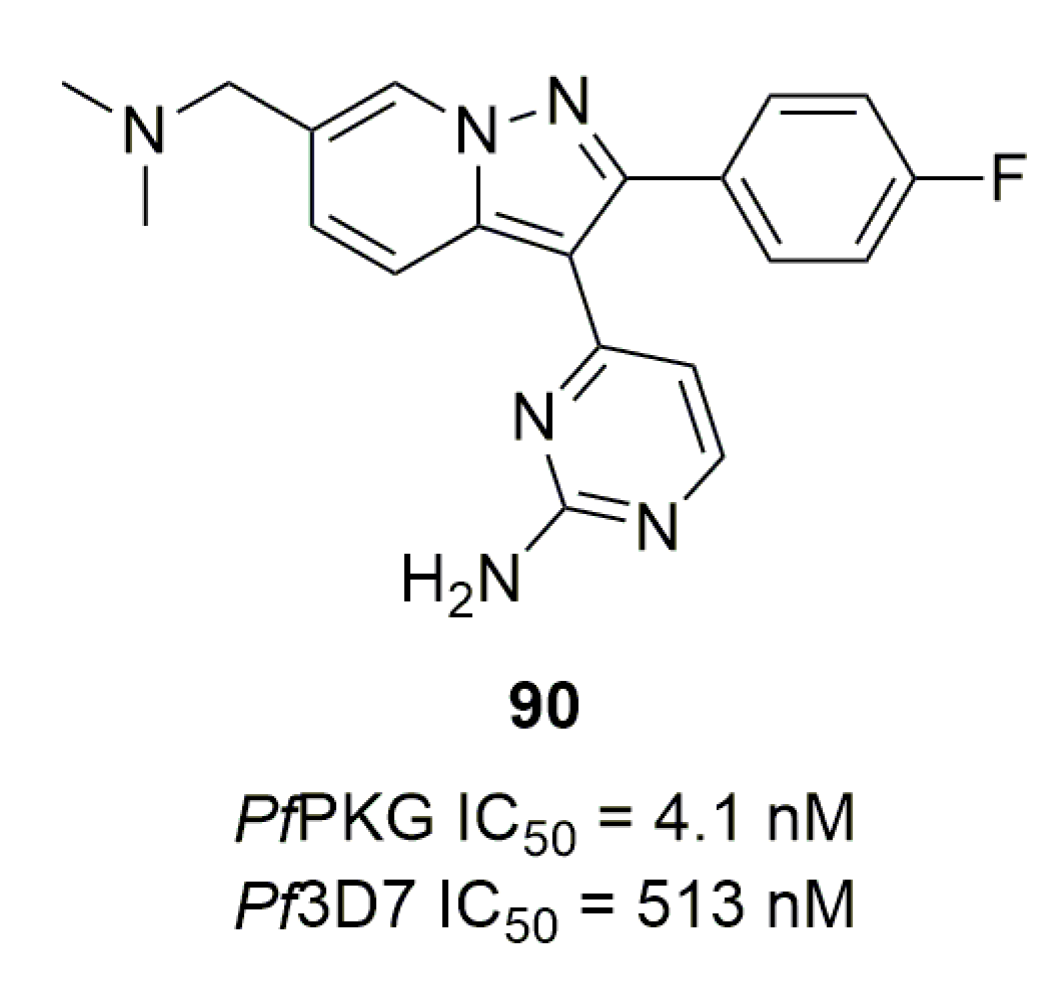{kind=link}
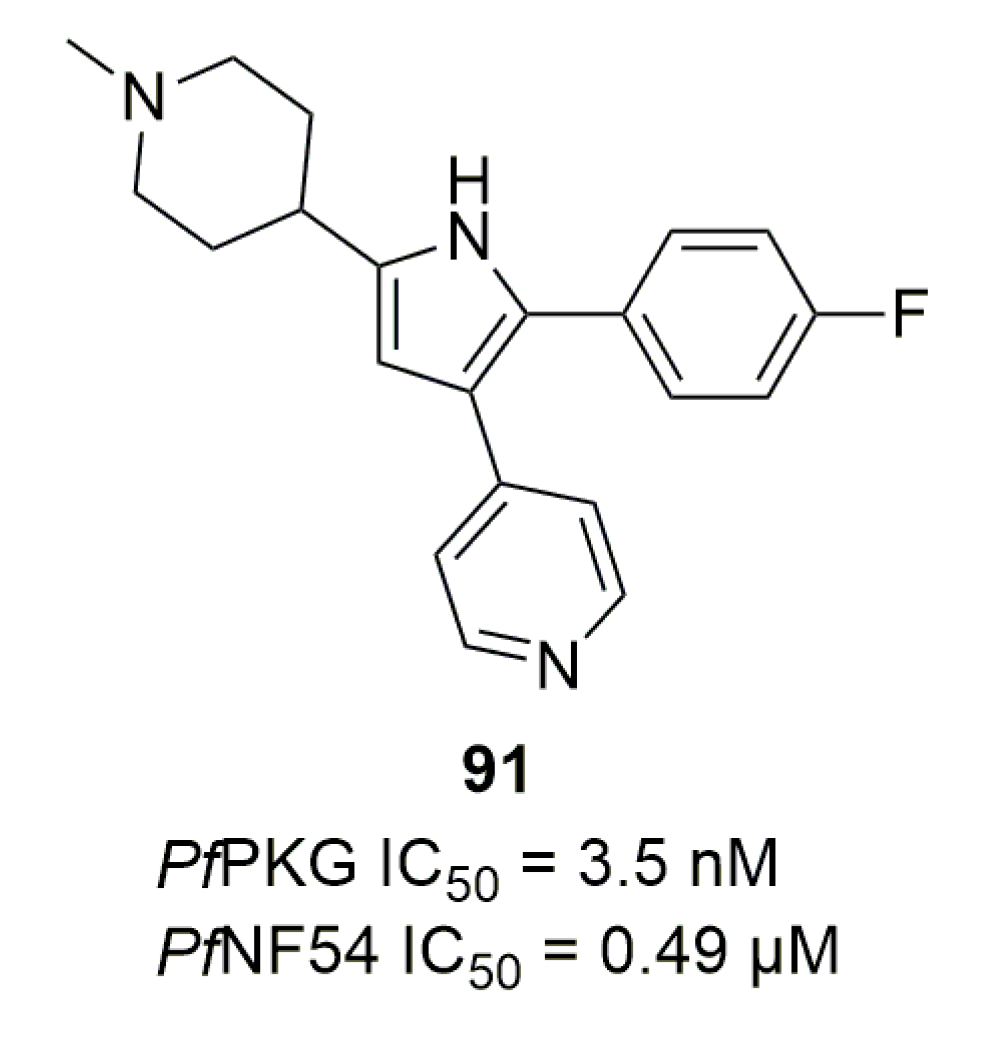{kind=link}
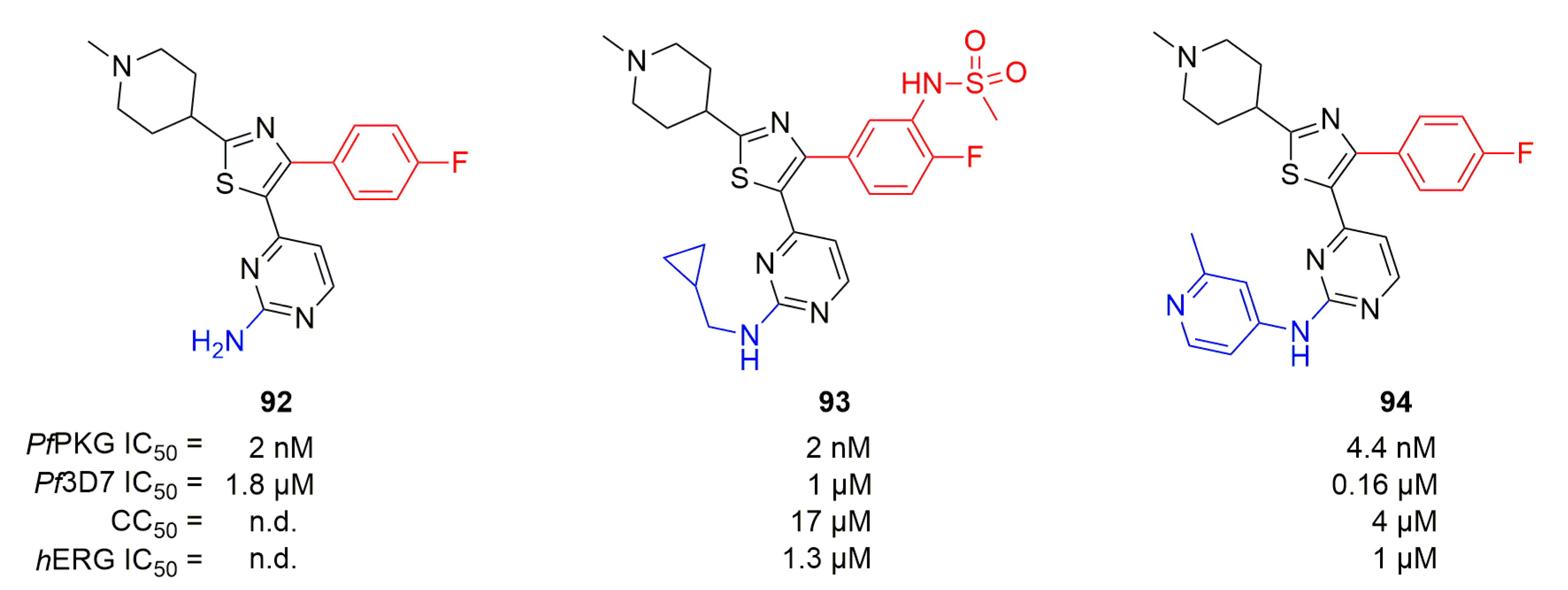{kind=link}
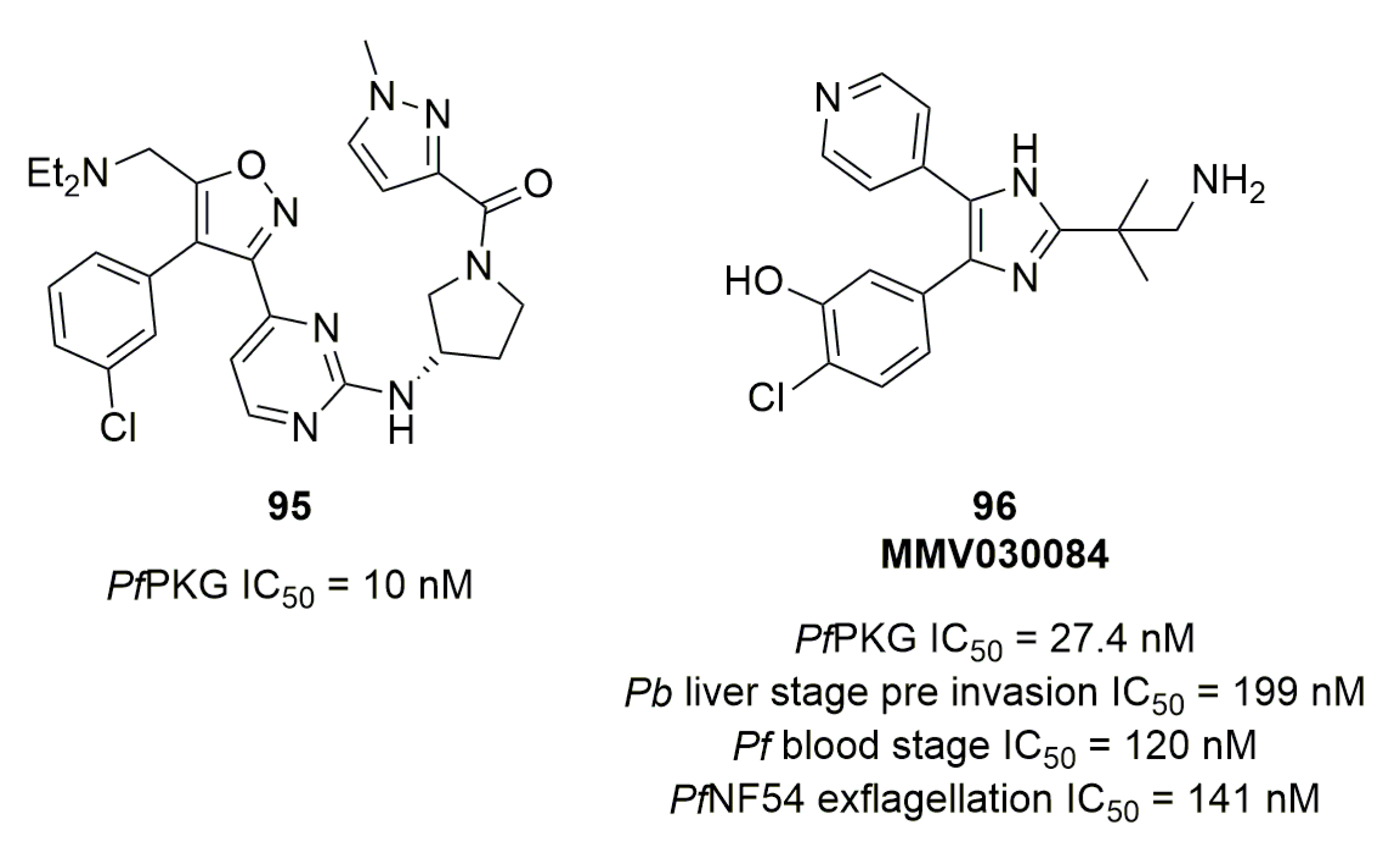{kind=link}
{kind=link}
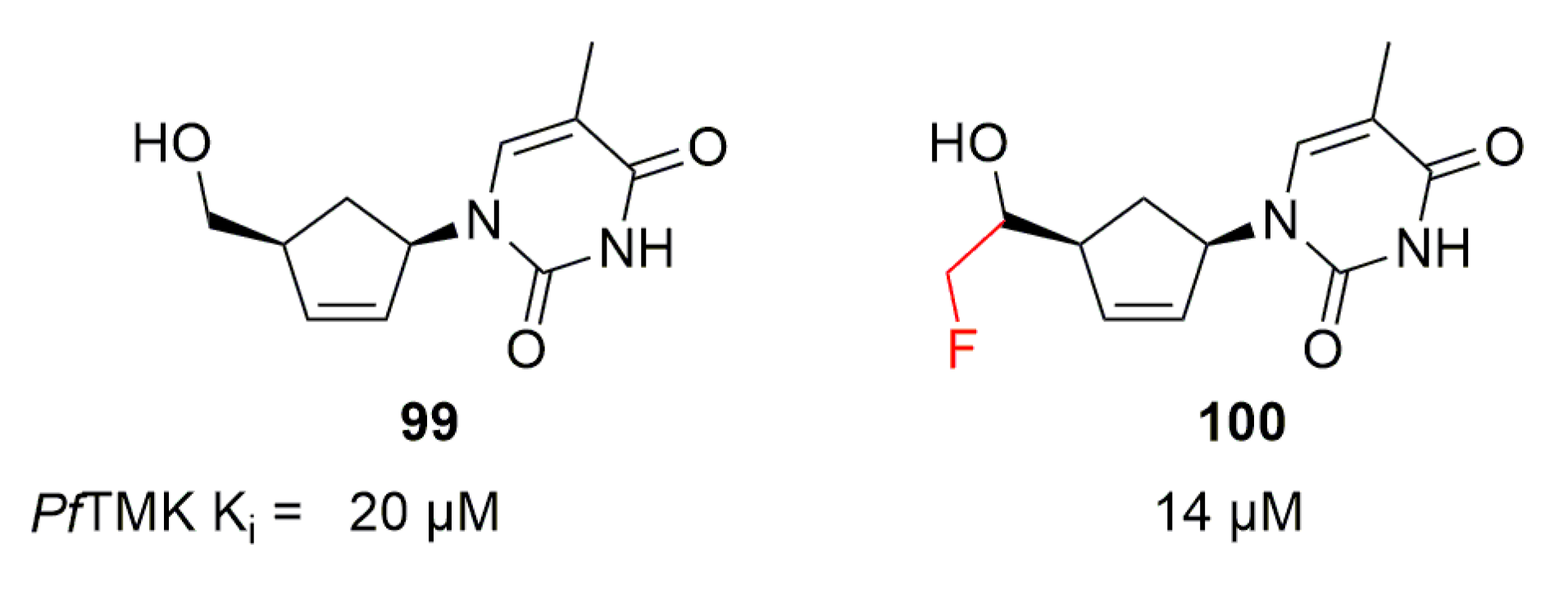{kind=link}
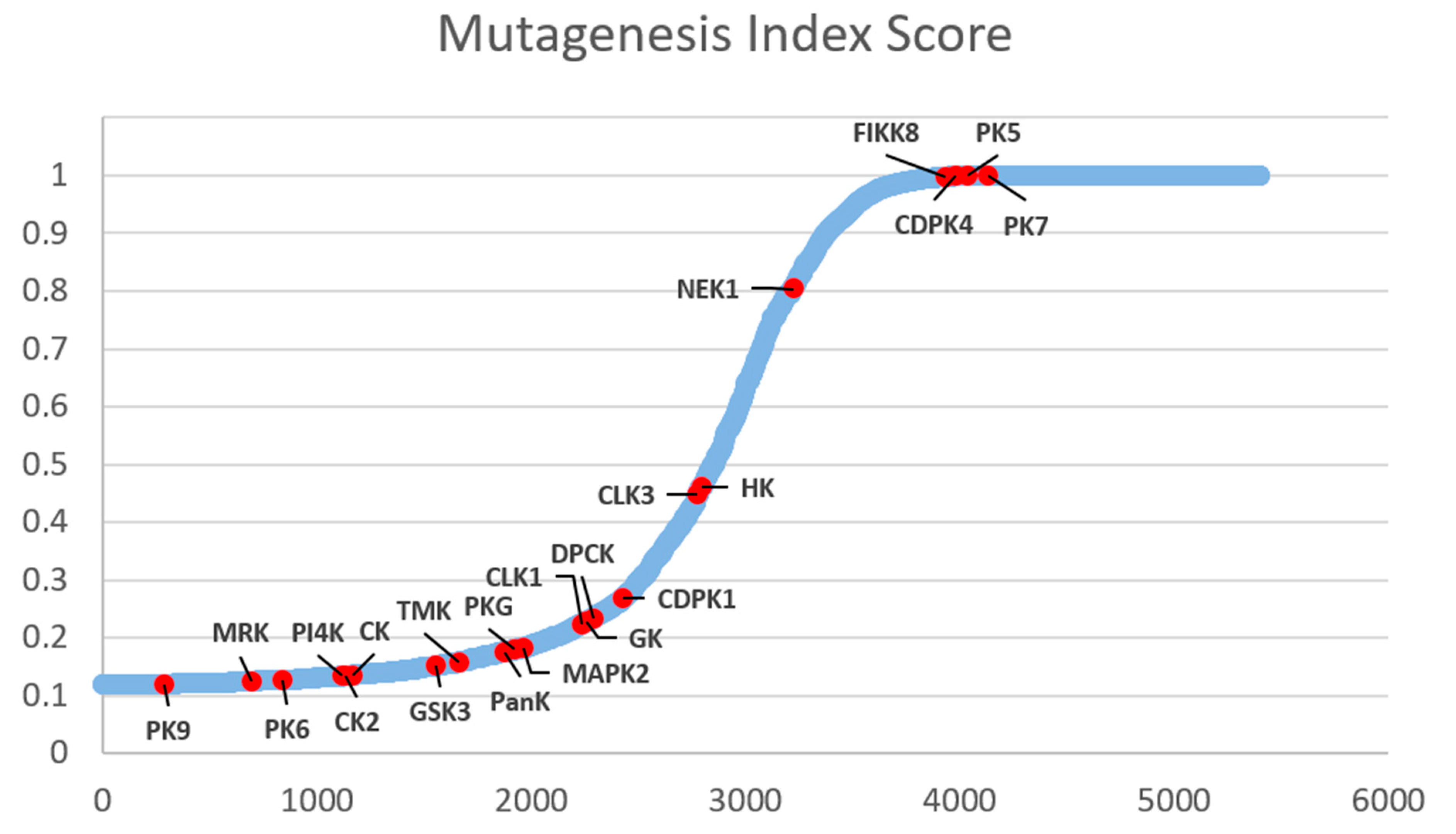{kind=link}
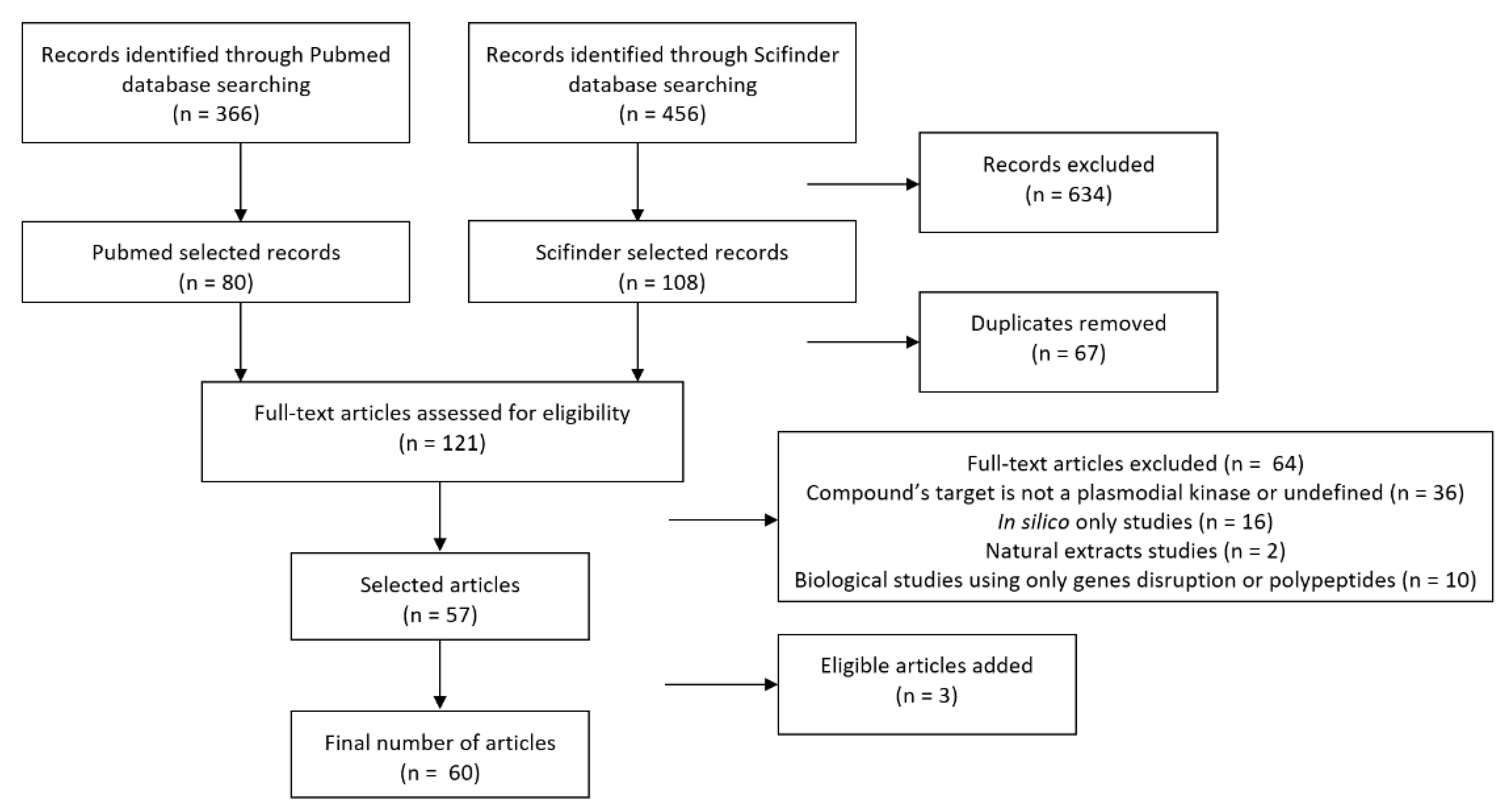{kind=link}
Table 1.
Inclusion and exclusion criteria used.
| Parameter | Inclusion | Exclusion |
|---|---|---|
| Compound’s target | A plasmodial kinase clearly defined as target | Target is not a plasmodial kinase or target is undefined |
| Study type | Medicinal chemistry studies Biology studies listing properties of one or more chemical compounds | In silico studies without biological tests Natural extract studies without active compounds determined Biological studies on protein pathways/phenotypic changes using only genes disruption or polypeptides |
Table 2.
Imidazopyridazine inhibitors of PfCDPK1.

| Compound | R1 | R2 | PfCDPK1 IC50 (nM) | Pf3D7 IC50 (nM) | In Vivo Parasitemia Reduction 1 |
|---|---|---|---|---|---|
| 5 |  |  | 13 | 400 | 46 |
| 6 |  |  | 70 | 103 | - |
| 7 |  |  | <10 | 12 | 4% |
| 8 |  |  | 12 | 80 | 51% |
1 Four-day Peter’s test [27] at an oral dose of 50 mg/kg/day.
Table 3.
ADMET parameters of compound 13 and 14 [37].
Table 3.
ADMET parameters of compound 13 and 14 [37].
| In Vitro | In Vivo | ||||
|---|---|---|---|---|---|
| Compound | Solubility (µM) in Buffer pH 6.5 | t1/2 (min) Human Liver Microsome | AUC 1 (µM·min) at 10 mg/kg per os | AUC 1 (µM·min) at 10 mg/kg Intraperitoneal | t1/2 (h) at 100 mg/kg per os |
| 13 | 47 | - | 57 | 317 | - |
| 14 | 82 | >60 | 430 | 863 | 13.5 |
1 AUC = Area Under Curve.
Table 4.
Bispyridinium bromide salt and bisquinolinium bromide salt inhibitors of PfCK.
| Compound | PfCKCho IC50 (µM) | Pf3D7 IC50 (nM) | HsCKα1 IC50 |
|---|---|---|---|
 | 276 | 3 | - |
 | 103 | 3 | - |
 | 2.4 | 142 | 1.7 |
Table 5.
Summary of in vitro properties of PfCLK inhibitors from Kern et al. [53].
Table 5.
Summary of in vitro properties of PfCLK inhibitors from Kern et al. [53].
| Compound | Pf3D7 IC50 (µM) | EIA 1 IC50 (µM) | % Inhibition (at Pf3D7 IC50 Concentration) | |||
|---|---|---|---|---|---|---|
| PfCLK1 | PfCLK2 | PfCLK3 | PfCLK4 | |||
 | 4.4 | 13.8 | 73% | 45.6% | 35.1% | 71.6% |
 | 0.6 | 19.8 | 68.4% | 76.4% | 74.2% | 76.1% |
1 Exflagellation Inhibitory Assay.
Table 6.
Pantothenate and pantothenate analog inhibitors of PfPanK.
| Compound | P. falciparum IC50 (µM) 1,2 | Pantothenate Phosphorylation Inhibition % | PfPanK IC50 |
|---|---|---|---|
 | - | - | - |
 | 76 | >85% (at 1 mM) | - |
 | 36 | - | 14 µM |
 | 13 | >60% (at 10 µM) | 140 nM |
 | 0.03 | - | - |
1Pf3D7 strain except for 39, where the value shown is on PfW2 strain. 2 Pantothenate concentration in growth medium was 1 µM except for 39, where it was 1.14 µM.
Table 7.
Thieno[2,3-b]pyridine inhibitors of PfGSK3.

| Compound | R | % Inhibition PfNF54LUC at 3 µM | PfGSK3 IC50 (µM) | HsGSK3 IC50 (µM) | PfNF54LUC IC50 (µM) | CC50 (HEK293, µM) |
|---|---|---|---|---|---|---|
| 48 |  | n.d. 1 | 0.24 | 9.08 | 5.5 | 35.2 |
| 49 |  | 99.9 | 0.72 | 40.2 | 1.2 | 5.56 |
| (+)-50 |  | 73.0 | n.d. | n.d. | 1.1 | n.d. |
| (−)-50 |  | −24.4 | n.d. | n.d. | n.d. | n.d. |
1 n.d. = not determined
Table 8.
Isobenzothiazolinones and ebselen as inhibitors of PfHK.

| Compound | X | R1 | R2 | PfHK IC50 (nM) | Pf3D7 EC50 (µM) |
|---|---|---|---|---|---|
| 51 | S | F | Cl | 160 | 0.62 |
| 52 | S | H | Cl | 270 | 1.1 |
| 53 | S | H | H | 230 | 5.2 |
| 54 | S | H | H | 10 | 6.8 |
Table 9.
Dimethyl 2-amido-cyclopenta[b]thiophene-3,4-dicarboxylates as inhibitors of PfHK.

| Compound | R | PfHK IC50 (µM) |
|---|---|---|
| 55 |  | 1.09 |
| 56 |  | 1.94 |
Table 10.
Three of the inhibitors of PfPI4K described by McNamara et al. [85].
Table 10.
Three of the inhibitors of PfPI4K described by McNamara et al. [85].
| Compound | P. yoelii liver stage IC50 (nM) | P. falciparum Asexual Blood-Stage IC50 (nM) | PfNF54 Gametocyte III IC50 (nM) |
|---|---|---|---|
 | 36 | 29.9 | 220 |
 | 9 | 3.9 | n.d. 1 |
 | n.d. | 71 | n.d. |
1 n.d. = not determined
Table 11.
Taketinib and its analog as inhibitors of PfPK9.

| Compound | R | Kd(app) PfPK9 (µM) | HuH7 P. berghei Infected Cell Load EC50 (µM) |
|---|---|---|---|
| 85 (taketinib) | NH2 | 0.46 | 7.3 |
| 86 | OH | 4.1 | 43 |
Table 12.
Imidazopyridines as inhibitors of PfPKG.

| Compound | R1 | R2 | R3 | R4 | PfPKG IC50 (nM) | Pf3D7 IC50 (nM) | % MLM Remaining 1 |
|---|---|---|---|---|---|---|---|
| 87 |  | H | H | H | 3.1 | 395 | 52 |
| 88 |  | Me | -NHSO2Me |  | 0.16 | 2.1 | - |
| 89 |  | H | H | Me | 2.8 | 525 | 93 |
1 % remaining after 30 min incubation with mouse liver microsomes.
Table 13.
Plasmodial kinases targeted by inhibiting compounds described in this review.
| Protein | Role of Protein—Consequence(s) of Inhibition 1 | Development Stage of the Most Advanced Compound(s) | Studies Reviewed |
|---|---|---|---|
| CDPK1 | Egress and invasion processes regulator—Inhibition of asexual blood-stage growth and gametogenesis, reduced mosquito infection | Hit to lead | [23,24,28,29,30,31,32,33] |
| CDPK4 | Exflagellation regulator—Reduced mosquito infection | Lead optimization | [33,36,37,38,39] |
| CK | Transformation of choline into phosphocholine | Hit discovery—Target validation needed | [41,45,46,47] |
| CK2 | Serine/threonine kinase, possibly phosphorylating many proteins during asexual blood-stage | Hit discovery—In vitro activity to be determined | [50] |
| CLK1 | Phosphorylation of spliceosomes proteins—Inhibition of asexual and sexual blood-stage growth | Hit discovery—In vitro activity to be determined | [53,55] |
| CLK3 | Phosphorylation of spliceosomes proteins—All-stage activity | Hit to lead | [53,54,56] |
| PanK | Transformation of pantothenate into 4′-phosphopantothenate | Hit discovery | [58,59,60] |
| DPCK | Transformation of dephospho-coenzyme A into coenzyme A | Hit discovery | [61] |
| FKk8 | Not defined | Hit discovery | [64,65] |
| GK | Transformation of (deoxy) guanosine-monophosphate into (deoxy) guanosine-diphosphate | Hit discovery—Target validation needed | [46] |
| GSK3 | Not defined | Hit to lead | [70,71] |
| HK | Transformation of glucose into glucose-6-phosphate | Hit discovery | [74,75] |
| MAP2 | Involved in many key cellular processes—Inhibition of asexual blood-stage and exflagellation | Hit discovery—Target validation needed | [77] |
| MRK | Regulation of gene expression and DNA replication | Hit discovery | [79,80,81] |
| NEK1 | Regulation of cell division processes | Hit discovery | [83,84] |
| PI4K | Transformation of phosphatidyl-inositol into phosphatidyl-inositol 4 phosphate—All-stage activity | Phase II clinical trial | [85,87,89,90,91,92] |
| PK5 | Cyclin-dependent like kinase, possibly regulating cell division | Hit discovery | [95] |
| PK6 | Cyclin-dependent like kinase, role not defined | Hit discovery - In vitro activity to be determined | [33] |
| PK7 | Orphan kinase, role not defined—Inhibition of asexual blood-stage and reduced mosquito infection | Hit to lead | [101,102,103] |
| PK9 | Orphan kinase, role unclear, able to phosphorylate PfUBC13—Deregulation of asexual hepatic stage growth | Hit discovery | [100] |
| PKG | Serine/threonine kinase—All-stage activity | Hit to lead | [109,110,112,113,114,116,117,118] |
| TMK | Transformation of thymidine monophosphate into thymidine diphosphate | Hit to lead | [121,122,123] |
1 When not specified, each of these proteins is assumed to at least inhibit asexual blood-stage growth.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mustière, R.; Vanelle, P.; Primas, N. Plasmodial Kinase Inhibitors Targeting Malaria: Recent Developments. Molecules 2020, 25, 5949. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245949
AMA Style
Mustière R, Vanelle P, Primas N. Plasmodial Kinase Inhibitors Targeting Malaria: Recent Developments. Molecules. 2020; 25(24):5949. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245949
Chicago/Turabian StyleMustière, Romain, Patrice Vanelle, and Nicolas Primas. 2020. "Plasmodial Kinase Inhibitors Targeting Malaria: Recent Developments" Molecules 25, no. 24: 5949. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245949