
From Discrete Complexes to Metal–Organic Layered Materials: Remarkable Hydrogen Bonding Frameworks


Abstract
:1. Introduction
2. Results and Discussion
2.1. Syntheses of the Complexes and Materials
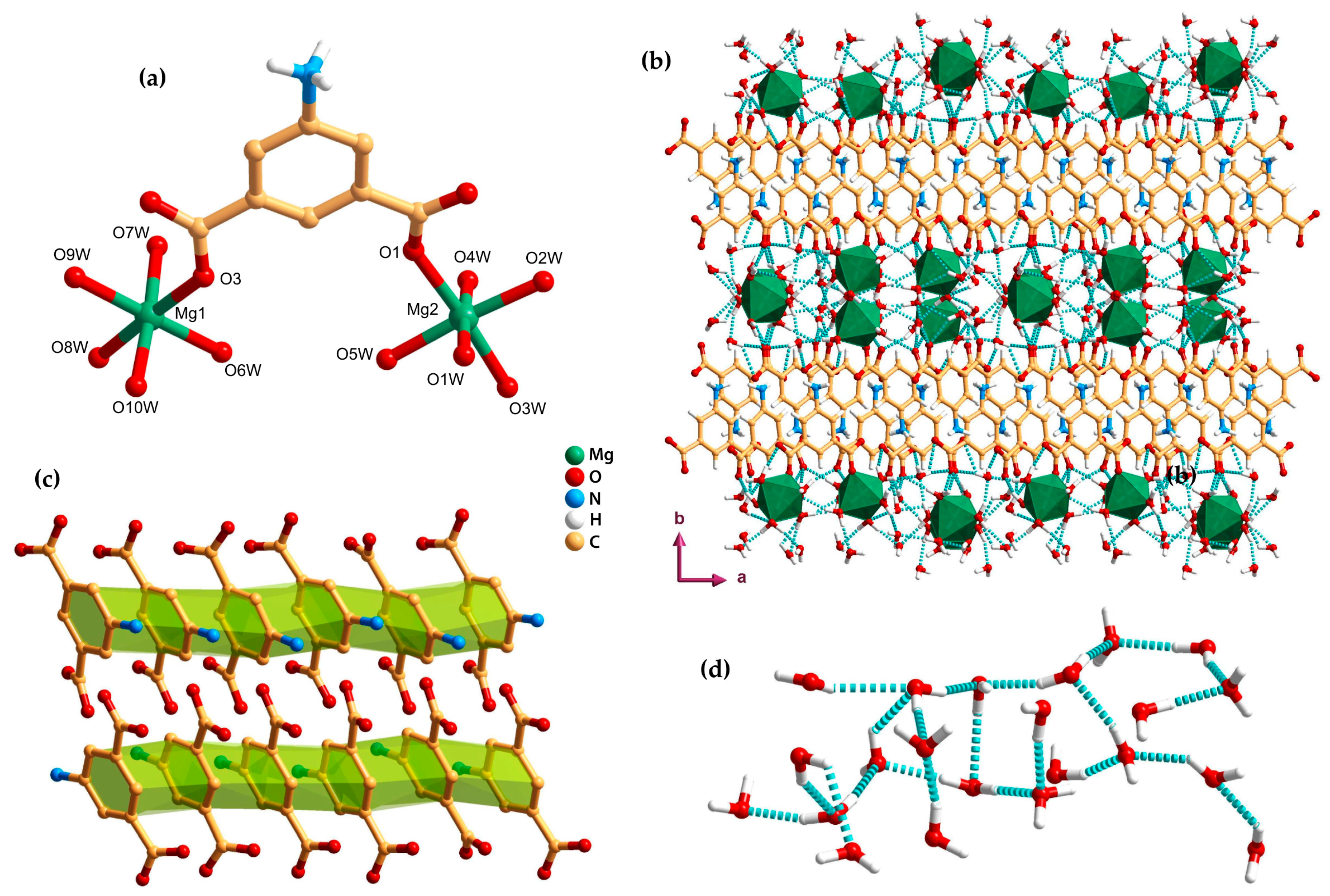
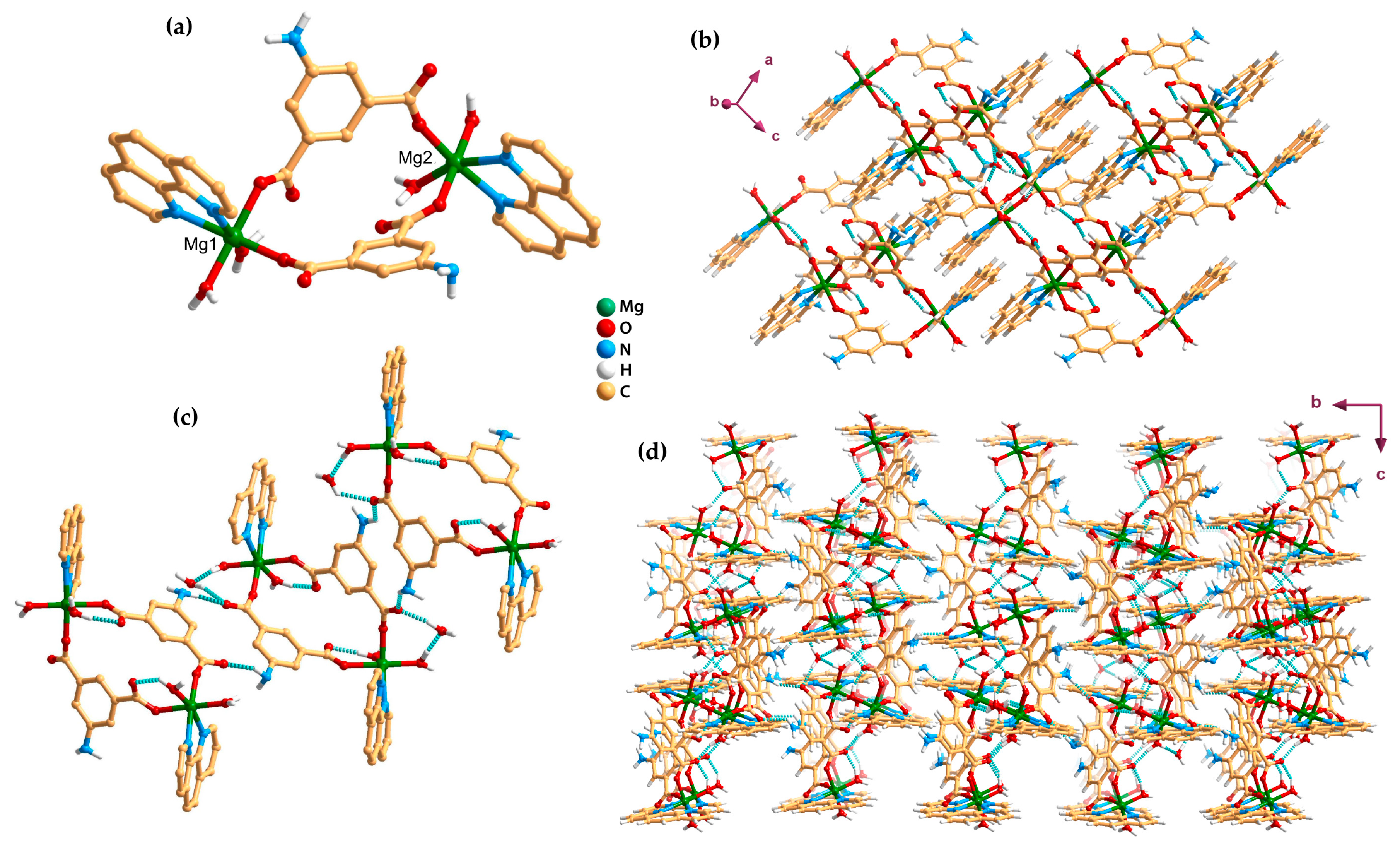
2.2. Discrete Complexes (0D)
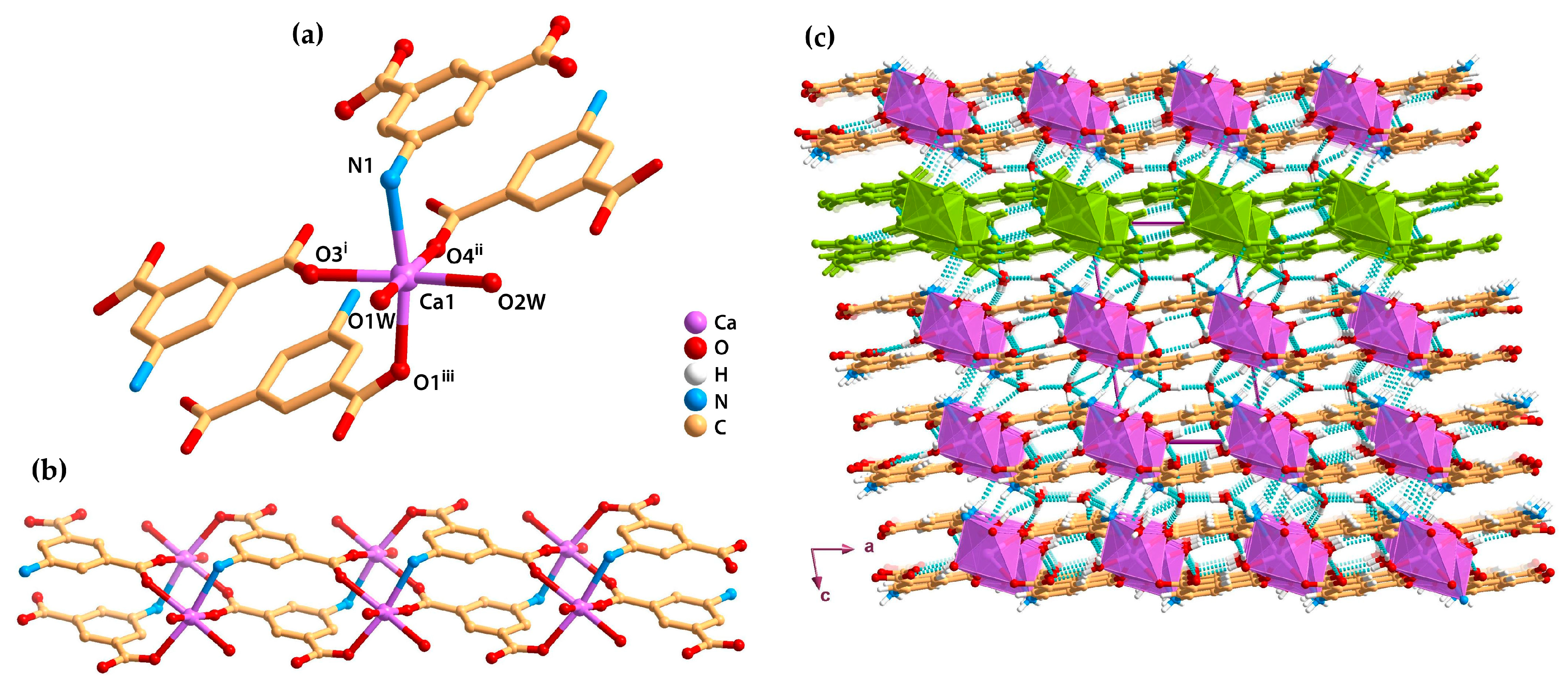
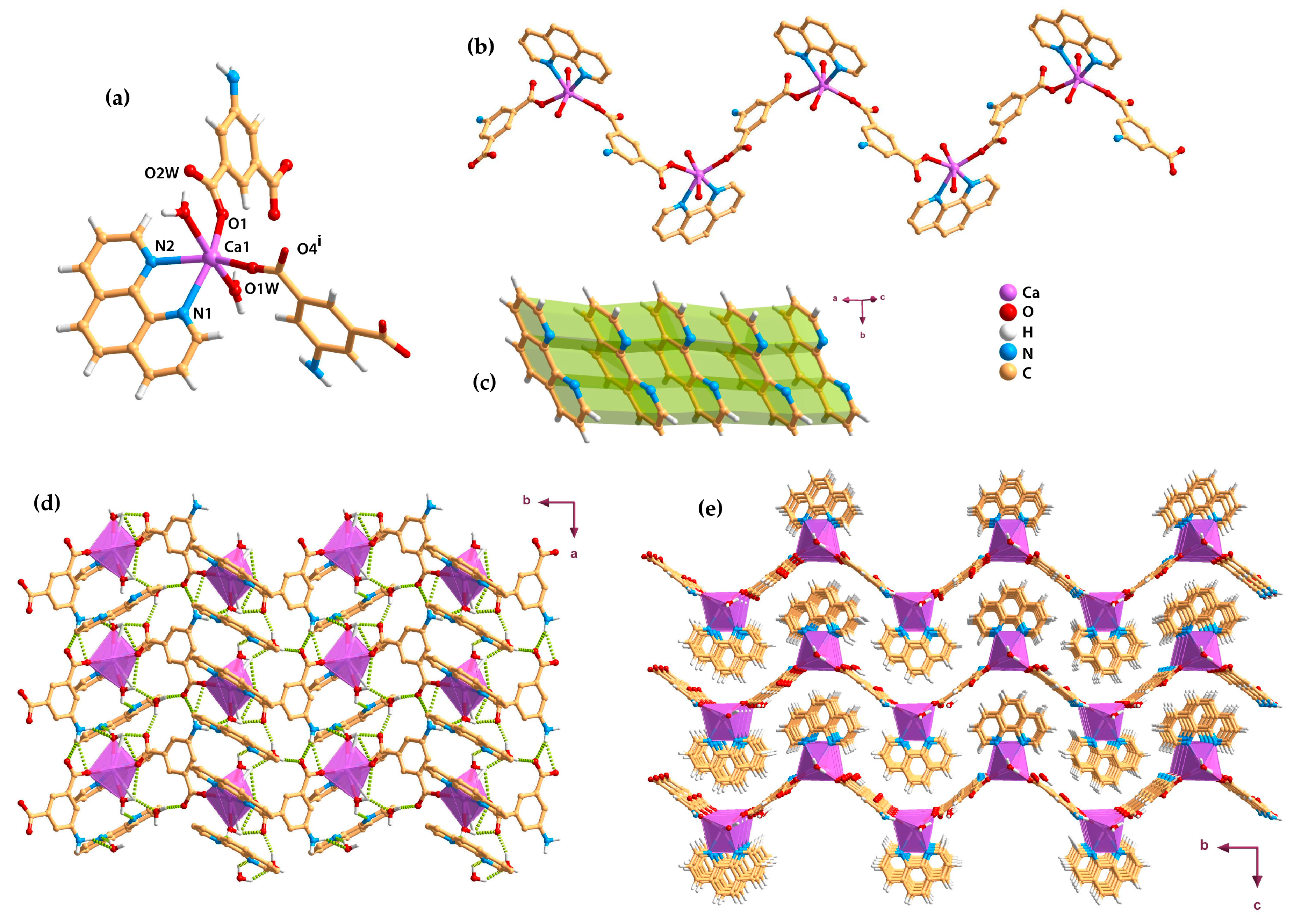
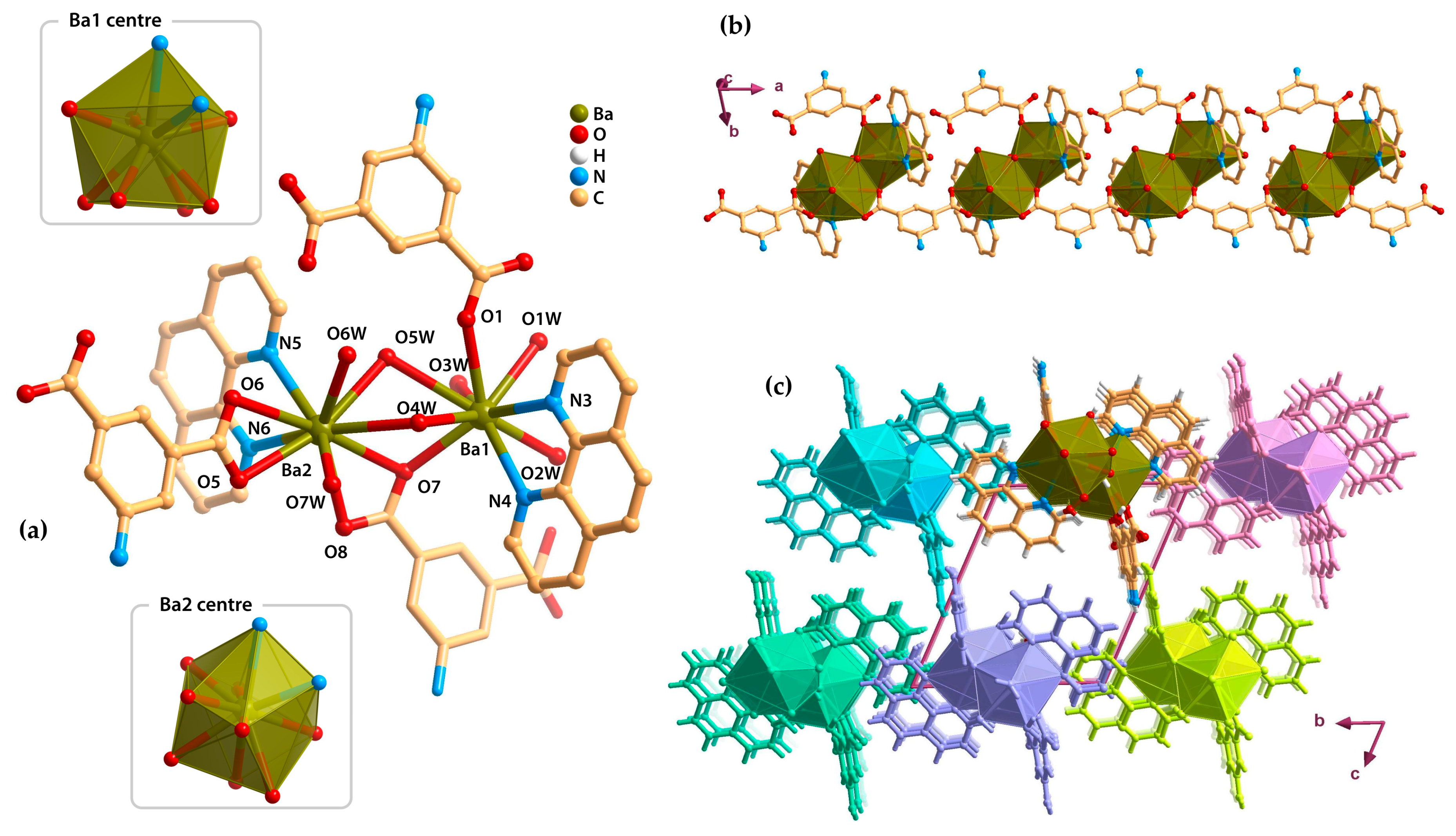
2.3. Metal–Organic Chains (1D)
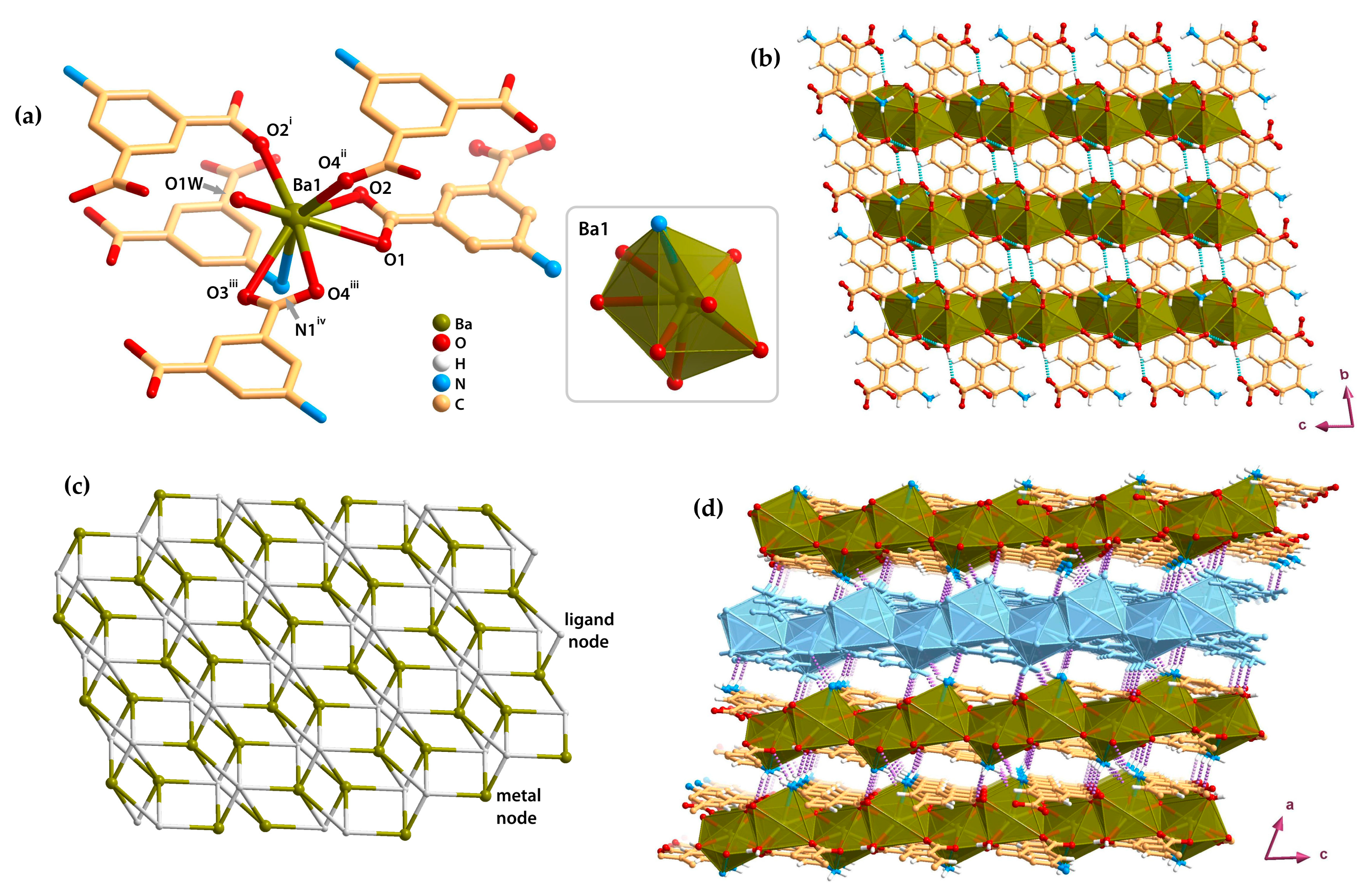
2.4. Metal–Organic Layers (2D)
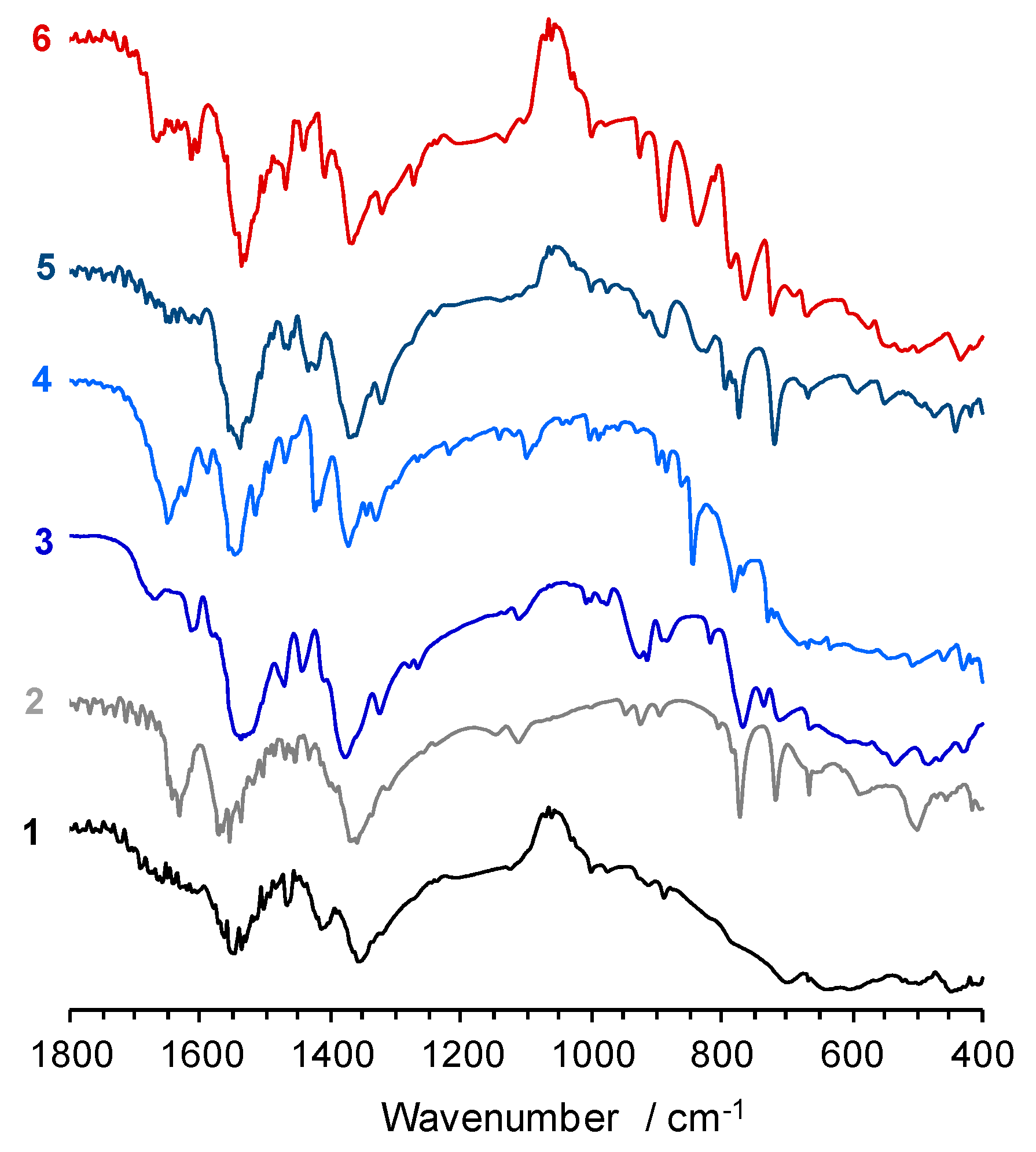
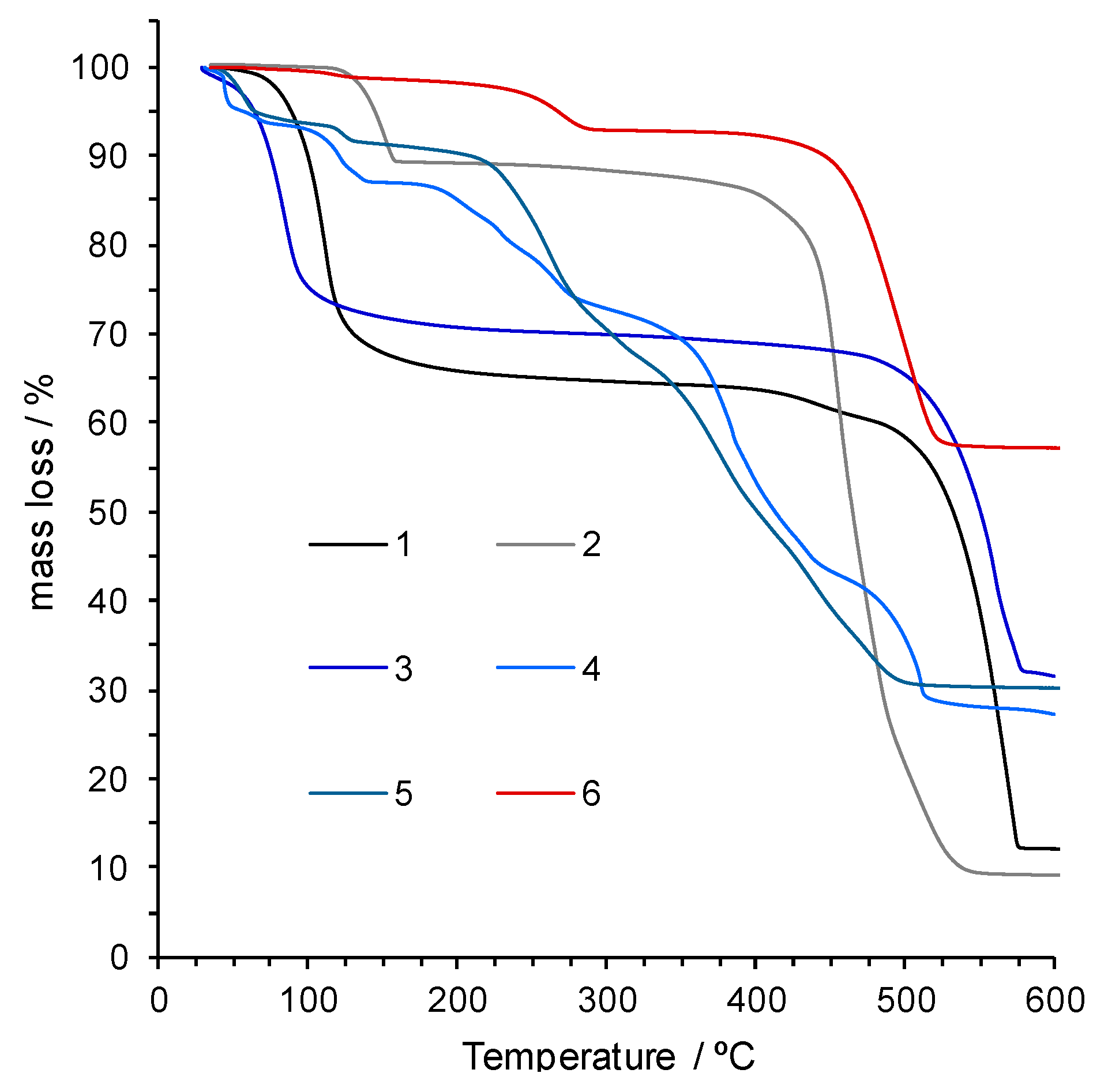
2.5. Vibrational Spectroscopy and Thermal Stability
3. Experimental Section
3.1. Materials and Methods
3.2. Preparation of Materials
3.3. X-ray Diffraction
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cao, R.; Lu, J.; Batten, S. Copper 5-sulfoisophthalato quasi-planar squares in coordination polymers modulated by alkaline-earth metals: A way to molecular squares? CrystEngComm 2008, 10, 784. [Google Scholar] [CrossRef]
- Tong, M.L.; Chen, X.M. Chapter 8—Synthesis of Coordination Compounds and Coordination Polymers. In Modern Inorganic Synthetic Chemistry, 2nd ed.; Xu, R., Xu, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 189–217. [Google Scholar]
- Cheong, V.F.; Moh, P.Y. Recent advancement in metal–organic framework: Synthesis, activation, functionalisation, and bulk production. Mater. Sci. Technol. 2018, 34, 1025–1045. [Google Scholar] [CrossRef]
- Gu, J.; Wen, M.; Liang, X.; Shi, Z.-F.; Kirillova, M.V.; Kirillov, A.M. Multifunctional Aromatic Carboxylic Acids as Versatile Building Blocks for Hydrothermal Design of Coordination Polymers. Crystals 2018, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Zhang, W.; Chen, Y.; Wang, Y. Uranium (VI) adsorption by copper and copper/iron bimetallic central MOFs. Colloids Surfaces A Physicochem. Eng. Asp. 2020, 587, 124334. [Google Scholar] [CrossRef]
- Li, J.; Li, W.-J.; Xu, S.-C.; Li, B.; Tang, Y.; Lin, Z.-F. Porous metal-organic framework with Lewis acid−base bifunctional sites for high efficient CO2 adsorption and catalytic conversion to cyclic carbonates. Inorg. Chem. Commun. 2019, 106, 70–75. [Google Scholar] [CrossRef]
- Yu, M.-H.; Space, B.; Franz, D.M.; Zhou, W.; He, C.; Li, L.; Krishna, R.; Chang, Z.; Li, W.; Hu, T.-L.; et al. Enhanced Gas Uptake in a Microporous Metal–Organic Framework via a Sorbate Induced-Fit Mechanism. J. Am. Chem. Soc. 2019, 141, 17703–17712. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.H.; Shi, L.; Ying, S.M.; Yan, G.Y.; Liu, L.H.; Sun, Y.Q.; Chen, Y.P. Two lanthanide metal–organic frameworks as sensitive luminescent sensors for the detection of Cr2+ and Cr2O72− in aqueous solutions. CrystEngComm 2018, 20, 189–197. [Google Scholar] [CrossRef]
- Chiu, C.-C.; Shieh, F.-K.; Tsai, H.-H.G. Ligand Exchange in the Synthesis of Metal–Organic Frameworks Occurs Through Acid-Catalyzed Associative Substitution. Inorg. Chem. 2019, 58, 14457–14466. [Google Scholar] [CrossRef]
- Parmar, B.; Patel, P.; Pillai, R.S.; Tak, R.K.; Kureshy, R.; Khan, N.-U.H.; Suresh, E. Cycloaddition of CO2 with an Epoxide-Bearing Oxindole Scaffold by a Metal-Organic Framework-Based Heterogeneous Catalyst under Ambient Conditions. Inorg. Chem. 2019, 58, 10084–10096. [Google Scholar] [CrossRef]
- Xie, W.; Tian, M.; Luo, X.; Jiang, Y.; He, N.; Liao, X.; Liu, Y. A dual-mode fluorescent and colorimetric immunoassay based on in situ ascorbic acid-induced signal generation from metal-organic frameworks. Sensors Actuators B Chem. 2020, 302, 127180. [Google Scholar] [CrossRef]
- Wiwasuku, T.; Boonmak, J.; Siriwong, K.; Ervithayasuporn, V.; Youngme, S. Highly sensitive and selective fluorescent sensor based on a multi-responsive ultrastable amino-functionalized Zn(II)-MOF for hazardous chemicals. Sensors Actuators B Chem. 2019, 284, 403–413. [Google Scholar] [CrossRef]
- Wang, J.-J.; Bao, Q.-L.; Chen, J. Two 2-D layered coordination polymers based on 5-aminoisophthalate and 1,10-phenanthroline. J. Coord. Chem. 2013, 66, 2578–2586. [Google Scholar] [CrossRef]
- Kan, M.-Y.; Shin, J.H.; Yang, C.-T.; Chang, C.-K.; Lee, L.-W.; Chen, B.-H.; Lu, K.-L.; Lee, J.S.; Lin, L.-C.; Kang, D.-Y. Activation-Controlled Structure Deformation of Pillared-Bilayer Metal–Organic Framework Membranes for Gas Separations. Chem. Mater. 2019, 31, 7666–7677. [Google Scholar] [CrossRef]
- Peterson, G.W.; Au, K.; Tovar, T.M.; Epps, T.H.; Epps, T.H. Multivariate CuBTC Metal–Organic Framework with Enhanced Selectivity, Stability, Compatibility, and Processability. Chem. Mater. 2019, 31, 8459–8465. [Google Scholar] [CrossRef]
- Wang, J.J.; Si, P.-P.; Yang, J.; Zhao, S.-S.; Li, P.-P.; Li, B.; Wang, S.-Y.; Lu, M.; Yu, S.-X. La(III)-based MOFs with 5-aminoisophthalic acid for optical detection and degradation of organic molecules in water. Polyhedron 2019, 162, 255–262. [Google Scholar] [CrossRef]
- Xu, W.-Q.; He, S.; Liu, S.-J.; Liu, X.-H.; Qiu, Y.-X.; Liu, W.-T.; Jiang, L.-C.; Jiang, J.-J. Post-synthetic modification of a metal-organic framework based on 5-aminoisophthalic acid for mercury sorption. Inorg. Chem. Commun. 2019, 108, 107515. [Google Scholar] [CrossRef]
- Scholz, G.; Abdulkader, A.; Kemnitz, E. Mechanochemical synthesis and characterization of alkaline earth metal terephthalates: M(C8H4O4)·nH2O(M = Ca,Sr,Ba). Zeitschrift für Anorganische und Allgemeine Chemie 2014, 640, 317–324. [Google Scholar] [CrossRef]
- Niekiel, F.; Stock, N. Discovery of New Calcium Etidronates Employing Ultrasound Adapted High-Throughput Methods. Cryst. Growth Des. 2013, 14, 599–606. [Google Scholar] [CrossRef]
- Buchanan, W.D.; Allis, D.; Ruhlandt-Senge, K. Synthesis and stabilization—Advances in organoalkaline earth metal chemistry. Chem. Commun. 2010, 46, 4449. [Google Scholar] [CrossRef]
- Asgharnejad, L.; Abbasi, A.; Najafi, M.; Janczak, J. Synthesis and Structure of Three New Alkaline Earth Metal–Organic Frameworks with High Thermal Stability as Catalysts for Knoevenagel Condensation. Cryst. Growth Des. 2019, 19, 2679–2686. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, L.-N.; Shi, W.-J.; Lu, Y.-K.; Hou, L.; Wang, Y.-Y. Four alkaline earth metal (Mg, Ca, Sr, Ba)-based MOFs as multiresponsive fluorescent sensors for Fe3+, Pb2+ and Cu2+ ions in aqueous solution. J. Solid State Chem. 2019, 277, 636–647. [Google Scholar] [CrossRef]
- Matlinska, M.A.; Ha, M.; Hughton, B.; Oliynyk, A.O.; Iyer, A.K.; Bernard, G.; Lambkin, G.R.; Lawrence, M.C.; Katz, M.J.; Mar, A.; et al. Alkaline Earth Metal-Organic Frameworks with Tailorable Ion Release: A Path for Supporting Biomineralization. ACS Appl. Mater. Interfaces 2019, 11, 32739–32745. [Google Scholar] [CrossRef] [PubMed]
- Harder, S. From Limestone to Catalysis: Application of Calcium Compounds as Homogeneous Catalysts. Chem. Rev. 2010, 110, 3852–3876. [Google Scholar] [CrossRef]
- Das, M.C.; Ghosh, S.K.; Sañudo, E.C.; Bharadwaj, P.K. Coordination polymers with pyridine-2,4,6-tricarboxylic acid and alkaline-earth/lanthanide/transition metals: Synthesis and X-ray structures. Dalton Trans. 2009, 9, 1644–1658. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, S.A.; Pournara, A.D.; Hatzidimitriou, A.G.; Manos, M.J.; Papaefstathiou, G.S.; Lazarides, T. Two new alkaline earth metal organic frameworks with the diamino derivative of biphenyl-4,4′-dicarboxylate as bridging ligand: Structures, fluorescence and quenching by gas phase aldehydes. Polyhedron 2018, 153, 173–180. [Google Scholar] [CrossRef]
- Li, Z.; Li, Z.; Li, S.; Wang, K.; Ma, F.; Tang, B. Potential application development of Sr/HCOOH metal organic framework in osteoarthritis. Microporous Mesoporous Mater. 2020, 294, 109835. [Google Scholar] [CrossRef]
- Margariti, A.; Pournara, A.D.; Manos, M.J.; Lazarides, T.; Papaefstathiou, G.S. Towards white-light emission by Tb3+/Eu3+ substitution in a Ca2+ framework. Polyhedron 2018, 153, 24–30. [Google Scholar] [CrossRef]
- Mazaj, M.; Mali, G.; Rangus, M.; Žunkovič, E.; Kaučič, V.; Logar, N.Z. Spectroscopic Studies of Structural Dynamics Induced by Heating and Hydration: A Case of Calcium-Terephthalate Metal–Organic Framework. J. Phys. Chem. C 2013, 117, 7552–7564. [Google Scholar] [CrossRef]
- Wang, X.; San, L.K.; Nguyen, H.; Shafer, N.M.; Hernandez, M.T.; Chen, Z. Alkaline earth metal–organic frameworks supported by ditopic carboxylates. J. Coord. Chem. 2013, 66, 826–835. [Google Scholar] [CrossRef]
- Platero-Prats, A.E.; Iglesias, M.; Snejko, N.; Monge, A.; Gutierrez-Puebla, E.; Monge, M.A. From Coordinatively Weak Ability of Constituents to Very Stable Alkaline-Earth Sulfonate Metal−Organic Frameworks. Cryst. Growth Des. 2011, 11, 1750–1758. [Google Scholar] [CrossRef]
- Saha, D.; Maity, T.; Das, S.; Koner, S. A magnesium-based multifunctional metal–organic framework: Synthesis, thermally induced structural variation, selective gas adsorption, photoluminescence and heterogeneous catalytic study. Dalton Trans. 2013, 42, 13912. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Maity, T.; Sen, R.; Koner, S. Heterogeneous catalysis over a barium carboxylate framework compound: Synthesis, X-ray crystal structure and aldol condensation reaction. Polyhedron 2012, 43, 63–70. [Google Scholar] [CrossRef]
- Sen, R.; Saha, D.; Koner, S. Controlled Construction of Metal-Organic Frameworks: Hydrothermal Synthesis, X-ray Structure, and Heterogeneous Catalytic Study. Chem. A Eur. J. 2012, 18, 5979–5986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qu, X.-N.; Xie, G.; Wei, Q.; Chen, S. Syntheses, structural analyses and luminescent property of four alkaline-earth coordination polymers. J. Solid State Chem. 2014, 210, 36–44. [Google Scholar] [CrossRef]
- Barbosa, A.; Julião, D.; Fernandes, D.M.; Peixoto, A.F.; Freire, C.; De Castro, B.; Granadeiro, C.; Balula, S.; Cunha-Silva, L. Catalytic performance and electrochemical behaviour of Metal–organic frameworks: MIL-101(Fe) versus NH2-MIL-101(Fe). Polyhedron 2017, 127, 464–470. [Google Scholar] [CrossRef]
- Granadeiro, C.; Nogueira, L.; Julião, D.; Mirante, F.; Ananias, D.; Cunha-Silva, L.; Balula, S. Influence of a porous MOF support on the catalytic performance of Eu-polyoxometalate based materials: Desulfurization of a model diesel. Catal. Sci. Technol. 2016, 6, 1515–1522. [Google Scholar] [CrossRef]
- Granadeiro, C.; Ribeiro, S.; Karmaoui, M.; Valença, R.; Ribeiro, J.C.; De Castro, B.; Cunha-Silva, L.; Balula, S. Production of ultra-deep sulfur-free diesels using a sustainable catalytic system based on UiO-66(Zr). Chem. Commun. 2015, 51, 13818–13821. [Google Scholar] [CrossRef]
- Sousaraei, A.; Queiros, C.; Moscoso, F.G.; Lopes-Costa, T.; Pedrosa, J.M.; Silva, A.; Cunha-Silva, L.; Cabanillas-Gonzalez, J. Subppm Amine Detection via Absorption and Luminescence Turn-On Caused by Ligand Exchange in Metal Organic Frameworks. Anal. Chem. 2019, 91, 15853–15859. [Google Scholar] [CrossRef]
- Xu, L.; Liu, B.; Liu, S.-X.; Jiao, H.; De Castro, B.; Cunha-Silva, L. The influence of 1-alkyl-3-methyl imidazolium ionic liquids on a series of cobalt-1,4-benzenedicarboxylate metal–organic frameworks. CrystEngComm 2014, 16, 10649–10657. [Google Scholar] [CrossRef]
- Xu, L.; Kwon, Y.-U.; De Castro, B.; Cunha-Silva, L. Novel Mn(II)-Based Metal–Organic Frameworks Isolated in Ionic Liquids. Cryst. Growth Des. 2013, 13, 1260–1266. [Google Scholar] [CrossRef]
- Rocha, J.; Shi, F.-N.; Paz, F.A.A.; Mafra, L.; Sardo, M.; Cunha-Silva, L.; Chisholm, J.; Ribeiro-Claro, P.J.A.; Trindade, T. 3D-2D-0D Stepwise Deconstruction of a Water Framework Templated by a Nanoporous Organic-Inorganic Hybrid Host. Chem. A Eur. J. 2010, 16, 7741–7749. [Google Scholar] [CrossRef] [PubMed]
- Soares-Santos, P.C.R.; Cunha-Silva, L.; Paz, F.A.A.; Ferreira, R.A.S.; Rocha, J.; Carlos, L.D.; Nogueira, H.I.S. Photo luminescent Lanthanide-Organic Bilayer Networks with 2,3-Pyrazinedicarboxylate and Oxalate. Inorg. Chem. 2010, 49, 3428–3440. [Google Scholar] [CrossRef] [PubMed]
- Murugavel, R.; Kumar, P.; Walawalkar, M.G.; Mathialagan, R. A Double Helix Is the Repeating Unit in a Luminescent Calcium 5-Aminoisophthalate Supramolecular Edifice with Water-Filled Hexagonal Channels. Inorg. Chem. 2007, 46, 6828–6830. [Google Scholar] [CrossRef] [PubMed]
- Kuang, R.Y.; Zhou, X.C.; Liu, L.J.; Lin, J.Y. A new 1D coordination polymer constructed by CaCl2 with 5-aminoiosphtalic acid (H2AIP) and 1,10-phenanthroline ligands using the water bath method. Russ. J. Coord. Chem. 2010, 36, 918–922. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Lin, C.-H. Poly[([mu]5-5-aminoisophthalato)aquabarium]. Acta Cryst. B 2011, 67, 1413. [Google Scholar]
- Kottke, T.; Stalke, D. Crystal handling at low temperatures. J. Appl. Crystallogr. 1993, 26, 615–619. [Google Scholar] [CrossRef] [Green Version]
- APEX2, Data Collection Software Version 2.1-RC13; Bruker AXS: Delft, The Netherlands, 2006.
- SAINT+, Data Integration Engine v. 7.23a©; Bruker AXS: Madison, WI, USA, 1997–2005.
- Sheldrick, G.M. SADABS v.2.01, Bruker/Siemens Area Detector Absorption Correction Program; Bruker AXS: Madison, WI, USA, 1998. [Google Scholar]
- Sheldrick, G.M. A short history ofSHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2007, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXS-97, Program for Crystal Structure Solution; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL-97, Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
Sample Availability: Most of the samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound/ Dimensionality | Heating Method | Metal Ion/ Co-Ligand | Conditions |
|---|---|---|---|
| 1/0D | Conventional | Mg(II) | 323 K, 65 h |
| 2/0D | Hydrothermal | Mg(II)/phen | 403 K, 72 h |
| 3/1D | Conventional | Ca(II) | 323 K, 68 h |
| 4/1D | Conventional | Ca(II)/phen | 323 K, 3 h |
| 5/1D | Conventional | Ba(II)/phen | 323 K, 68 h |
| 6/2D | Conventional | Ba(II) | 323 K, 65 h |
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| Formula | C40H81Mg3N5O46 | C40 H36Mg2N6O13 | C8H11CaNO7 | C32H27CaN5O7 | C64H60Ba2N10O17 | C8H7BaNO5 |
| Mr | 1441.03 | 857.37 | 273.26 | 633.67 | 1515.90 | 334.49 |
| Crystal morphology | orange/prism | Orange/plate | orange/prism | orange/prism | orange/prism | orange/prism |
| Crystal size/mm | 0.15 × 0.12 × 0.08 | 0.32 × 0.20 × 0.03 | 0.17 × 0.10 × 0.05 | 0.27 × 0.15 × 0.10 | 0.21 × 0.16 × 0.10 | 0.19 × 0.07 × 0.06 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic | Triclinic | Triclinic |
| Space group | P21/c | P21/c | P−1 | P21 | P1 | P−1 |
| a/Å | 17.6784 (10) | 11.7432 (9) | 8.1375 (9) | 8.2128 (14) | 10.9458 (4) | 7.76210 (10) |
| b/Å | 29.6003 (18) | 47.433 (3) | 10.2726 (10) | 17.373 (3) | 11.9419 (4) | 7.96520 (10) |
| c/Å | 11.7133 (7) | 13.9297 (9) | 13.3429 (14) | 10.127 (2) | 13.4085 (5) | 8.34160 (10) |
| α/° | 90 | 90 | 74.135 (5) | 90 | 64.1780 (10) | 79.6180 (10) |
| β/° | 98.049 (4) | 97.143 (4) | 79.343 (5) | 93.105(10) | 79.870 (2) | 65.5740 (10) |
| γ/° | 90 | 90 | 84.520 (5) | 90 | 77.1790 (10) | 83.5750 (10) |
| Volume/Å3 | 6069.0 (6) | 7698.9 (10) | 1053.19 (19) | 1442.8(5) | 1532.06 (10) | 461.483 (10) |
| Z | 4 | 8 | 4 | 2 | 1 | 2 |
| ρcalc/g cm−3 | 1.577 | 1.479 | 1.723 | 1.459 | 1.643 | 2.407 |
| F(000) | 3040 | 3568 | 568 | 660 | 762 | 316 |
| µ/mm−1 | 0.171 | 0.141 | 0.621 | 0.277 | 1.359 | 4.303 |
| θ range/° | 3.73–25.68 | 3.65–23.26 | 3.67–25.03 | 3.90–25.02 | 3.68–27.48 | 3.67–27.46 |
| Reflections collected | 87367 | 51678 | 27959 | 13902 | 36614 | 7516 |
| Independent relections | 11,485 (Rint = 0.1469) | 10,997 (Rint = 0.0930) | 3680 (Rint = 0.0294) | 5042 (Rint = 0.0703) | 13,072 (Rint = 0.0330) | 2099 (Rint = 0.0225) |
| Parameters/Restraints | 1045/110 | 1178/45 | 355/20 | 430/13 | 904/36 | 148/5 |
| Final R indices [I > 2σ(I)] | R1 = 0.0793 wR2 = 0.1939 | R1 = 0.108 wR2 = 0.2839 | R1 = 0.0292 wR2 = 0.0806 | R1 = 0.0648 wR2 = 0.1404 | R1 = 0.0296 wR2 = 0.0580 | R1 = 0.0152 wR2 = 0.0316 |
| Final R indices (all data) | R1 = 0.1603 wR2 = 0.2399 | R1 = 0.1583 wR2 = 0.3209 | R1 = 0.0418 wR2 = 0.0899 | R1 = 0.0918 wR2 = 0.1554 | R1 = 0.0330 wR2 = 0.0597 | R1 = 0.0163 wR2 = 0.0321 |
| Min. and max. Residual electr. density/e Å3 | 0.9864 and −0.9748 | 0.890 and −1.099 | 0.352 and −0.266 | 1.211 and −0.343 | 0.845 and −0.410 | 0.454 and −0.420 |
| Deposition code | CCDC-1981181 | CCDC-1981183 | CCDC-1981178 | CCDC-1981179 | CCDC-1981182 | CCDC-1981180 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Queirós, C.; Silva, A.M.G.; de Castro, B.; Cunha-Silva, L. From Discrete Complexes to Metal–Organic Layered Materials: Remarkable Hydrogen Bonding Frameworks. Molecules 2020, 25, 1353. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25061353
Queirós C, Silva AMG, de Castro B, Cunha-Silva L. From Discrete Complexes to Metal–Organic Layered Materials: Remarkable Hydrogen Bonding Frameworks. Molecules. 2020; 25(6):1353. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25061353
Chicago/Turabian StyleQueirós, Carla, Ana M. G. Silva, Baltazar de Castro, and Luís Cunha-Silva. 2020. "From Discrete Complexes to Metal–Organic Layered Materials: Remarkable Hydrogen Bonding Frameworks" Molecules 25, no. 6: 1353. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25061353