Role of the Neutrophil in the Pathogenesis of Advanced Cancer and Impaired Responsiveness to Therapy †

 , ,
, ,
Abstract
:1. Introduction
2. Pro-Oxidative, Pro-Carcinogenic Mechanisms of Neutrophils
3. Recruitment and Exploitation of Neutrophils by Tumors
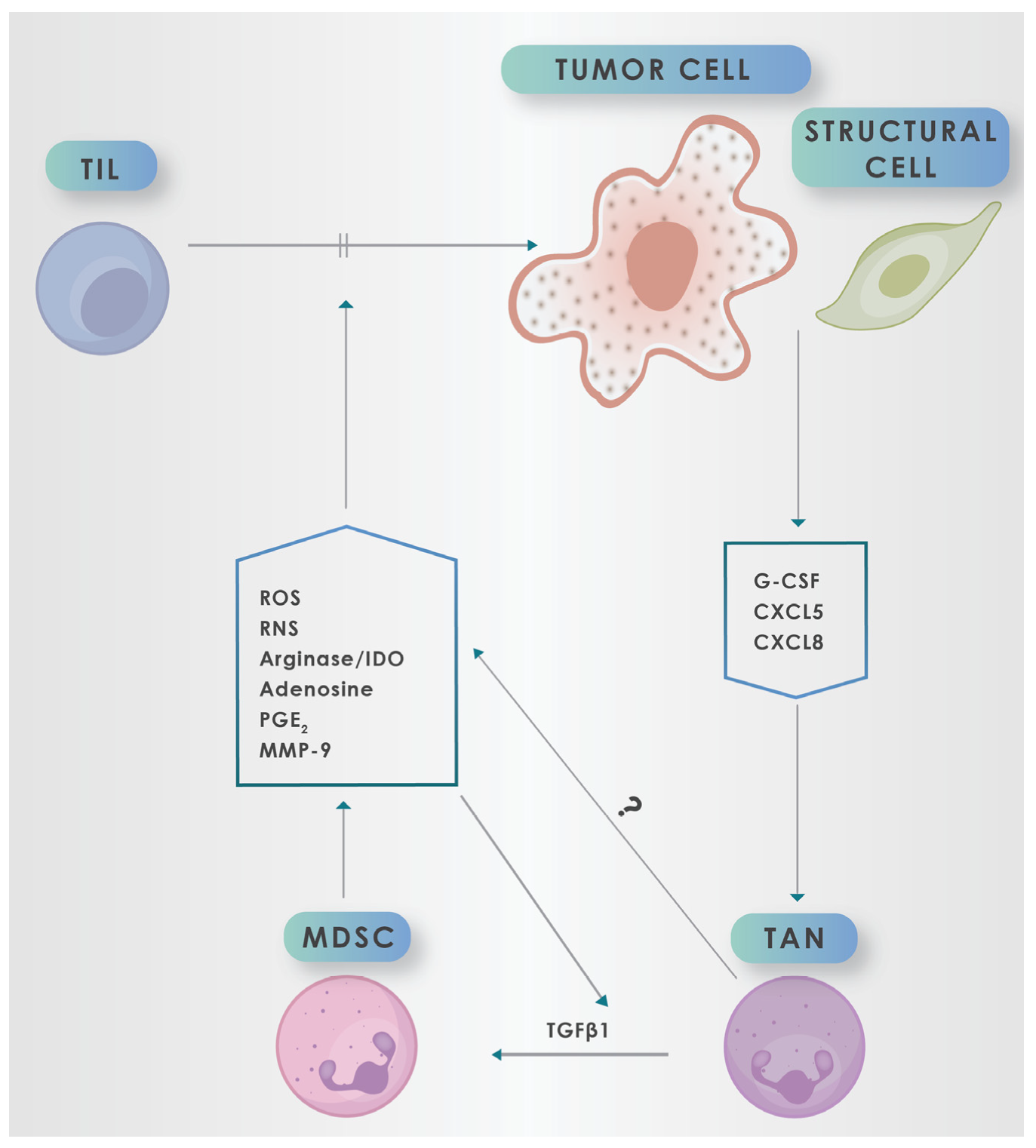
4. Myeloid-Derived Suppressor Cells (MDSCs)
5. Mechanisms by which MDSCs Promote an Immunosuppressive TME
5.1. Pro-Oxidative Mechanisms
5.2. Non-Oxidative Mechanisms
5.2.1. Amino Acid Deprivation
5.2.2. Differentiation and Expansion of Tregs
5.2.3. Proteases
5.2.4. Expression of Programmed Death Ligand-1 (PD-L1) and Fas ligand (FasL)
5.2.5. Production of Adenosine and Prostaglandin E2
5.2.6. Other Mechanisms
6. Role of Neutrophils in Tumor Metastasis
6.1. Role of Neutrophil Extracellular Traps (NETs) in Metastasis
6.2. Neutrophil-Derived Pro-Metastatic Cytokines, Growth factors and Granule Enzymes
7. Adjunctive Therapeutic Targeting of MDSCs of Neutrophilic Origin
7.1. Cytokine Targeting
7.2. Targeting CXCR1/CXCR2
7.3. Other Strategies
8. Conclusions
Author Contributions
Conflicts of Interest
References
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Anderson, R.; Tintinger, G.R.; Feldman, C. Inflammation and cancer: The role of the human neutrophil. S. Afr. J. Sci. 2014, 110, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Schraufstatter, I.U.; Hyslop, P.A.; Hinshaw, D.B.; Spragg, R.G.; Sklar, L.A.; Cochrane, C.G. Hydrogen peroxide-induced injury of cells and its prevention by inhibitors of poly(ADP-ribose) polymerase. Proc. Natl. Acad. Sci. USA 1986, 83, 4908–4912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schraufstätter, I.; Hyslop, P.A.; Jackson, J.H.; Cochrane, C.G. Oxidant-induced DNA damage of target cells. J. Clin. Investig. 1988, 82, 1040–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.H.; Gajewski, E.; Schraufstatter, I.U.; Hyslop, P.A.; Fuciarelli, A.F.; Cochrane, C.G.; Dizdaroglu, M. Damage to the bases in DNA induced by stimulated human neutrophils. J. Clin. Investig. 1989, 84, 1644–1649. [Google Scholar] [CrossRef] [Green Version]
- Weitzman, S.A.; Gordon, L.I. Inflammation and cancer: Role of phagocyte-generated oxidants in carcinogenesis. Blood 1990, 76, 655–663. [Google Scholar] [CrossRef]
- Van Rensburg, C.E.; Van Staden, A.M.; Anderson, R. Inactivation of poly (ADP-ribose) polymerase by hypochlorous acid. Free Radic. Biol. Med. 1991, 11, 285–291. [Google Scholar] [CrossRef]
- Jaiswal, M.; LaRusso, N.F.; Nishioka, N.; Nakabeppu, Y.; Gores, G.J. Human Ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001, 61, 6388–6393. [Google Scholar]
- Cai, Y.J.; Lu, J.J.; Zhu, H.; Xie, H.; Huang, M.; Lin, L.P.; Zhang, X.W.; Ding, J. Salvicine triggers DNA double-strand breaks and apoptosis by GSH-depletion-driven H2O2 generation and topoisomerase II inhibition. Free Radic. Biol. Med. 2008, 45, 627–635. [Google Scholar] [CrossRef]
- Sato, T.; Takeda, H.; Otake, S.; Yokozawa, J.; Nishise, S.; Fujishima, S.; Orii, T.; Fukui, T.; Takano, J.; Sasaki, Y.; et al. Increased plasma levels of 8-hydroxydeoxyguanosine are associated with development of colorectal tumors. J. Clin. Biochem. Nutr. 2010, 47, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Li, X.; Wang, R.; Yu, J.; Ye, M.; Mao, L.; Zhang, S.; Zheng, S. Association between oxidative DNA damage and risk of colorectal cancer: Sensitive determination of urinary 8-hydroxy-2’-deoxyguanosine by UPLC-MS/MS analysis. Sci. Rep. 2016, 6, 32581. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, J.; Zhang, J.; Li, S.; Wang, H.; Du, J. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int. J. Cancer 2019, 145, 1946–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, X.; Zhao, H.; Wang, J.; Zhang, Q. CXCL5 secreted from adipose tissue-derived stem cells promotes cancer cell proliferation. Oncol. Lett. 2018, 15, 1403–1410. [Google Scholar] [CrossRef]
- Verbeke, H.; Struyf, S.; Laureys, G.; Van Damme, J. The expression and role of CXC chemokines in colorectal cancer. Cytokine Growth Factor Rev. 2011, 22, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, H.; Geboes, K.; Van Damme, J.; Struyf, S. The role of CXC chemokines in the transition of chronic inflammation to esophageal and gastric cancer. Biochim. Biophys. Acta 2012, 1825, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, T.; Clarke, M.; Steele, C.W.; Samuel, M.S.; Neumann, J.; Jung, A.; Huels, D.; Olson, M.F.; Das, S.; Nibbs, R.J.; et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Investig. 2012, 122, 3127–3144. [Google Scholar] [CrossRef]
- Miyazaki, H.; Patel, V.; Wang, H.; Edmunds, R.K.; Gutkind, J.S.; Yeudall, W.A. Down-regulation of CXCL5 inhibits squamous carcinogenesis. Cancer Res. 2006, 66, 4279–4284. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; King, J.; Moro, A.; Sugi, M.D.; Dawson, D.W.; Kaplan, J.; Li, G.; Lu, X.; Strieter, R.M.; Burdick, M.; et al. Overexpression of CXCL5 is associated with poor survival in patients with pancreatic cancer. Am. J. Pathol. 2011, 178, 1340–1349. [Google Scholar] [CrossRef]
- Ma, S.; Cheng, Q.; Cai, Y.; Gong, H.; Wu, Y.; Yu, X.; Shi, L.; Wu, D.; Dong, C.; Liu, H. IL-17A produced by γδ T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res. 2014, 74, 1969–1982. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Ou, B.; Han, D.; Wang, P.; Zong, Y.; Zhu, C.; Liu, D.; Zheng, M.; Sun, J.; Feng, H.; et al. Tumor-derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk-1/Snail and AKT/GSK3β/β-catenin pathways. Mol. Cancer 2017, 16, 70. [Google Scholar] [CrossRef] [Green Version]
- Najjar, Y.G.; Rayman, P.; Jia, X.; Pavicic, P.G., Jr.; Rini, B.I.; Tannenbaum, C.; Ko, J.; Haywood, S.; Cohen, P.; Hamilton, T.; et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1β, IL8, CXCL5, and Mip-1α. Clin. Cancer Res. 2017, 23, 2346–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, D.; Zhao, Y.; Xu, J. Activation of CXCL5-CXCR2 axis promotes proliferation and accelerates G1 to S phase transition of papillary thyroid carcinoma cells and activates JNK and p38 pathways. Cancer Biol. Ther. 2019, 20, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Haider, C.; Hnat, J.; Wagner, R.; Huber, H.; Timelthaler, G.; Grubinger, M.; Coulouarn, C.; Schreiner, W.; Schlangen, K.; Sieghart, W.; et al. Transforming growth factor-β and Axl induce CXCL5 and neutrophil recruitment in hepatocellular carcinoma. Hepatology 2019, 69, 222–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsthuber, A.; Lipp, K.; Andersen, L.; Ebersberger, S.; Graña-Castro, O.; Ellmeier, W.; Petzelbauer, P.; Lichtenberger, B.M.; Loewe, R. CXCL5 as regulator of neutrophil function in cutaneous melanoma. J. Investig. Dermatol. 2019, 139, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, S.P.; Kessler, J.F.; Miller, T.P. Leukocyte concentrations in discrimination of benign from malignant lung lesions. Am. J. Med. 1986, 80, 1035–1040. [Google Scholar] [CrossRef]
- Almand, B.; Clark, J.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Kasuga, I.; Makino, S.; Kiyokawa, H.; Katoh, H.; Ebihara, Y.; Ohyashiki, K. Tumor-related leukocytosis is linked with poor prognosis in patients with lung carcinoma. Cancer 2001, 92, 2399–2405. [Google Scholar] [CrossRef]
- Connolly, G.C.; Khorana, A.A.; Kuderer, N.M.; Culakova, E.; Francis, C.W.; Lyman, G.H. Leukocytosis, thrombosis and early mortality in cancer patients initiating chemotherapy. Thromb. Res. 2010, 126, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Ning, H.; Liu, M.; Lin, J.; Luo, S.; Zhu, W.; Xu, J.; Wu, W.C.; Liang, J.; Shao, C.K.; et al. Spleen mediates a distinct hematopoietic progenitor response supporting tumor-promoting myelopoiesis. J. Clin. Investig. 2018, 128, 3425–3438. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, M.; Asada, Y.; Terada, T.; Takehara, A.; Munemoto, Y.; Fujisawa, K.; Mitsui, T.; Iida, Y.; Miura, S.; Sudo, Y. Aggressive recurrence of gastric cancer as a granulocyte-colony-stimulating factor-producing tumor. Int. J. Clin. Oncol. 2010, 15, 191–195. [Google Scholar] [CrossRef]
- Morris, K.T.; Khan, H.; Ahmad, A.; Weston, L.L.; Nofchissey, R.A.; Pinchuk, I.V.; Beswick, E.J. G-CSF and G-CSFR are highly expressed in human gastric and colon cancers and promote carcinoma cell proliferation and migration. Br. J. Cancer 2014, 110, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Aliper, A.M.; Frieden-Korovkina, V.P.; Buzdin, A.; Roumiantsev, S.A.; Zhavoronkov, A. A role for G-CSF and GM-CSF in nonmyeloid cancers. Cancer Med. 2014, 3, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Hayakawa, N.; Nakamura, S. Granulocyte colony-stimulating factor-producing upper urinary tract carcinoma: Systematic review of 46 cases reported in Japan. Clin. Oncol. 2014, 26, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Kanetaka, K.; Kobayashi, S.; Nagata, Y.; Kinosita, N.; Fukuoka, J.; Murakami, S.; Fujita, F.; Takatsuk, M.; Eguchi, S. Severe neutrophilic leukocytosis as a progression marker in granulocyte colony-stimulating factor-producing squamous cell carcinoma of the esophagus. Clin. Case Rep. 2017, 5, 688–693. [Google Scholar] [CrossRef]
- Tavakkoli, M.; Wilkins, C.R.; Mones, J.V.; Mauro, M.J. A novel paradigm between leukocytosis, G-CSF secretion, neutrophil-to-lymphocyte ratio, myeloid-derived suppressor cells, and prognosis in non-small cell lung cancer. Front. Oncol. 2019, 9, 295. [Google Scholar] [CrossRef] [Green Version]
- Lecot, P.; Sarabi, M.; Pereira Abrantes, M.; Mussard, J.; Koenderman, L.; Caux, C.; Bendriss-Vermare, N.; Michallet, M.C. Neutrophil heterogeneity in cancer: From biology to therapies. Front. Immunol. 2019, 10, 2155. [Google Scholar] [CrossRef] [Green Version]
- Granot, Z. Neutrophils as a therapeutic target in cancer. Front. Immunol. 2019, 10, 1710. [Google Scholar] [CrossRef] [Green Version]
- Kargl, J.; Zhu, X.; Zhang, H.; Yang, G.H.Y.; Friesen, T.J.; Shipley, M.; Maeda, D.Y.; Zebala, J.A.; McKay-Fleisch, J.; Meredith, G.; et al. Neutrophil content predicts lymphocyte depletion and anti-PD1 treatment failure in NSCLC. JCI Insight 2019, 4, 130850. [Google Scholar] [CrossRef] [Green Version]
- Wang, K. Targeting IL-17 for cancer-associated inflammation and immunity. J. Immunol. 2017, 198, 66.5. Available online: https://www.jimmunol.org/content/198/1_Supplement/66.5 (accessed on 6 January 2020).
- Akbay, E.A.; Koyama, S.; Liu, Y.; Dries, R.; Bufe, L.E.; Silkes, M.; Alam, M.M.; Magee, D.M.; Jones, R.; Jinushi, M.; et al. Interleukin-17A promotes lung tumor progression through neutrophil attraction to tumor sites and mediating resistance to PD-1 blockade. J. Thorac. Oncol. 2017, 12, 1268–1279. [Google Scholar] [CrossRef] [Green Version]
- Ravindranathan, S.; Nguyen, K.G.; Kurtz, S.L.; Frazier, H.N.; Smith, S.G.; Koppolu, B.P.; Rajaram, N.; Zaharoff, D.A. Tumor-derived granulocyte colony-stimulating factor diminishes efficacy of breast tumor cell vaccines. Breast Cancer Res. 2018, 20, 126. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, A.J.; Hong, C.S.; Lin, A.J.; Xiang, Y.; Cooper, D.E.; Zhang, J.; Xu, E.S.; Kuo, H.C.; Mowery, Y.M.; Carpenter, D.J.; et al. Neutrophils promote tumor resistance to radiation therapy. Proc. Natl. Acad. Sci. USA 2019, 116, 18584–18589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carus, A.; Ladekarl, M.; Hager, H.; Nedergaard, B.S.; Donskov, F. Tumour-associated CD66b+ neutrophil count is an independent prognostic factor for recurrence in localised cervical cancer. Br. J. Cancer 2013, 108, 2116–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.; Kim, K.H.; Yoon, H.I.; Kim, G.E.; Kim, Y.B. Tumor-related leucocytosis is associated with poor radiation response and clinical outcome in uterine cervical cancer patients. Ann. Oncol. 2016, 27, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- American Society of Clinical Oncology. Recommendations for the use of hematopoietic colony-stimulating factors: Evidence-based, clinical practice guidelines. J. Clin. Oncol. 1994, 12, 2471–2508. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Trabelsi, M.; Farah, F.; Zouari, B.; Jaafoura, M.H.; Kharrat, M. An immunoscore system based on CD3+ and CD8+ infiltrating lymphocytes densities to predict the outcome of patients with colorectal adenocarcinoma. Onco Targets Ther. 2019, 12, 8663–8673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.D.; Huang, C.W.; Liu, Z.F.; Jiang, L.J.; Chen, J.W.; Xie, D.; Zhou, F.J.; Lu, H.M.; Liu, Z.W. Prognostic role of the immunoscore for patients with urothelial carcinoma of the bladder who underwent radical cystectomy. Ann. Surg. Oncol. 2019, 26, 4148–4156. [Google Scholar] [CrossRef]
- Nie, R.C.; Yuan, S.Q.; Wang, Y.; Chen, Y.B.; Cai, Y.Y.; Chen, S.; Li, S.M.; Zhou, J.; Chen, G.M.; Luo, T.Q.; et al. Robust immunoscore model to predict the response to anti-PD1 therapy in melanoma. Aging (Albany NY) 2019, 11, 11576–11590. [Google Scholar] [CrossRef]
- Chen, H.; Xia, B.; Zheng, T.; Lou, G. Immunoscore system combining CD8 and PD-1/PD-L1: A novel approach that predicts the clinical outcomes for cervical cancer. Int. J. Biol. Markers 2019. [CrossRef] [Green Version]
- Bergenfelz, C.; Leandersson, K. The Generation and Identity of Human Myeloid-Derived Suppressor Cells. Front Oncol. 2020, 10, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Nefedova, Y.; Lei, A.; Gabrilovich, D. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin. Immunol. 2018, 35, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Aarts, C.E.M.; Hiemstra, I.H.; Tool, A.T.J.; van den Berg, T.K.; Mul, E.; van Bruggen, R.; Kuijpers, T.W. Neutrophils as suppressors of T cell proliferation: Does age matter? Front. Immunol. 2019, 10, 2144. [Google Scholar] [CrossRef] [PubMed]
- Aarts, C.E.M.; Hiemstra, I.H.; Béguin, E.P.; Hoogendijk, A.J.; Bouchmal, S.; van Houdt, M.; Tool, A.T.J.; Mul, E.; Jansen, M.H.; Janssen, H.; et al. Activated neutrophils exert myeloid-derived suppressor cell activity damaging T cells beyond repair. Blood Adv. 2019, 3, 3562–3574. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, R.V.; Kuznetsova, V.; Connelly, A.N.; Hel, Z.; Szalai, A.J. C-reactive protein promotes the expansion of myeloid derived cells with suppressor functions. Front. Immunol. 2019, 10, 2183. [Google Scholar] [CrossRef]
- Weber, J.S.; Tang, H.; Hippeli, L.; Qian, M.; Wind-Rotolo, M.; Larkin, J.M.G.; Wolchok, J.D.; Sznol, M.; Robert, C.; Woods, D.M.; et al. Serum IL-6 and CRP as prognostic factors in melanoma patients receiving single agent and combination checkpoint inhibition. J. Clin. Oncol. 2019, 37, 100. [Google Scholar] [CrossRef]
- Gray, S.; Axelsson, B. The prevalence of deranged C-reactive protein and albumin in patients with incurable cancer approaching death. PLoS ONE 2018, 13, e0193693. [Google Scholar] [CrossRef] [Green Version]
- Shrotriya, S.; Walsh, D.; Nowacki, A.S.; Lorton, C.; Aktas, A.; Hullihen, B.; Benanni-Baiti, N.; Hauser, K.; Ayvaz, S.; Estfan, B. Serum C-reactive protein is an important and powerful prognostic biomarker in most adult solid tumors. PLoS ONE 2018, 13, e0202555. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Piccard, H.; Muschel, R.J.; Opdenakker, G. On the dual roles and polarized phenotypes of neutrophils in tumor development and progression. Crit. Rev. Oncol. Hematol. 2012, 82, 296–309. [Google Scholar] [CrossRef]
- Wang, X.; Qiu, L.; Li, Z.; Wang, X.Y.; Yi, H. Understanding the multifaceted role of neutrophils in cancer and autoimmune diseases. Front. Immunol. 2018, 9, 2456. [Google Scholar] [CrossRef] [PubMed]
- Giese, M.A.; Hind, L.E.; Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 2019, 133, 2159–2167. [Google Scholar] [CrossRef] [PubMed]
- Treffers, L.W.; Hiemstra, I.H.; Kuijpers, T.W.; van den Berg, T.K.; Matlung, H.L. Neutrophils in cancer. Immunol. Rev. 2016, 273, 312–328. [Google Scholar] [CrossRef]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Pillay, J.; Kamp, V.M.; van Hoffen, E.; Visser, T.; Tak, T.; Lammers, J.W.; Ulfman, L.H.; Leenen, L.P.; Pickkers, P.; Koenderman, L. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J. Clin. Investig. 2012, 122, 327–336. [Google Scholar] [CrossRef]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Dilek, N.; de Silly, R.V.; Blancho, G.; Vanhove, B. Myeloid-derived suppressor cells: Mechanisms of action and recent advances in their role in transplant tolerance. Front. Immunol. 2012, 3, 208. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ye, Q.J.; Xing, Y.F.; Lin, J.X.; Lin, Q.; Wu, X.Y. Expansion of Lox-1+CD15+ myeloid-derived suppressor cells in hepatocellular carcinoma patients. J. Clin. Oncol. 2017, 35, 124. [Google Scholar] [CrossRef]
- Zhong, L.M.; Liu, Z.G.; Zhou, X.; Song, S.H.; Weng, G.Y.; Wen, Y.; Liu, F.B.; Cao, D.L.; Liu, Y.F. Expansion of PMN-myeloid derived suppressor cells and their clinical relevance in patients with oral squamous cell carcinoma. Oral Oncol. 2019, 95, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, J.; Guo, G.; Zhou, J. Norepinephrine-induced myeloid-derived suppressor cells block T-cell responses via generation of reactive oxygen species. Immunopharmacol. Immunotoxicol. 2015, 37, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhang, M.; Zhou, J. Myeloid-derived suppressor cells in major depression patients suppress T-cell responses through the production of reactive oxygen species. Psychiatry Res. 2015, 228, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Schmielau, J.; Finn, O.J. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of T-cell function in advanced cancer patients. Cancer Res. 2001, 61, 4756–4760. [Google Scholar]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid. Med. Cell. Longev. 2016, 2016, 1580967. [Google Scholar] [CrossRef] [Green Version]
- Kono, K.; Salazar-Onfray, F.; Petersson, M.; Hansson, J.; Masucci, G.; Wasserman, K.; Nakazawa, T.; Anderson, P.; Kiessling, R. Hydrogen peroxide secreted by tumor-derived macrophages down-modulates signal-transducing zeta molecules and inhibits tumor-specific T cell- and natural killer cell-mediated cytotoxicity. Eur. J. Immunol. 1996, 26, 1308–1313. [Google Scholar] [CrossRef]
- Malmberg, K.J.; Arulampalam, V.; Ichihara, F.; Petersson, M.; Seki, K.; Andersson, T.; Lenkei, R.; Masucci, G.; Pettersson, S.; Kiessling, R. Inhibition of activated/memory (CD45RO+) T cells by oxidative stress associated with block of NF-κB activation. J. Immunol. 2001, 167, 2595–2601. [Google Scholar] [CrossRef] [Green Version]
- Kiemke, M.; Wabnitz, G.H.; Funke, F.; Funk, B.; Kirchgessner, H.; Samstag, Y. Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity 2008, 29, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Ball, J.A.; Vlisidou, I.; Blunt, M.D.; Wood, W.; Ward, S.H. Hydrogen peroxide triggers a dual signalling axis to selectively suppress activated human T lymphocyte migration. J. Immunol. 2017, 198, 3679–3689. [Google Scholar] [CrossRef] [Green Version]
- Özkan, B.; Lim, H.; Park, S.G. Immunomodulatory function of myeloid-derived suppressor cells during B cell-mediated immune responses. Int. J. Mol. Sci. 2018, 19, 1468. [Google Scholar] [CrossRef] [Green Version]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.; Schneck, J.; Gabrilovich, S.I. Altered recognition of antigen is a novel mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasic, T.; Colombo, P.; Soldani, C.; Wang, C.M.; Miranda, E.; Roncalli, M.; Bronte, V.; Viola, A. Modulation of human T-cell functions by reactive nitrogen species. Eur. J. Immunol. 2011, 4, 1843–1849. [Google Scholar] [CrossRef]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, J.; Wang, J.; Vangundy, Z.; You, J.; Yildiz, V.; Yu, L.; Foote, I.P.; Branson, O.E.; Stiff, A.R.; Brooks, T.R.; et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4+T cells in cancer and measurement of STAT1 nitration. Sci. Rep. 2017, 7, 15424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Ramakrishnan, R.; Altiok, S.; Youn, J.I.; Cheng, P.; Celis, E.; Pisarev, V.; Sherman, S.; Sporn, M.B.; Gabrilovich, D. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J. Clin. Investig. 2011, 121, 4015–4029. [Google Scholar] [CrossRef] [Green Version]
- Zea, A.H.; Rodriguez, P.C.; Culotta, K.S.; Hernandez, C.; DeSalvo, J.; Ochoa, J.B.; Park, H.J.; Zabaleta, J.; Ochoa, A.C. L-Arginine modulates CD3ζ expression and T cell function in activated human T lymphocytes. Cell. Immunol. 2004, 232, 21–31. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Ochoa, A.C.; Al-Khami, A. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front. Immunol. 2017, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Facciabene, A.; Motz, G.T.; Coukos, G. T regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef] [Green Version]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, P.Y.; Ma, G.; Weber, K.J.; Ozao-Choy, J.; Wang, G.; Yin, B.; Divino, C.M.; Chen, S.H. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010, 70, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okła, K.; Czerwonka, A.; Wawruszak, A.; Bobiński, M.; Bilska, M.; Tarkowski, R.; Bednarek, W.; Wertel, I.; Kotarski, J. Clinical relevance and immunosuppressive pattern of circulating and infiltrating subsets of myeloid-derived suppressor cells (MDSCs) in epithelial ovarian cancer. Front. Immunol. 2019, 10, 691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlecker, E.; Stojanovic, A.; Eisen, C.; Quack, C.; Falk, C.S.; Umansky, V.; Cerwenka, A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favouring tumor growth. J. Immunol. 2012, 189, 5602–5611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mougiakakos, D.; Johansson, C.C.; Kiessling, R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood 2009, 113, 3542–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Germann, M.; Zangger, N.; Sauvain, M.O.; Sempoux, C.; Bowler, A.D.; Wirapati, P.; Kandalaft, L.E.; Delorenzi, M.; Tejpar, S.; Coukos, G.; et al. Neutrophils suppress tumor-infiltrating T cells in colon cancer via matrix metalloproteinase-mediated activation of TGFβ. EMBO Mol. Med. 2020, 12, e10681. [Google Scholar] [CrossRef]
- Lu, C.; Red, P.S.; Lee, J.R.; Savage, N.; Liu, K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1247135. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Powis de Tenbossche, C.G.; Cané, S.; Colau, D.; van Baren, N.; Lurquin, C.; Schmitt-Verhulst, A.M.; Liljeström, P.; Uyttenhove, C.; Van den Eynde, B.J. Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat. Commun. 2017, 8, 1404. [Google Scholar] [CrossRef] [Green Version]
- Limagne, E.; Euvrard, R.; Thibaudin, M.; Rébé, C.; Derangère, V.; Chevriaux, A.; Boidot, R.; Végran, F.; Bonnefoy, N.; Vincent, J.; et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-Bevacizumab drug treatment regimen. Cancer Res. 2016, 76, 5241–5252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Aparicio, M.; Alfaro, C. Influence of interleukin-8 and neutrophil extracellular trap (NET) formation in the tumor microenvironment: Is there a pathogenic role? J. Immunol. Res. 2019, 2019, 6252138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.J.; Lee, S.H.; Kim, E.K.; Lee, E.J.; Baek, J.A.; Park, S.H.; Kwok, S.K.; Cho, M.L. Interleukin-10 produced by myeloid-derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci. Rep. 2018, 8, 3753. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-derived suppressor cells: Immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.; Abdelaal, A.M.; Shi, L.; Liang, H.; Xiong, L.; Kidder, K.; Venkataramani, M.; Culpepper, C.; Zen, K.; Liu, Y. Arginase-1 is neither constitutively expressed in nor required for myeloid-derived suppressor cell-mediated inhibition of T-cell proliferation. Eur. J. Immunol. 2018, 48, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- Centuori, S.M.; Trad, M.; LaCasse, C.J.; Alizadeh, D.; Larmonier, C.B.; Hanke, N.T.; Kartchner, J.; Janikashvili, N.; Bonnotte, B.; Larmonier, N.; et al. Myeloid-derived suppressor cells from tumor-bearing mice impair TGF-β-induced differentiation of CD4+CD25+FoxP3+ Tregs from CD4+CD25–FoxP3– T cells. J. Leukoc. Biol. 2012, 92, 987–997. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.L.; Klement, J.D.; Lu, C.; Redd, P.S.; Xiao, W.; Yang, D.; Browning, D.D.; Savage, N.M.; Buckhaults, P.J.; Morse, H.C., III; et al. Myeloid-derived suppressor cells produce IL-10 to elicit DNMT3b-dependent IRF8 silencing to promote colitis-associated colon tumorigenesis. Cell Rep. 2018, 25, 3036–3046.e6. [Google Scholar] [CrossRef] [Green Version]
- Ku, A.W.; Muhitch, J.B.; Powers, C.A.; Diehl, M.; Kim, M.; Fisher, D.T.; Sharda, A.P.; Clements, V.K.; O’Loughlin, K.; Minderman, H.; et al. Tumor-induced MDSC act via remote control to inhibit L-selectin-dependent adaptive immunity in lymphnodes. eLife 2016, 5, e17375. [Google Scholar] [CrossRef]
- Stiff, A.; Trikha, P.; Mundy-Bosse, B.; McMichael, E.; Mace, T.A.; Benner, B.; Kendra, K.; Campbell, A.; Gautam, S.; Abood, D.; et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor-mediated natural killer cell function. Clin. Cancer Res. 2018, 24, 1891–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jablonska, J.; Lang, S.; Sionov, R.V.; Granot, Z. The regulation of pre-metastatic niche formation by neutrophils. Oncotarget 2017, 8, 112132–112144. [Google Scholar] [CrossRef] [Green Version]
- Mouchmore, K.A.; Anderson, R.L.; Hamilton, J.A. Neutrophils, G-CSF and their contribution to breast cancer metastasis. FEBS J. 2018, 285, 665–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, J.; Morton, J.P.; Sansom, O.J. Neutrophils: Homing in on the myeloid mechanism of metastasis. Mol. Immunol. 2019, 110, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Wong, S.L.; Martinod, K.; Gallant, M.; Cabral, J.E.; Wang, Y.; Wagner, D.D. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 2016, 5, e1134073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Raftery, M.J.; Lalwani, P.; Krautkrmer, E.; Peters, T.; Scharffetter-Kochanek, K.; Kruger, R.; Hofmann, J.; Seeger, K.; Krüger, D.H.; Schönrich, G. β2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. J. Exp. Med. 2014, 211, 1485–1497. [Google Scholar] [CrossRef]
- Demers, M.; Krause, D.S.; Schatzberg, D.; Martinod, K.; Voorhees, J.R.; Fuchs, T.A.; Scadden, D.T.; Wagner, D.D. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13076–13081. [Google Scholar] [CrossRef] [Green Version]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef]
- Demers, M.; Wagner, D.D. NETosis: A new factor in tumor progression and cancer-associated thrombosis. Semin. Thromb. Hemost. 2014, 40, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Najmeh, S.; Cools-Lartigue, J.; Rayes, R.F.; Gowing, S.; Vourtzoumis, P.; Bourdeau, F.; Giannias, B.; Berube, J.; Rousseau, S.; Ferri, L.E.; et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int. J. Cancer 2017, 140, 2321–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, S.J.; Liang, S.; Sharma, A.; Dong, C.; Robertson, G.P. Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res. 2010, 70, 6071–6082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cedervall, J.; Hamidi, A.; Olsson, A.K. Platelets, NETs and cancer. Thromb. Res. 2018, 164, S148–S152. [Google Scholar] [CrossRef]
- Thålin, C.; Lundström, S.; Seignez, C.; Daleskog, M.; Lundström, A.; Henriksson, P.; Helleday, T.; Phillipson, M.; Wallén, H.; Demers, M. Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS ONE 2018, 13, e0191231. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Tecchio, C.; Scapini, P.; Pizzolo, G.; Cassatella, M.A. On the cytokines produced by human neutrophils in tumors. Semin. Cancer Biol. 2013, 23, 159–170. [Google Scholar] [CrossRef]
- Horikawa, N.; Abiko, K.; Matsumura, N.; Hamanishi, J.; Baba, T.; Yamaguchi, K.; Yoshioka, Y.; Koshiyama, M.; Konishi, I. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin. Cancer Res. 2017, 23, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Campbell, I.K.; Leong, D.; Edwards, K.M.; Rayzman, V.; Ng, M.; Goldberg, G.L.; Wilson, N.J.; Scalzo-Inguanti, K.; Mackenzie-Kludas, C.; Lawlor, K.E.; et al. Therapeutic targeting of the G-CSF receptor reduces neutrophil trafficking and joint inflammation in antibody-mediated inflammatory arthritis. J. Immunol. 2016, 197, 4392–4402. [Google Scholar] [CrossRef]
- Ouyang, W.; Kolls, J.K.; Zheng, Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 2008, 28, 454–467. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.; Girault, A.; Ohresser, M.; Lereclus, E.; Paintaud, G.; Lecomte, T.; Raoul, W. Monoclonal antibodies targeting the IL-17/IL-17RA axis: An opportunity to improve the efficiency of anti-VEGF therapy in fighting metastatic colorectal cancer? Clin. Colorectal Cancer 2018, 17, e109–e113. [Google Scholar] [CrossRef] [Green Version]
- Fabre, J.A.S.; Giustinniani, J.; Garbar, C.; Merrouche, Y.; Antonicelli, F.; Bensussan, A. The interleukin-17 family of cytokines in breast cancer. Int. J. Mol. Sci. 2018, 19, 3880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanaudenaerde, B.M.; Wuyts, W.A.; Geudens, N.; Dupont, L.J.; Schoofs, K.; Smeets, S.; Van Raemdonck, D.E.; Verleden, G.M. Macrolides inhibit IL17-induced IL8 and 8-isoprostane release from human airway smooth muscle cells. Am. J. Transplant. 2007, 7, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Hagiwara, K.; Honda, Y.; Gomi, K.; Kobayashi, T.; Takahashi, H.; Tokue, Y.; Watanabe, A.; Nukiwa, T. Clarithromycin suppresses lipopolysaccharide-induced interleukin-8 production by human monocytes through AP-1 and NF-kappa B transcription factors. J. Antimicrob. Chemother. 2002, 49, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Van Nuffel, A.M.; Sukhatme, V.; Pantziarka, P.; Meheus, L.; Sukhatme, V.P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)-clarithromycin as an anti-cancer agent. Ecancermedicalscience 2015, 9, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, B.L.; Anderson, R. Realizing the clinical potential of immunogenic cell death in cancer chemotherapy and radiotherapy. Int. J. Mol. Sci. 2019, 20, 959. [Google Scholar] [CrossRef] [Green Version]
- Aricò, E.; Castiello, L.; Capone, I.; Gabriele, L.; Belardelli, F. Type I interferons and cancer: An evolving story demanding novel clinical applications. Cancers 2019, 11, 1943. [Google Scholar] [CrossRef] [Green Version]
- Bertini, R.; Allegretti, M.; Bizzarri, C.; Moriconi, A.; Locati, M.; Zampella, G.; Cervellera, M.N.; Di Cioccio, V.; Cesta, M.C.; Galliera, E.; et al. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: Prevention of reperfusion injury. Proc. Natl. Acad. Sci. USA 2004, 101, 11791–11796. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chan, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Rennard, S.I.; Dale, D.C.; Donohue, J.F.; Kanniess, F.; Magnussen, H.; Sutherland, E.R.; Watz, H.; Lu, S.; Stryszak, P.; Rosenberg, E.; et al. CXCR2 antagonist MK-7123. A phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2015, 191, 1001–1011. [Google Scholar] [CrossRef]
- Lau, T.S.; Chan, L.K.Y.; Man, G.C.W.; Kwong, J. Abstract 1232: Paclitaxel induces immunogenic cell death in ovarian cancer via TLR4-independent and dependent pathways. Cancer Res. 2019, 79, 1232. [Google Scholar] [CrossRef]
- Le, D.; Gutierrez, M.E.; Saleh, M.; Chen, E.; Mallick, A.B.; Pishvaian, M.J.; Krauss, J.; O’Dwyer, P.; Garrido-Laguna, I.; Zhao, Q.; et al. Abstract CT124: A phase Ib/II study of BMS-813160, a CC chemokine receptor (CCR) 2/5 dual antagonist, in combination with chemotherapy or nivolumab in patients (pts) with advanced pancreatic or colorectal cancer. Cancer Res. 2018, 78, CT124. [Google Scholar] [CrossRef]
- Juric, V.; O’Sullivan, C.; Stefanutti, E.; Kovalenko, M.; Greenstein, A.; Barry-Hamilton, V.; Mikaelian, I.; Degenhardt, J.; Yue, P.; Smith, V.; et al. MMP-9 inhibition promotes anti-tumor immunity through disruption of biochemical and physical barriers to T-cell trafficking to tumors. PLoS ONE 2018, 13, e0207255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.H.; Minton, S.E.; Han, H.S.; Ismail-Khan, R.; Neuger, A.; Khambati, F.; Noyes, D.; Lush, R.; Chiappori, A.A.; Roberts, J.D.; et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget 2016, 7, 22928–22938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, G.A.; Condamine, T.; Mony, S.; Hashimoto, A.; Wang, F.; Liu, Q.; Forero, A.; Bendell, J.; Witt, R.; Hockstein, N.; et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin. Cancer Res. 2017, 23, 2942–2950. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Type of Malignancy | Associated Infective Agent |
|---|---|
| Squamous cell carcinoma of the bone, sinuses and skin | Chronic osteomyelitis most commonly caused by Staphylococcus aureus |
| Urinary bladder cancer | Schistosoma haematobium |
| Ovarian cancer | Pelvic inflammatory disease most commonly caused by Chlamydia trachomatis and Neisseria gonorrhoeae |
| Gastric cancer | Gastritis caused by Helicobacter pylori |
| MALT lymphoma | Helicobacter pylori |
| Lung carcinomas | Chronic and recurrent pulmonary infection as a result of various bacterial pathogens |
| Testicular cancer | Orchitis caused by mumps virus |
| Hepatocellular carcinoma | Hepatitis viruses B and C |
| Cervical cancer | Human papilloma virus |
| Kaposi’s sarcoma | Human herpes virus type 8 |
| Type of Malignancy | Associated Conditions |
|---|---|
| Colon carcinomas | Inflammatory bowel disease (Crohn’s disease, colitis) |
| Urinary bladder cancer | Long-term indwelling catheters, stones |
| Gall bladder cancer | Chronic cholecystitis, cholelithiasis |
| Oesophageal squamous cell carcinoma and adenocarcinoma | Chronic exposure to chemical irritants and acid reflux oesophagitis, respectively |
| Lung carcinomas | Cigarette smoking, pulmonary fibrosis, sarcoidosis |
| Mesothelioma | Asbestos inhalation |
| Head and neck cancer | Cigarette smoking |
| Skin cancer (basal cell/squamous cell carcinoma, melanoma) | Exposure to sunlight |
| Mediators and Mechanisms of Pro-Oxidative Activity | ||
|---|---|---|
| Mediator | Mechanism of Immunosuppression | Ref |
| H2O2 | Activation of poly(ADP-ribose) polymerase resulting in depletion of nicotinamide adenine dinucleotide and adenosine-5′-triphosphate | [3] |
| H2O2 | Trogocytosis | [54] |
| H2O2 | Decreased expression of the TCR zeta-chain, resulting in decreased T cell activation and anti-tumor cytokine production, especially interferon-gamma | [68,70,71,74] |
| H2O2 | Induction of T cell apoptosis | [75] |
| H2O2 | Attenuation of activation of NFκB resulting in decreased production of T cell cytokines | [77] |
| H2O2 | Oxidation of cofilin resulting in impaired T cell activation and recruitment | [63,78] |
| H2O2 | Decreased expression of T cell CXCR3, resulting in failure of responsiveness to CXCL11 | [79] |
| Peroxynitrite | Nitration of the TCR resulting in the failure to interact with MHC/antigenic peptides presented by APCs | [82] |
| Peroxynitrite/RNS | Induction of T cell apoptosis | [83] |
| Peroxynitrite/RNS | Nitrative inactivation of T cell chemokines such as CCL2 | [84] |
| Peroxynitrite/RNS | Nitrative inhibition of antigen presentation by dendritic cells to T cells | [85] |
| Non-Oxidative Mechanisms | ||
| Arginase 1 | Depletes arginine necessary for many anti-tumor activities of T cells | [52,61,63,81,87,88] |
| IL-10 and TGFβ1 | Differentiation and expansion of pro-tumorigenic of Foxp3+ regulatory T cells | [89,90,91,92,93,94,95] |
| Indoleamine-2,3-dioxygenase | Depletes tryptophan necessary for T cell proliferation | [96] |
| Sequestration of cystine and cysteine | Compromises T cell intracellular anti-oxidant defences | [97] |
| Proteases | Proteolytic inactivation of T cell-derived immunostimulatory cytokines such as IL-2, IL-6 and TNF-α; activation of latent TGFß1 | [63,98] |
| Expression of PD-L1 | Suppression of T cell activation via interaction with PD-1 | [99] |
| Expression of FasL | Induces apoptosis of TILs | [100] |
| EctonucleotidasesCD39 and CD73 | Promote formation of immunosuppressive adenosine | [101] |
| Cyclooxygenases 1 and 2 | Promote formation of immunosuppressive PGE2 | [102] |
| Intra-tumoral NETs | Impede access of TILs to tumor cells | [103] |
| Type of Adjunctive Therapy | Status | Ref |
|---|---|---|
| mAb targeting of G-CSF or its receptor | Seemingly impracticable | [35,129] |
| mAb targeting of IL-17A or its receptor | Promising pre-clinical findings in colorectal, breast and non-small cell lung cancers | [40,131,132] |
| Macrolide antibiotic targeting of IL-8 and IL-17A production | Uncertain | [135] |
| mAb targeting of TGFβ1 | Promising, but poses the risk of dysregulated immune homeostasis | [59] |
| Administration of type I interferons to prevent N1→N2 TAN reprogramming | Uncertain, but administration of ICD-inducing strategies may be preferable | [59,137] |
| Administration of CXCR1/2 antagonists | Very promising, undergoing advanced clinical evaluation | [138,139,140,141] |
| mAb targeting of MMP-9 | Unproven, but may represent an alternative strategy to prevent activation of latent TGFβ1 in the TME | [98] |
| Small molecule inhibitors of arginase-1 and IDO to preserve arginine and tryptophan, respectively, in the TME | Unproven | [144,145] |
| Small molecule antagonists of adenosine A2A receptors, as well as inhibitors of ATP ectonucleotidases and cyclooxygenases to prevent activation of T cell adenylyl cyclase via production of adenosine and PGE2, respectively | Unproven | [146] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rapoport, B.L.; Steel, H.C.; Theron, A.J.; Smit, T.; Anderson, R. Role of the Neutrophil in the Pathogenesis of Advanced Cancer and Impaired Responsiveness to Therapy. Molecules 2020, 25, 1618. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071618
Rapoport BL, Steel HC, Theron AJ, Smit T, Anderson R. Role of the Neutrophil in the Pathogenesis of Advanced Cancer and Impaired Responsiveness to Therapy. Molecules. 2020; 25(7):1618. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071618
Chicago/Turabian StyleRapoport, Bernardo L., Helen C. Steel, Annette J. Theron, Teresa Smit, and Ronald Anderson. 2020. "Role of the Neutrophil in the Pathogenesis of Advanced Cancer and Impaired Responsiveness to Therapy" Molecules 25, no. 7: 1618. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071618