
Coordination Polymers Based on a Biphenyl Tetraphosphonate Linker: Synthesis Control and Photoluminescence

 , ,
, ,
Abstract
:1. Introduction
- (i)
- layered [Ln7(H5btp)4(H5.5btp)2(H6btp)2(H2O)12]∙23.5H2O∙MeOH [where Ln3+ = Eu3+ (1Eu) and Gd3+ (1Gd)] (prepared using the same self-assembly methodology previously reported for the materials based on La3+ and H8btp, i.e., microwave-irradiation followed by slow evaporation);
- (ii)
- 3D [Ln4(H3btp)(H4btp)(H5btp)(H2O)8]∙3H2O [where Ln3+ = Ce3+ (2Ce), Pr3+ (2Pr), and Nd3+ (2Nd)], obtained from conventional hydro(solvo)thermal synthesis. This approach allowed us to obtain a material with increased dimensionality (3D) in a much less synthesis time.
2. Results and Discussion
2.1. Crystal Structure Details
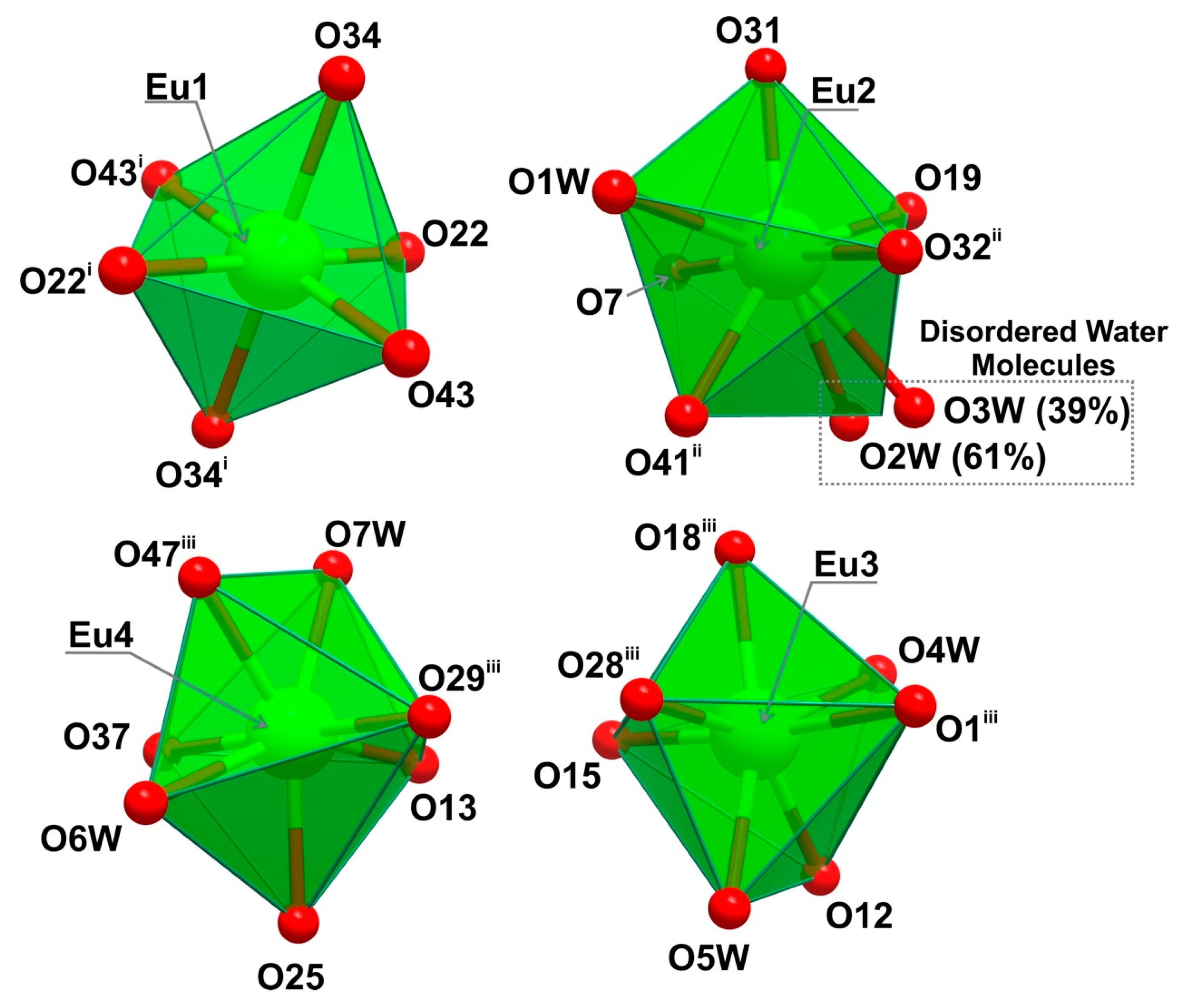
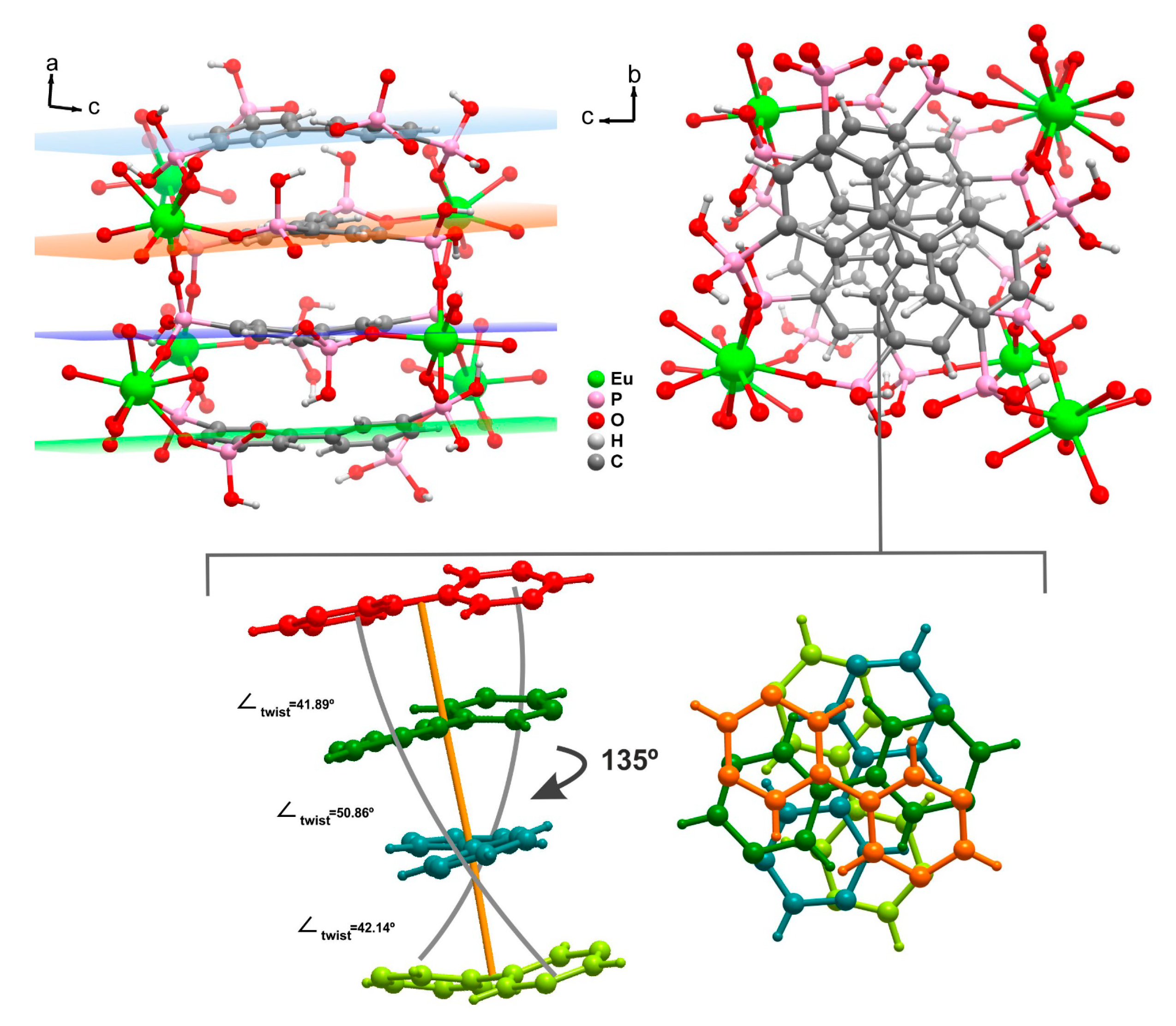
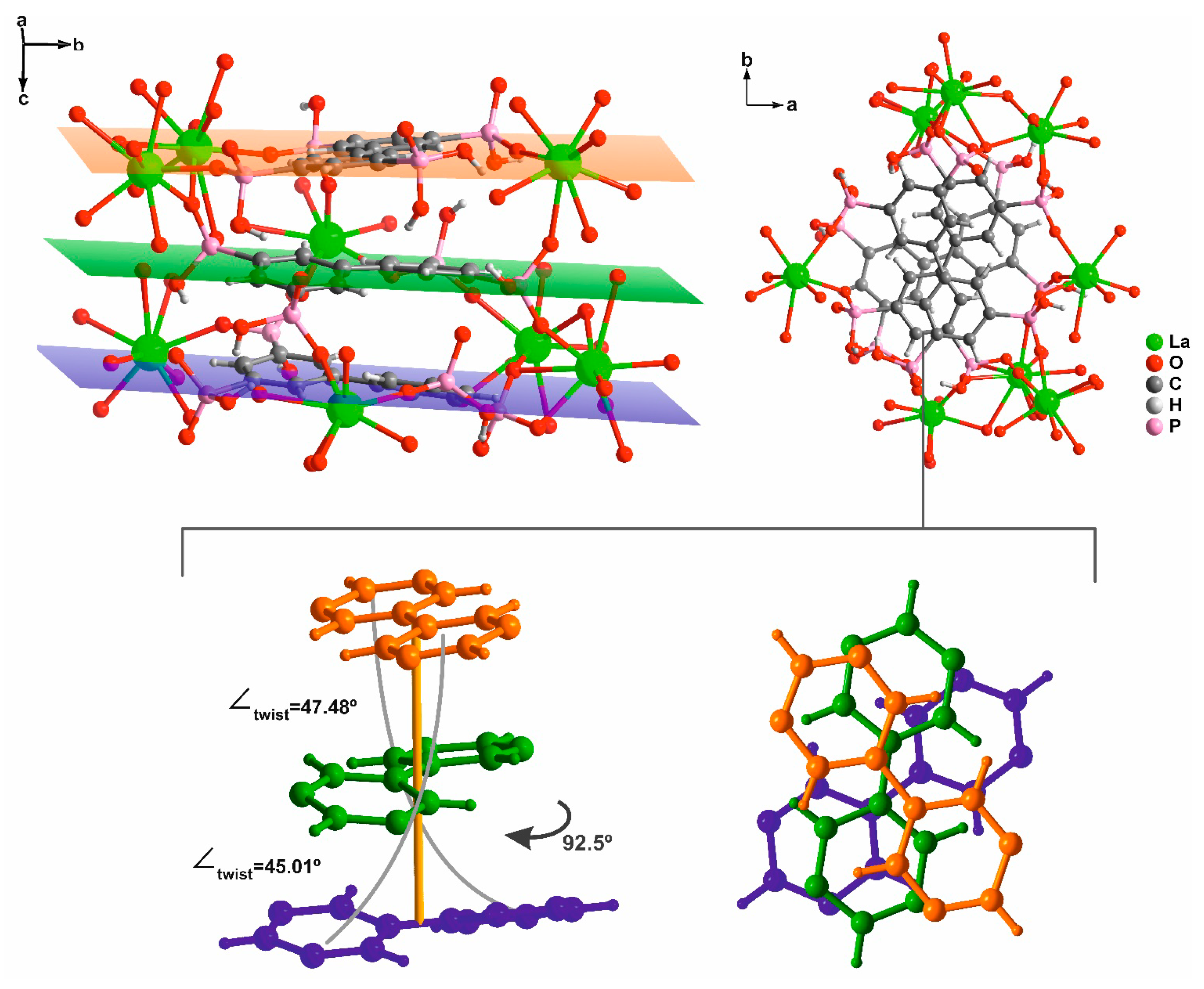
2.1.1. [Ln7(H5btp)4(H5.5btp)2(H6btp)2(H2O)12]∙23.5H2O∙MeOH (1)
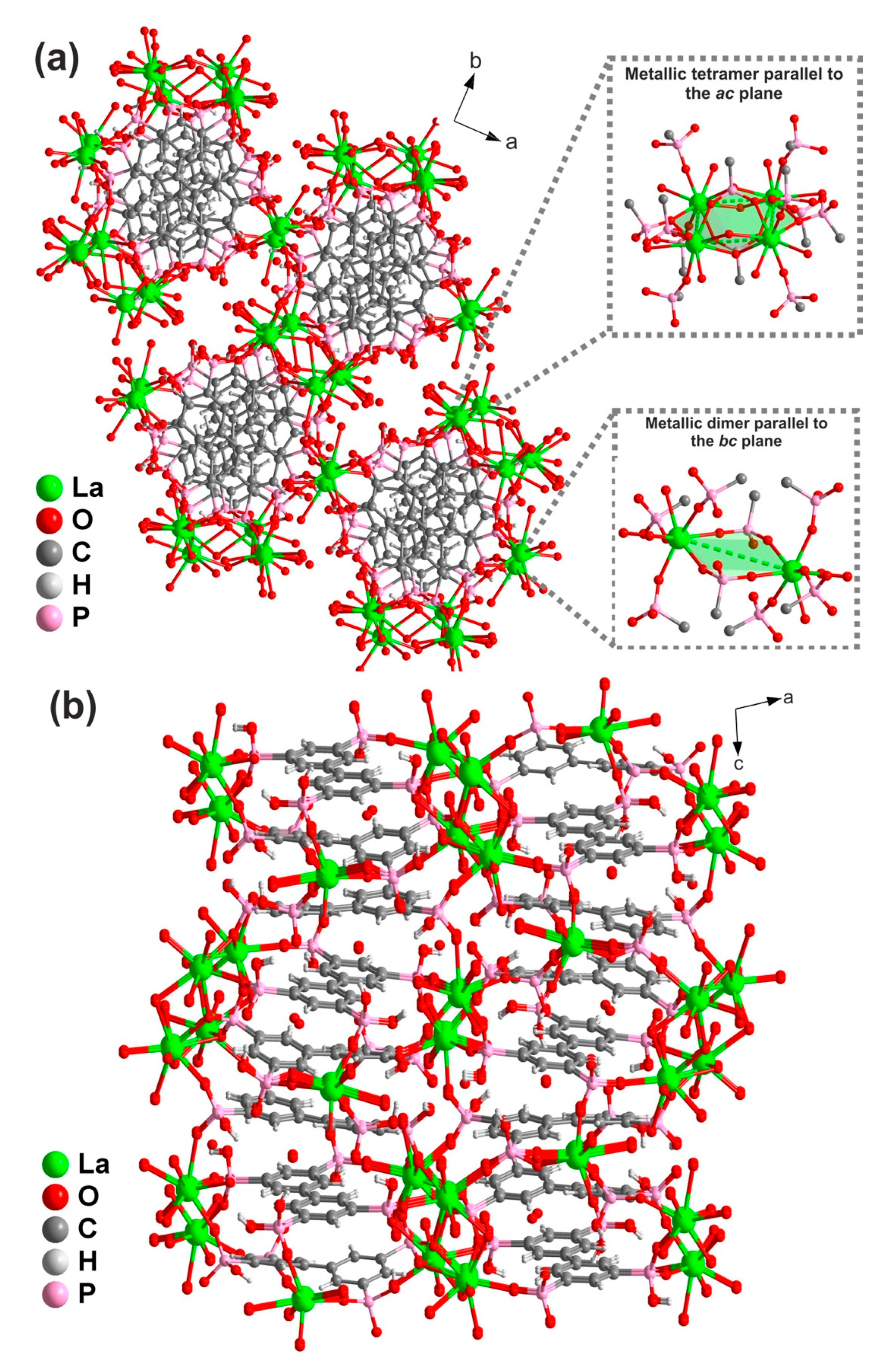
2.1.2. [Ln4(H3btp)(H4btp)(H5btp)(H2O)8]∙3H2O (2)
2.2. Structural Characterization
2.2.1. Thermogravimetry
2.2.2. FT-IR Spectroscopic Studies
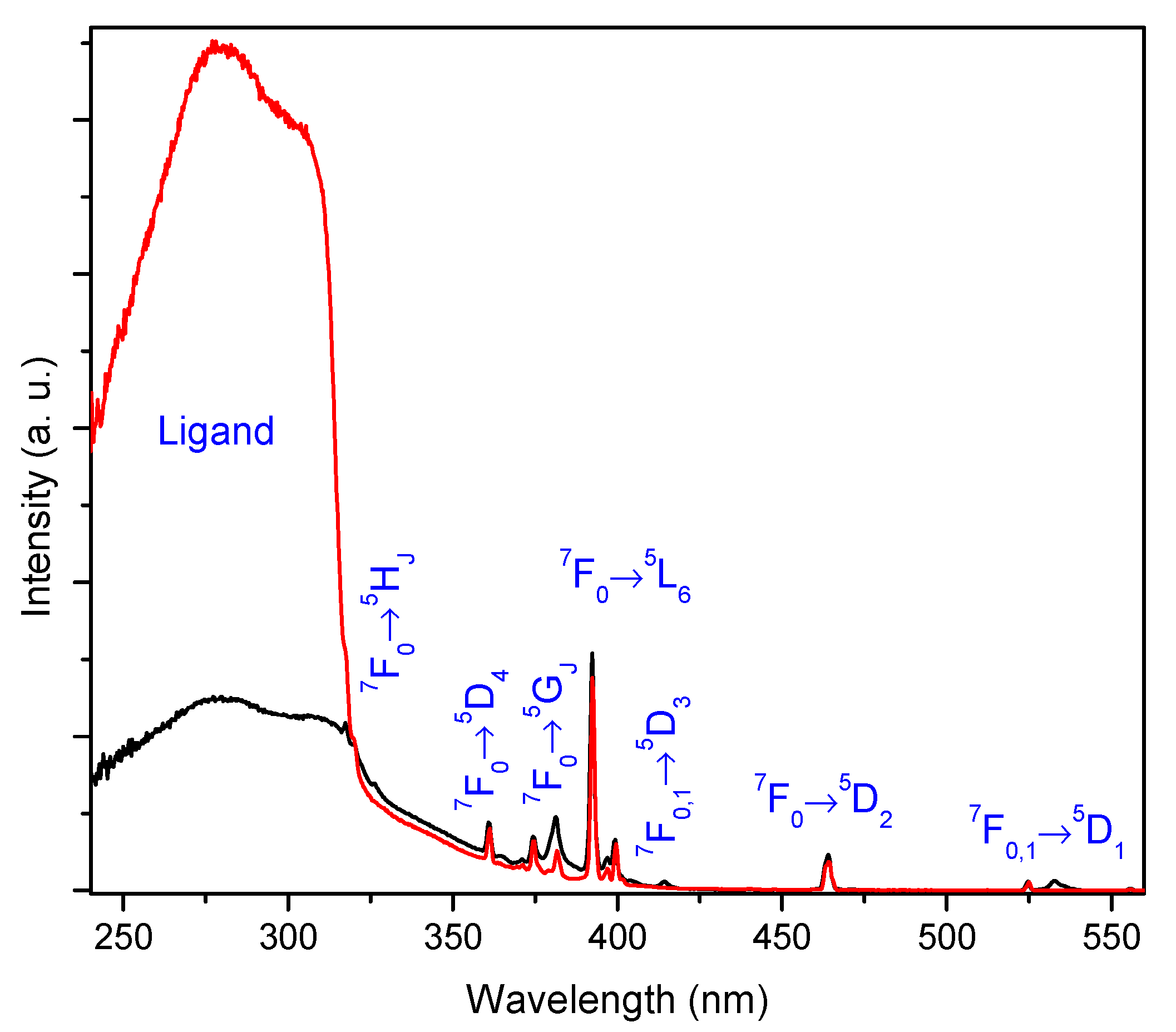
2.3. Photoluminescence Studies
3. Materials and Methods
3.1. Synthesis of [Ln7(H5btp)4(H5.5btp)2(H6btp)2(H2O)12]∙23.5H2O∙MeOH (1)
3.2. Synthesis of [Ln4(H3btp)(H4btp)(H5btp)(H2O)8]∙3H2O (2)
- (i)
- heating during 48 h up to 140 °C;
- (ii)
- 24 h uphold of the temperature (140 °C);
- (iii)
- cooling down during 48 h until ambient temperature.
3.3. Single-Crystal X-ray Diffraction Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Silva, P.; Vilela, S.M.F.; Tomé, J.P.C.; Paz, F.A.A. Multifunctional metal-organic frameworks: From academia to industrial applications. Chem. Soc. Rev. 2015, 44, 6774–6803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Wen, H.M.; Cui, Y.J.; Zhou, W.; Qian, G.D.; Chen, B.L. Emerging multifunctional metal-organic framework materials. Adv. Mater. 2016, 28, 8819–8860. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Perez, E.S.; Murdock, C.R.; Didas, S.A.; Jones, C.W. Direct capture of co2 from ambient air. Chem. Rev. 2016, 116, 11840–11876. [Google Scholar] [CrossRef] [PubMed]
- Suh, M.P.; Park, H.J.; Prasad, T.K.; Lim, D.W. Hydrogen storage in metal-organic frameworks. Chem. Rev. 2012, 112, 782–835. [Google Scholar] [CrossRef] [PubMed]
- Joharian, M.; Morsali, A.; Tehrani, A.A.; Carlucci, L.; Proserpio, D.M. Water-stable fluorinated metal-organic frameworks (f-mofs) with hydrophobic properties as efficient and highly active heterogeneous catalysts in aqueous solution. Green Chem. 2018, 20, 5336–5345. [Google Scholar] [CrossRef]
- Huang, Y.B.; Liang, J.; Wang, X.S.; Cao, R. Multifunctional metal-organic framework catalysts: Synergistic catalysis and tandem reactions. Chem. Soc. Rev. 2017, 46, 126–157. [Google Scholar] [CrossRef]
- Tehrani, A.A.; Esrafili, L.; Abedi, S.; Morsali, A.; Carlucci, L.; Proserpio, D.M.; Wang, J.; Junk, P.C.; Liu, T.F. Urea metal-organic frameworks for nitro-substituted compounds sensing. Inorg. Chem. 2017, 56, 1446–1454. [Google Scholar] [CrossRef]
- Smith, J.A.; Singh-Wilmot, M.A.; Carter, K.P.; Cahill, C.L.; Ridenour, J.A. Lanthanide-2,3,5,6-tetrabromoterephthalic acid metal-organic frameworks: Evolution of halogen center dot center dot center dot halogen interactions across the lanthanide series and their potential as selective bifunctional sensors for the detection of fe3+, cu2+, and nitroaromatics. Cryst. Growth Des. 2019, 19, 305–319. [Google Scholar]
- Lustig, W.P.; Mukherjee, S.; Rudd, N.D.; Desai, A.V.; Li, J.; Ghosh, S.K. Metal-organic frameworks: Functional luminescent and photonic materials for sensing applications. Chem. Soc. Rev. 2017, 46, 3242–3285. [Google Scholar] [CrossRef]
- Pereira, C.F.; Figueira, F.; Mendes, R.F.; Rocha, J.; Hupp, J.T.; Farha, O.K.; Simões, M.M.Q.; Tomé, J.P.C.; Paz, F.A.A. Bifunctional porphyrin-based nano-metal-organic frameworks: Catalytic and chemosensing studies. Inorg. Chem. 2018, 57, 3855–3864. [Google Scholar] [CrossRef]
- Venkatramaiah, N.; Pereira, C.F.; Mendes, R.F.; Paz, F.A.A.; Tomé, J.P.C. Phosphonate appended porphyrins as versatile chemosensors for selective detection of trinitrotoluene. Anal. Chem. 2015, 87, 4515–4522. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wu, C.S. Synthesis, functionalization, and applications of metal-organic frameworks in biomedicine. Dalton Trans. 2018, 47, 2114–2133. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, P.; Gref, R.; Baati, T.; Allan, P.K.; Maurin, G.; Couvreur, P.; Férey, G.; Morris, R.E.; Serre, C. Metal-organic frameworks in biomedicine. Chem. Rev. 2012, 112, 1232–1268. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zhao, D.; Lu, Y.; Sun, W.Y. Photoluminescent metal-organic frameworks and their application for sensing biomolecules. J. Mater. Chem. A 2019, 7, 22744–22767. [Google Scholar] [CrossRef]
- He, J.; Xu, J.L.; Yin, J.C.; Li, N.; Bu, X.H. Recent advances in luminescent metal-organic frameworks for chemical sensors. Sci. China-Mater. 2019, 62, 1655–1678. [Google Scholar] [CrossRef] [Green Version]
- Kuyuldar, S.; Genna, D.T.; Burda, C. On the potential for nanoscale metal-organic frameworks for energy applications. J. Mater. Chem. A 2019, 7, 21545–21576. [Google Scholar] [CrossRef]
- Zhang, Z.; Sang, W.; Xie, L.S.; Dai, Y.L. Metal-organic frameworks for multimodal bioimaging and synergistic cancer chemotherapy. Coord. Chem. Rev. 2019, 399, 213022. [Google Scholar] [CrossRef]
- Bobbitt, N.S.; Mendonça, M.L.; Howarth, A.J.; Islamoglu, T.; Hupp, J.T.; Farha, O.K.; Snurr, R.Q. Metal-organic frameworks for the removal of toxic industrial chemicals and chemical warfare agents. Chem. Soc. Rev. 2017, 46, 3357–3385. [Google Scholar] [CrossRef]
- Wang, H.L.; Zhu, Q.L.; Zou, R.Q.; Xu, Q. Metal-organic frameworks for energy applications. Chem 2017, 2, 52–80. [Google Scholar] [CrossRef] [Green Version]
- Colodrero, R.M.P.; Salcedo, I.R.; Bazaga-Garcia, M.; Barouda, E.; Papadaki, M.; Papathanasiou, K.E.; Hernandez-Alonso, D.; Rius, J.; Aranda, M.A.G.; Losilla, E.R.; et al. High-throughput synthesis of pillared-layered magnesium tetraphosphonate coordination polymers: Framework interconversions and proton conductivity studies. Inorganics 2018, 6, 96. [Google Scholar] [CrossRef] [Green Version]
- Salcedo, I.R.; Colodrero, R.M.P.; Bazaga-Garcia, M.; Vasileiou, A.; Papadaki, M.; Olivera-Pastor, P.; Infantes-Molina, A.; Losilla, E.R.; Mezei, G.; Cabeza, A.; et al. From light to heavy alkali metal tetraphosphonates (m = li, na, k, rb, cs): Cation size-induced structural diversity and water-facilitated proton conductivity. Crystengcomm 2018, 20, 7648–7658. [Google Scholar] [CrossRef]
- Ji, B.M.; Deng, D.S.; Ma, J.Y.; Sun, C.W.; Zhao, B. Two- and three-dimensional lanthanide-based coordination polymers assembled by the synergistic effect of various lanthanide radii and flexibility of a new binicotinate-containing ligand: In situ synthesis, structures, and properties. RSC Adv. 2015, 5, 2239–2248. [Google Scholar] [CrossRef]
- Xiang, S.C.; Hu, S.M.; Sheng, T.L.; Chen, J.S.; Wu, X.T. Structural diversity of infinite 3d-4f heterometallic cluster compounds driven by various lanthanide radii. Chem.-Eur. J. 2009, 15, 12496–12502. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.H.; Li, Y.F.; Wang, Q.; Huang, X.G.; Zhang, Y.; Gao, C.J.; Liu, W.S.; Tang, Y.; Zhang, H.R.; Shao, Y.L. Two- to one-dimensional: Radii-dependent self-assembly crystal structures and luminescent properties of lanthanide coordination polymers with an amide type semirigid bridging ligand. Cryst. Growth Des. 2011, 11, 4205–4212. [Google Scholar] [CrossRef]
- Wang, H.S.; Li, J.; Li, J.Y.; Wang, K.; Ding, Y.; Xia, X.H. Lanthanide-based metal-organic framework nanosheets with unique fluorescence quenching properties for two-color intracellular adenosine imaging in living cells. NPG Asia Mater. 2017, 9, e354. [Google Scholar] [CrossRef] [Green Version]
- Zareba, J.K. Tetraphenylmethane and tetraphenylsilane as building units of coordination polymers and supramolecular networks—A focus on tetraphosphonates. Inorg. Chem. Commun. 2017, 86, 172–186. [Google Scholar] [CrossRef]
- Zheng, T.; Yang, Z.X.; Gui, D.X.; Liu, Z.Y.; Wang, X.X.; Dai, X.; Liu, S.T.; Zhang, L.J.; Gao, Y.; Chen, L.H.; et al. Overcoming the crystallization and designability issues in the ultrastable zirconium phosphonate framework system. Nat. Commun. 2017, 8, 15369. [Google Scholar] [CrossRef]
- Firmino, A.D.G.; Mendes, R.F.; Ananias, D.; Vilela, S.M.F.; Carlos, L.D.; Tomé, J.P.C.; Rocha, J.; Paz, F.A.A. Microwave synthesis of a photoluminescent metal-organic framework based on a rigid tetraphosphonate linker. Inorg. Chim. Acta 2017, 455, 584–594. [Google Scholar] [CrossRef]
- Mendes, R.F.; Ananias, D.; Carlos, L.D.; Rocha, J.; Paz, F.A.A. Photoluminescent lanthanide–organic framework based on a tetraphosphonic acid linker. Cryst. Growth Des. 2017, 17, 5191–5199. [Google Scholar] [CrossRef]
- Vilela, S.M.F.; Firmino, A.D.G.; Mendes, R.F.; Fernandes, J.A.; Ananias, D.; Valente, A.A.; Ott, H.; Carlos, L.D.; Rocha, J.; Tomé, J.P.C.; et al. Lanthanide-polyphosphonate coordination polymers combining catalytic and photoluminescence properties. Chem. Commun. 2013, 49, 6400–6402. [Google Scholar] [CrossRef]
- Ananias, D.; Firmino, A.D.G.; Mendes, R.F.; Paz, F.A.A.; Nolasco, M.; Carlos, L.D.; Rocha, J. Excimer formation in a terbium metal-organic framework assists luminescence thermometry. Chem. Mat. 2017, 29, 9547–9554. [Google Scholar] [CrossRef]
- Mendes, R.F.; Antunes, M.M.; Silva, P.; Barbosa, P.; Figueiredo, F.; Linden, A.; Rocha, J.; Valente, A.A.; Paz, F.A.A. A lamellar coordination polymer with remarkable catalytic activity. Chem.-A Eur. J. 2016, 22, 13136–13146. [Google Scholar] [CrossRef]
- Mendes, R.F.; Silva, P.; Antunes, M.M.; Valente, A.A.; Paz, F.A.A. Sustainable synthesis of a catalytic active one-dimensional lanthanide–organic coordination polymer. Chem. Commun. 2015, 51, 10807–10810. [Google Scholar] [CrossRef] [PubMed]
- Firmino, A.D.G.; Mendes, R.F.; Antunes, M.M.; Barbosa, P.C.; Vilela, S.M.F.; Valente, A.A.; Figueiredo, F.M.L.; Tomé, J.P.C.; Paz, F.A.A. Robust multifunctional yttrium-based metal organic frameworks with breathing effect. Inorg. Chem. 2017, 56, 1193–1208. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, E.V.; Blatov, V.A.; Kochetkov, A.V.; Proserpio, D.M. Underlying nets in three-periodic coordination polymers: Topology, taxonomy and prediction from a computer-aided analysis of the cambridge structural database. Crystengcomm 2011, 13, 3947–3958. [Google Scholar] [CrossRef]
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied topological analysis of crystal structures with the program package topospro. Cryst. Growth Des. 2014, 14, 3576–3586. [Google Scholar] [CrossRef]
- In RCSR (Reticular Chemistry Structure Resource). Available online: http://rcsr.anu.edu.au/ (accessed on 15 January 2020).
- In EPINET. Available online: http://epinet.anu.edu.au (accessed on 15 January 2020).
- Bonneau, C.; O’Keeffe, M.; Proserpio, D.M.; Blatov, V.A.; Batten, S.R.; Bourne, S.A.; Lah, M.S.; Eon, J.G.; Hyde, S.T.; Wiggin, S.B.; et al. Deconstruction of crystalline networks into underlying nets: Relevance for terminology guidelines and crystallographic databases. Cryst. Growth Des. 2018, 18, 3411–3418. [Google Scholar] [CrossRef]
- Kottke, T.; Stalke, D.J. Crystal handling at low temperatures. Appl. Crystallogr. 1993, 26, 615–619. [Google Scholar] [CrossRef] [Green Version]
- SAINT+. Data Integration Engine v. 8.37a©; Bruker AXS: Madison, WI, USA, 1997–2015. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus x-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Shelxt - integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with shelxl. Acta Crystallogr. Sect. C-Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittric, B.J. ShelXle: A Qt graphical user interface for SHELXL. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar]
- Brandenburg, K. DIAMOND, Version 3.2f; Crystal Impact GbR: Bonn, Germany, 1997–2010. [Google Scholar]
Sample Availability: Limited amount of samples of the compounds may be available from the authors upon request. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1Eu | 2La | |
|---|---|---|
| Formula | C97H166Eu7O132.5P32 | C36H52La4O47P12 |
| Formula weight | 5507.05 | 2164.05 |
| Temperature/K | 150(2) | 150(2) |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c |
| a/Å | 13.6342(19) | 12.5332(16) |
| b/Å | 34.172(5) | 24.147(3) |
| c/Å | 19.049(3) | 20.852(3) |
| b/° | 93.213(5) | 100.037(3) |
| Volume/Å3 | 8861(2) | 6213.9(14) |
| Z | 2 | 4 |
| ρcalc/g cm−3 | 2.064 | 2.313 |
| m(Mo Ka)/mm−1 | 2.854 | 3.122 |
| Crystal type | Colorless plate | Colorless plate |
| Crystal size/mm | 0.20 × 0.10 × 0.02 | 0.17 × 0.06 × 0.02 |
| θ range (°) | 3.52–25.35 | 3.518–29.13 |
| Index ranges | −16 ≤ h ≤ 16 −41 ≤ k ≤ 41 −22 ≤ l ≤ 22 | −17 ≤ h ≤ 17 −33 ≤ k ≤ 33 −26 ≤ l ≤ 28 |
| Collected Reflections | 243,730 | 167,012 |
| Independent Reflections | 16,181 (Rint = 0.1633) | 16,687 (Rint = 0.0779) |
| Completeness | 99.7% (θ = 25.24) | 99.7% (θ = 25.24) |
| Final R indices [I > 2σ(I)] | R1 = 0.0639 wR2 = 0.1327 | R1 = 0.0549 wR2 = 0.1101 |
| Final R indices (all data) | R1 = 0.1088 wR2 = 0.1507 | R1 = 0.0709 wR2 = 0.1153 |
| Largest diff. peak and hole/eÅ−3 | 2.005 and −2.108 | 2.468 and −1.888 |
| ; where | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Firmino, A.D.G.; Mendes, R.F.; Ananias, D.; Barbosa, J.S.; Tomé, J.P.C.; Almeida Paz, F.A. Coordination Polymers Based on a Biphenyl Tetraphosphonate Linker: Synthesis Control and Photoluminescence. Molecules 2020, 25, 1835. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25081835
Firmino ADG, Mendes RF, Ananias D, Barbosa JS, Tomé JPC, Almeida Paz FA. Coordination Polymers Based on a Biphenyl Tetraphosphonate Linker: Synthesis Control and Photoluminescence. Molecules. 2020; 25(8):1835. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25081835
Chicago/Turabian StyleFirmino, Ana D. G., Ricardo F. Mendes, Duarte Ananias, Jéssica S. Barbosa, João P. C. Tomé, and Filipe A. Almeida Paz. 2020. "Coordination Polymers Based on a Biphenyl Tetraphosphonate Linker: Synthesis Control and Photoluminescence" Molecules 25, no. 8: 1835. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25081835