
PDADMAC/PSS Oligoelectrolyte Multilayers: Internal Structure and Hydration Properties at Early Growth Stages from Atomistic Simulations

 , ,
, ,  , and
, and 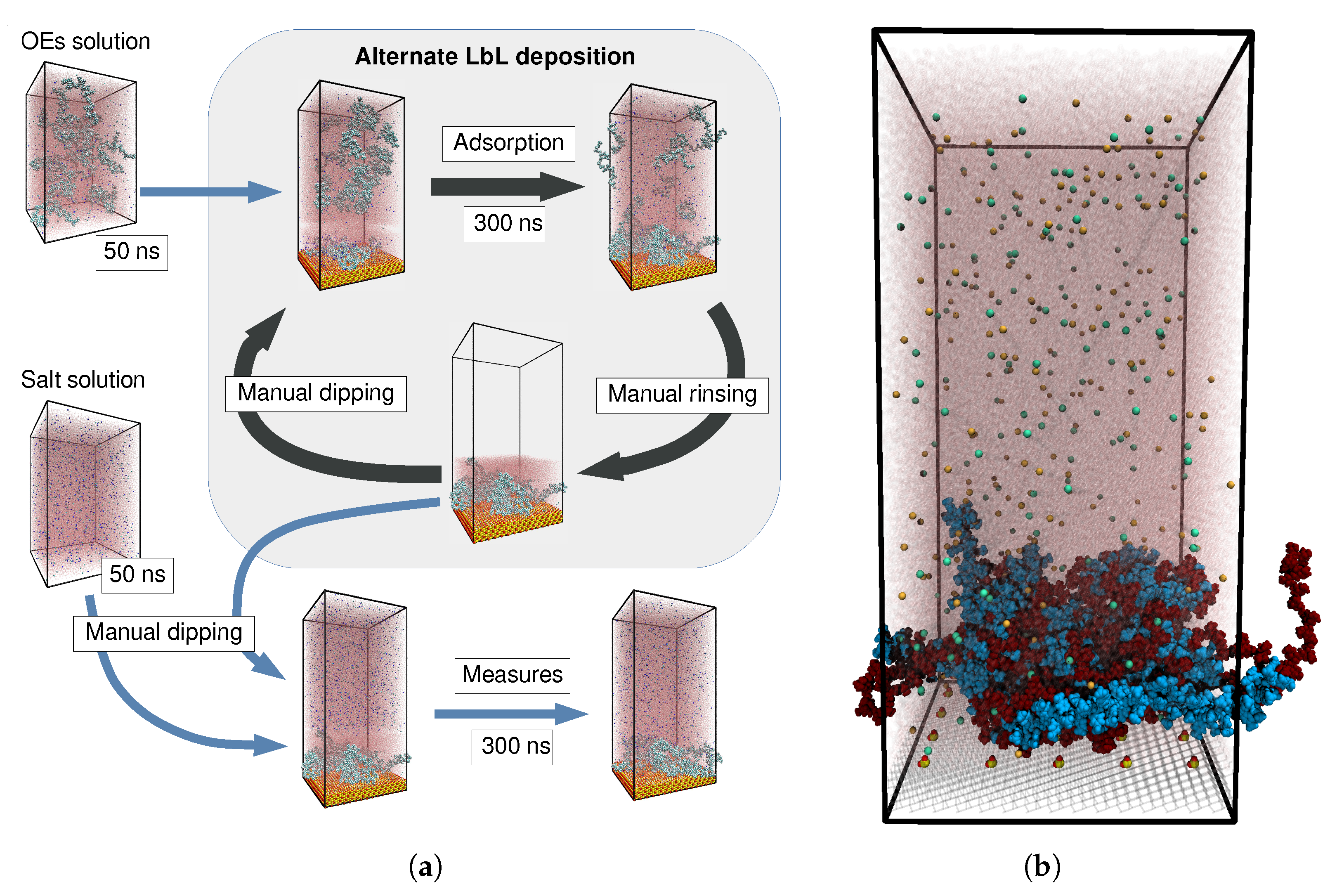{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
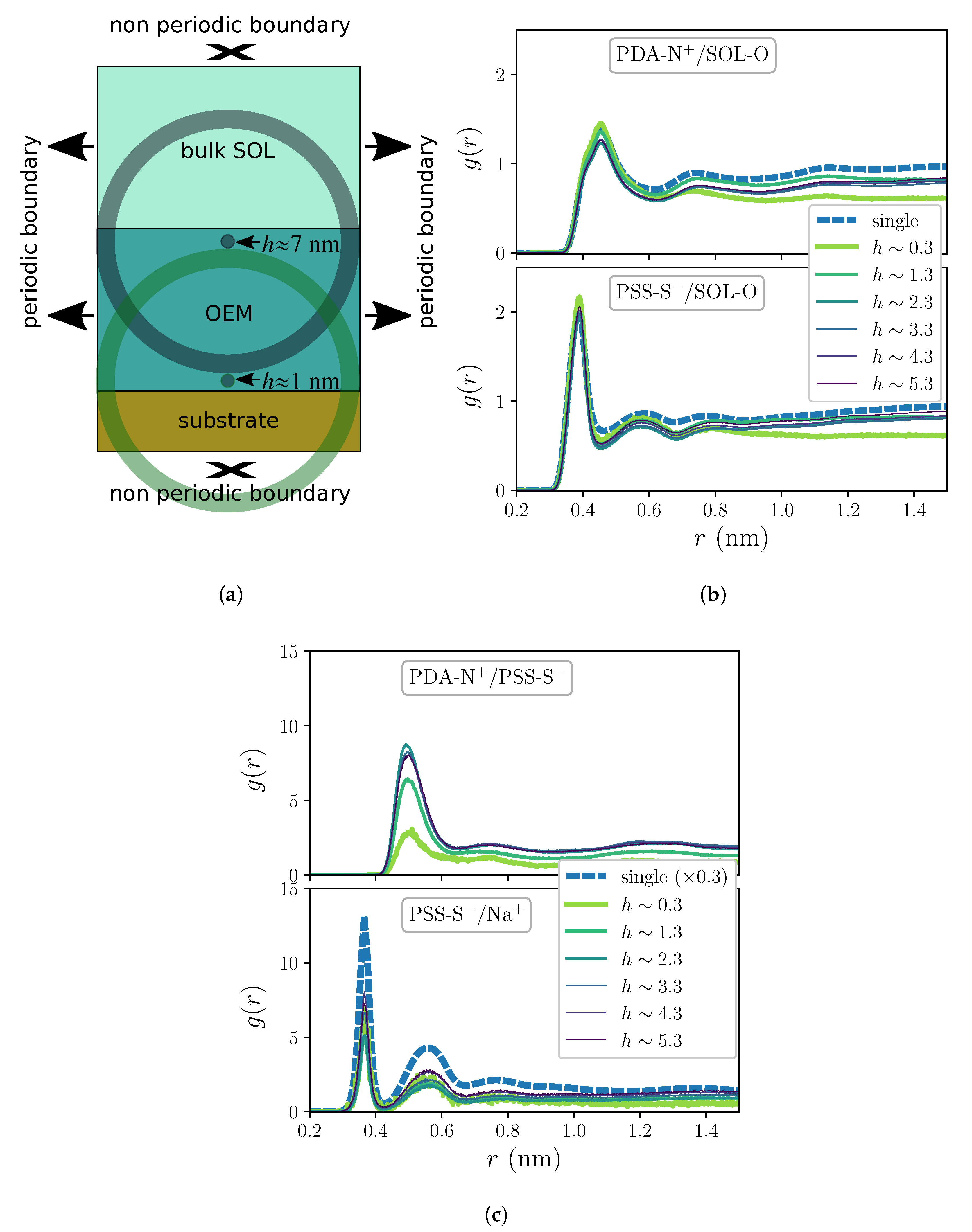
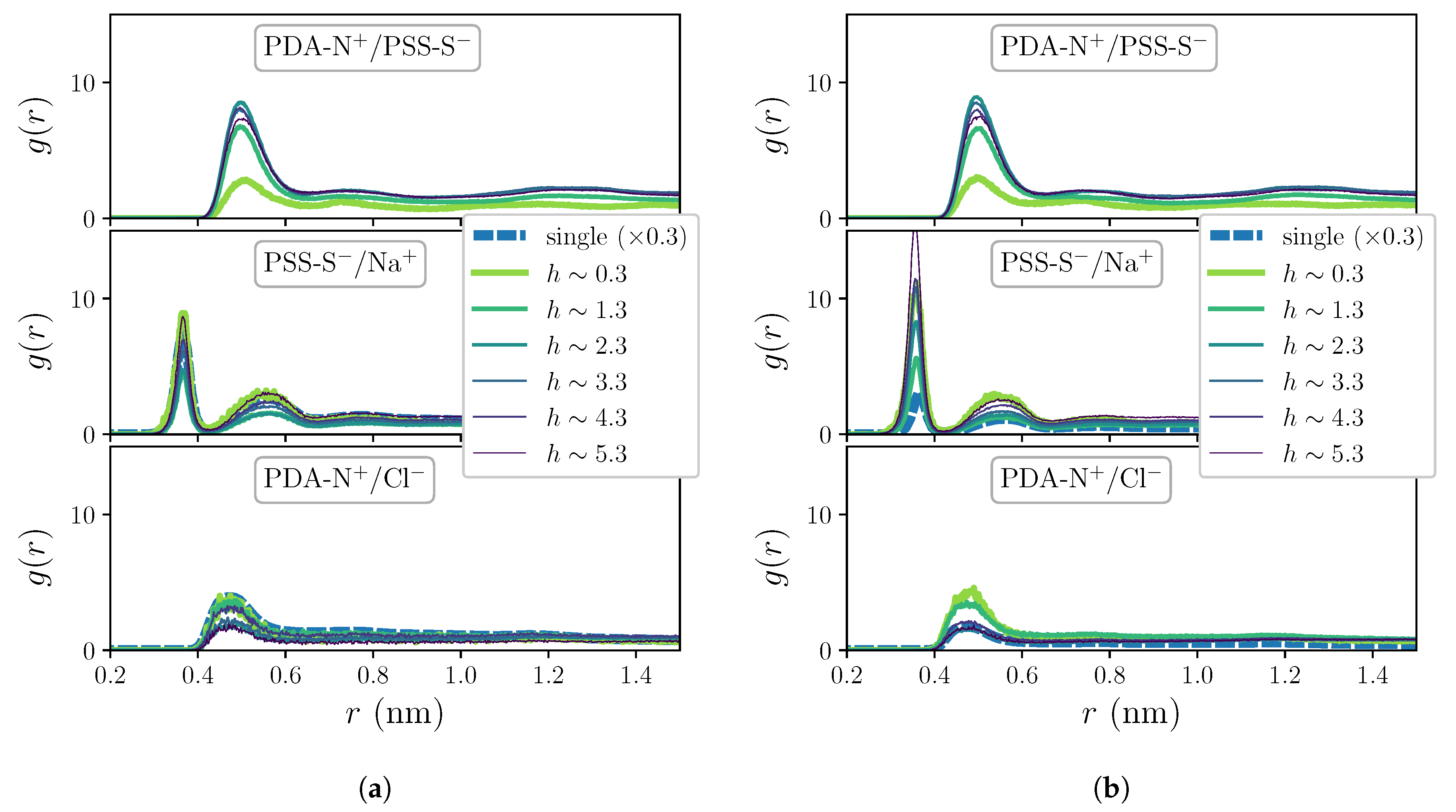
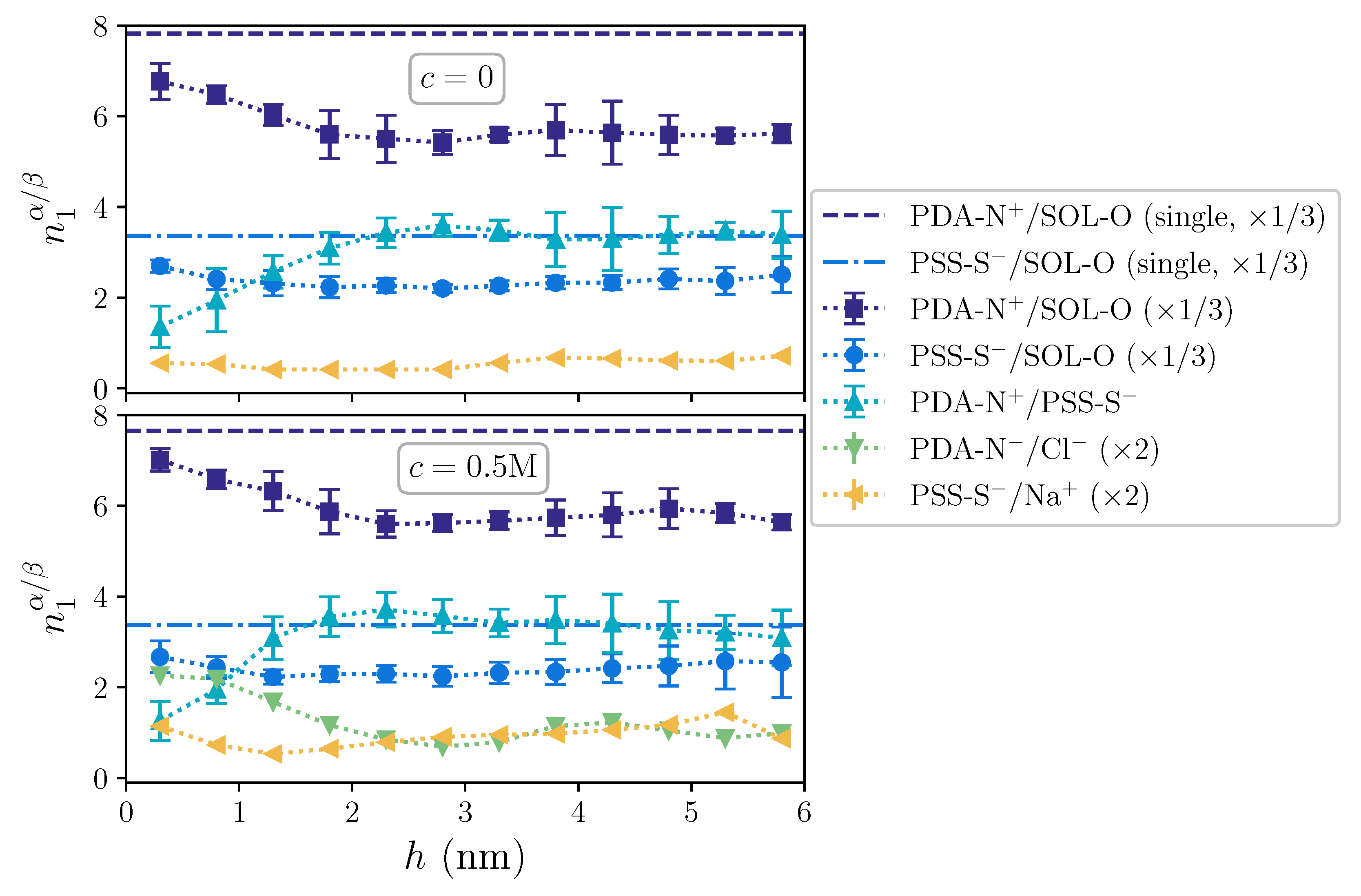
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LbL | layer-by-layer |
| PEM | polyelectrolyte multilayer |
| OEM | oligoelectrolyte multilayer |
| PDADMAC | poly(diallyl dimethyl ammonium chloride) |
| PSS | poly(styrene sulfonate sodium salt) |
| RDF | radial distribution function |
References
- Iler, R. Multilayers of colloidal particles. J. Colloid Interface Sci. 1966, 21, 569–594. [Google Scholar] [CrossRef]
- Decher, G.; Hong, J.; Schmitt, J. Buildup of ultrathin multilayer films by a self-assembly process: III. Consecutively alternating adsorption of anionic and cationic polyelectrolytes on charged surfaces. Thin Solid Films 1992, 210–211, 831–835. [Google Scholar] [CrossRef]
- Decher, G. Fuzzy Nanoassemblies: Toward Layered Polymeric Multicomposites. Science 1997, 277, 1232–1237. [Google Scholar] [CrossRef]
- Ariga, K.; Yamauchi, Y.; Rydzek, G.; Ji, Q.; Yonamine, Y.; Wu, K.C.W.; Hill, J.P. Layer-by-layer Nanoarchitectonics: Invention, Innovation, and Evolution. Chem. Lett. 2014, 43, 36–68. [Google Scholar] [CrossRef]
- Xiao, F.X.; Pagliaro, M.; Xu, Y.J.; Liu, B. Layer-by-layer assembly of versatile nanoarchitectures with diverse dimensionality: A new perspective for rational construction of multilayer assemblies. Chem. Soc. Rev. 2016, 45, 3088–3121. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.R.; Mano, J.F. Polyelectrolyte multilayered assemblies in biomedical technologies. Chem. Soc. Rev. 2014, 43, 3453–3479. [Google Scholar] [CrossRef]
- Gentile, P.; Carmagnola, I.; Nardo, T.; Chiono, V. Layer-by-layer assembly for biomedical applications in the last decade. Nanotechnology 2015, 26, 422001. [Google Scholar] [CrossRef]
- Ariga, K.; Hill, J.P.; Ji, Q. Layer-by-layer assembly as a versatile bottom-up nanofabrication technique for exploratory research and realistic application. Phys. Chem. Chem. Phys. 2007, 9, 2319–2340. [Google Scholar] [CrossRef]
- Schlenoff, J.B. Retrospective on the Future of Polyelectrolyte Multilayers. Langmuir 2009, 25, 14007–14010. [Google Scholar] [CrossRef]
- Decher, G.; Schlenoff, J.B. (Eds.) Multilayer Thin Films: Sequential Assembly of Nanocomposite Materials, 2nd ed.; Wiley-VCH: New York, NY, USA, 2012. [Google Scholar]
- Trau, D.; Renneberg, R. Encapsulation of glucose oxidase microparticles within a nanoscale layer-by-layer film: Immobilization and biosensor applications. Biosens. Bioelectron. 2003, 18, 1491–1499. [Google Scholar] [CrossRef]
- Arsenault, A.C.; Halfyard, J.; Wang, Z.; Kitaev, V.; Ozin, G.A.; Manners, I.; Mihi, A.; Miguez, H. Tailoring photonic crystals with nanometer-scale precision using polyelectrolyte multilayers. Langmuir 2005, 21, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Lu, F.; Chang, Q.; Liu, Y.; Liu, H.; Li, Y.; Xu, W.; Cui, G.; Zhuang, J.; Li, X. Fabrication of Ultrathin Films with Large Third-Order Nonlinear Optical Properties. ChemPhysChem 2005, 6, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Malaisamy, R.; Bruening, M.L. High-flux nanofiltration membranes prepared by adsorption of multilayer polyelectrolyte membranes on polymeric supports. Langmuir 2005, 21, 10587–10592. [Google Scholar] [CrossRef] [PubMed]
- Bruening, M.L.; Dotzauer, D.M.; Jain, P.; Ouyang, L.; Baker, G.L. Creation of functional membranes using polyelectrolyte multilayers and polymer brushes. Langmuir 2008, 24, 7663–7673. [Google Scholar] [CrossRef]
- Datta, S.; Cecil, C.; Bhattacharyya, D. Functionalized Membranes by Layer-By-Layer Assembly of Polyelectrolytes and In Situ Polymerization of Acrylic Acid for Applications in Enzymatic Catalysis. Ind. Eng. Chem. Res. 2008, 47, 4586–4597. [Google Scholar] [CrossRef]
- De Grooth, J.; Oborný, R.; Potreck, J.; Nijmeijer, K.; de Vos, W.M. The role of ionic strength and odd-even effects on the properties of polyelectrolyte multilayer nanofiltration membranes. J. Membr. Sci. 2015, 475, 311–319. [Google Scholar] [CrossRef]
- De Grooth, J.; Haakmeester, B.; Wever, C.; Potreck, J.; de Vos, W.M.; Nijmeijer, K. Long term physical and chemical stability of polyelectrolyte multilayer membranes. J. Membr. Sci. 2015, 489, 153–159. [Google Scholar] [CrossRef]
- Thierry, B.; Winnik, F.M.; Merhi, Y.; Tabrizian, M. Nanocoatings onto arteries via layer-by-layer deposition: Toward the in vivo repair of damaged blood vessels. J. Am. Chem. Soc. 2003, 125, 7494–7495. [Google Scholar] [CrossRef]
- Séon, L.; Lavalle, P.; Schaaf, P.; Boulmedais, F. Polyelectrolyte Multilayers: A Versatile Tool for Preparing Antimicrobial Coatings. Langmuir 2015, 31, 12856–12872. [Google Scholar] [CrossRef]
- Khopade, A.J.; Arulsudar, N.; Khopade, S.A.; Hartmann, J. Ultrathin Antibiotic Walled Microcapsules. Biomacromolecules 2005, 6, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Micciulla, S.; Soltwedel, O.; Lohmann, O.; von Klitzing, R. Temperature responsive behavior of polymer brush/polyelectrolyte multilayer composites. Soft Matter 2016, 12, 1176–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, S.; Helm, C.A. Single Polyelectrolyte Layers Adsorbed at High Salt Conditions: Polyelectrolyte Brush Domains Coexisting with Flatly Adsorbed Chains. Macromolecules 2009, 42, 6733–6740. [Google Scholar] [CrossRef]
- Roiter, Y.; Trotsenko, O.; Tokarev, V.; Minko, S. Single Molecule Experiments Visualizing Adsorbed Polyelectrolyte Molecules in the Full Range of Mono- and Divalent Counterion Concentrations. J. Am. Chem. Soc. 2010, 132, 13660–13662. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, D.; van der Burgh, S.; de Keizer, A.; Stuart, M.A.C. Kinetics of formation and dissolution of weak polyelectrolyte multilayers: Role of salt and free polyions. Langmuir 2002, 18, 5607–5612. [Google Scholar] [CrossRef]
- Cerdà, J.J.; Qiao, B.; Holm, C. Understanding polyelectrolyte multilayers: An open challenge for simulations. Soft Matter 2009, 5, 4412–4425. [Google Scholar] [CrossRef]
- Reddy, G.; Yethiraj, A. Solvent effects in polyelectrolyte adsorption: Computer simulations with explicit and implicit solvent. J. Chem. Phys. 2010, 132, 074903. [Google Scholar] [CrossRef]
- Messina, R. Polyelectrolyte Multilayering on a Charged Planar Surface. Macromolecules 2004, 37, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Messina, R.; Holm, C.; Kremer, K. Polyelectrolyte Adsorption and Multilayering on Charged Colloidal particles. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 3557. [Google Scholar] [CrossRef]
- Patel, P.A.; Jeon, J.; Mather, P.T.; Dobrynin, A.V. Molecular Dynamics Simulations of Layer-by-Layer Assembly of Polyelectrolytes at Charged Surfaces: Effects of Chain Degree of Polymerization and Fraction of Charged Monomers. Langmuir 2005, 21, 6113–6122. [Google Scholar] [CrossRef]
- Patel, P.A.; Jeon, J.; Mather, P.T.; Dobrynin, A.V. Molecular dynamics simulations of multilayer polyelectrolyte films: Effect of electrostatic and short-range interactions. Langmuir 2006, 22, 9994–10002. [Google Scholar] [CrossRef]
- Carrillo, J.M.Y.; Dobrynin, A.V. Molecular Dynamics Simulations of Polyelectrolyte Adsorption. Langmuir 2007, 23, 2472–2482. [Google Scholar] [CrossRef] [PubMed]
- Narambuena, C.; Leiva, E.; Pérez, E. Counterion condensation on polyelectrolyte chains adsorbed on charged surfaces. Colloids Surfaces A Physicochem. Eng. Asp. 2015, 487, 49–57. [Google Scholar] [CrossRef]
- Bucur, C.B.; Sui, Z.; Schlenoff, J.B. Ideal Mixing in Polyelectrolyte Complexes and Multilayers: Entropy Driven Assembly. J. Am. Chem. Soc. 2006, 128, 13690–13691. [Google Scholar] [CrossRef] [PubMed]
- Schlenoff, J.B.; Rmaile, A.H.; Bucur, C.B. Hydration Contributions to Association in Polyelectrolyte Multilayers and Complexes: Visualizing Hydrophobicity. J. Am. Chem. Soc. 2008, 130, 13589–13597. [Google Scholar] [CrossRef]
- Qiao, B.; Cerdà, J.J.; Holm, C. Poly(styrenesulfonate)-Poly(diallyldimethylammonium) Mixtures: Toward the Understanding of Polyelectrolyte Complexes and Multilayers via Atomistic Simulations. Macromolecules 2010, 43, 7828–7838. [Google Scholar] [CrossRef]
- Batys, P.; Zhang, Y.; Lutkenhaus, J.L.; Sammalkorpi, M. Hydration and Temperature Response of Water Mobility in Poly(diallyldimethylammonium)-Poly(sodium 4-styrenesulfonate) Complexes. Macromolecules 2018, 51, 8268–8277. [Google Scholar] [CrossRef] [Green Version]
- Qiao, B.; Cerdà, J.J.; Holm, C. Atomistic Study of Surface Effects on Polyelectrolyte Adsorption: Case Study of a Poly(styrenesulfonate) Monolayer. Macromolecules 2011, 44, 1707–1718. [Google Scholar] [CrossRef]
- Qiao, B.; Sega, M.; Holm, C. An atomistic study of a poly(styrene sulfonate)/poly(diallyldimethylammonium) bilayer: The role of surface properties and charge reversal. Phys. Chem. Chem. Phys. 2011, 13, 16336–16342. [Google Scholar] [CrossRef]
- Qiao, B.F.; Sega, M.; Holm, C. Properties of water in the interfacial region of a polyelectrolyte bilayer adsorbed onto a substrate studied by computer simulations. Phys. Chem. Chem. Phys. 2012, 14, 11425–11432. [Google Scholar] [CrossRef]
- Micciulla, S.; Sánchez, P.A.; Smiatek, J.; Qiao, B.; Sega, M.; Laschewsky, A.; Holm, C.; von Klitzing, R. Layer-by-layer formation of oligoelectrolyte multilayers: A combined experimental and computational study. Soft Mater. 2014, 12, S14–S21. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, P.A.; Vögele, M.; Smiatek, J.; Qiao, B.; Sega, M.; Holm, C. Atomistic simulation of PDADMAC/PSS oligoelectrolyte multilayers: Overall comparison of tri- and tetra-layer system. Soft Matter 2019, 15, 9437–9451. [Google Scholar] [CrossRef] [PubMed]
- Ladam, G.; Schaad, P.; Voegel, J.C.; Schaaf, P.; Decher, G.; Cuisinier, F. In Situ Determination of the Structural Properties of Initially Deposited Polyelectrolyte Multilayers. Langmuir 2000, 16, 1249–1255. [Google Scholar] [CrossRef]
- Porcel, C.; Lavalle, P.; Ball, V.; Decher, G.; Senger, B.; Voegel, J.C.; Schaaf, P. From Exponential to Linear Growth in Polyelectrolyte Multilayers. Langmuir 2006, 22, 4376–4383. [Google Scholar] [CrossRef]
- Schönhoff, M. Self-assembled polyelectrolyte multilayers. Curr. Opin. Colloid Interface Sci. 2003, 8, 86–95. [Google Scholar] [CrossRef]
- Ahrens, H.; Büscher, K.; Eck, D.; Förster, S.; Luap, C.; Papastavrou, G.; Schmitt, J.; Steitz, R.; Helm, C. Poly(styrene sulfonate) self-organization: Electrostatic and secondary interactions. Macromol. Symp. 2004, 211, 93–106. [Google Scholar] [CrossRef]
- Micciulla, S.; Dodoo, S.; Chevigny, C.; Laschewsky, A.; von Klitzing, R. Short versus long chain polyelectrolyte multilayers: A direct comparison of self-assembly and structural properties. Phys. Chem. Chem. Phys. 2014, 16, 21988–21998. [Google Scholar] [CrossRef] [Green Version]
- Record, M.T.; Anderson, C.F. Interpretation of preferential interaction coefficients of nonelectrolytes and of electrolyte ions in terms of a two-domain model. Biophys. J. 1995, 68, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Courtenay, E.; Capp, M.; Saecker, R.; Record, M., Jr. Thermodynamic analysis of interactions between denaturants and protein surface exposed on unfolding: Interpretation of urea and guanidinium chloride m-values and their correlation with changes in accessible surface area (ASA) using preferential interaction coefficients and the local-bulk domain model. Proteins 2000, 41, 72–85. [Google Scholar] [CrossRef]
- Smiatek, J. Aqueous ionic liquids and their effects on protein structures: An overview on recent theoretical and experimental results. J. Physics Condens. Matter 2017, 29, 233001. [Google Scholar] [CrossRef]
- Oprzeska-Zingrebe, E.A.; Smiatek, J. Preferential binding of urea to single-stranded DNA structures: A molecular dynamics study. Biophys. J. 2018, 114, 1551–1562. [Google Scholar] [CrossRef]
- Oprzeska-Zingrebe, E.A.; Smiatek, J. Aqueous ionic liquids in comparison with standard co-solutes. Biophys. Rev. 2018, 10, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.A.; Argyris, D.; Papavassiliou, D.V.; Striolo, A. Interfacial Water on Crystalline Silica: A Comparative Molecular Dynamics Simulation Study. Mol. Simul. 2011, 37, 172–195. [Google Scholar] [CrossRef]
- Behrens, S.H.; Grier, D.G. The charge of glass and silica surfaces. J. Chem. Phys. 2001, 115, 6716–6721. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Roberts, J.E.; Santore, M.M. The relationship between polymer/substrate charge density and charge overcompensation by adsorbed polyelectrolyte layers. J. Colloid Interface Sci. 2002, 247, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Skarba, M.; Galletto, P.; Cakara, D.; Borkovec, M. Effects of heat treatment on the aggregation and charging of Stöber-type silica. J. Colloid Interface Sci. 2005, 292, 139–147. [Google Scholar] [CrossRef]
- Smiatek, J.; Sega, M.; Holm, C.; Schiller, U.D.; Schmid, F. Mesoscopic simulations of the counterion-induced electro-osmotic flow: A comparative study. J. Chem. Phys. 2009, 130, 244702. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Hess, B.; Holm, C.; van der Vegt, N. Modeling multi-body effects in ionic solutions with a concentration dependent dielectric permittivity. Phys. Rev. Lett. 2006, 96, 147801. [Google Scholar] [CrossRef]
- Hess, B.; Holm, C.; van der Vegt, N. Osmotic Coeffcients of atomistic NaCl (aq) force-fields. J. Chem. Phys. 2006, 124, 164509. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L. A smooth Particle Mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez, P.A.; Vögele, M.; Smiatek, J.; Qiao, B.; Sega, M.; Holm, C. PDADMAC/PSS Oligoelectrolyte Multilayers: Internal Structure and Hydration Properties at Early Growth Stages from Atomistic Simulations. Molecules 2020, 25, 1848. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25081848
Sánchez PA, Vögele M, Smiatek J, Qiao B, Sega M, Holm C. PDADMAC/PSS Oligoelectrolyte Multilayers: Internal Structure and Hydration Properties at Early Growth Stages from Atomistic Simulations. Molecules. 2020; 25(8):1848. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25081848
Chicago/Turabian StyleSánchez, Pedro A., Martin Vögele, Jens Smiatek, Baofu Qiao, Marcello Sega, and Christian Holm. 2020. "PDADMAC/PSS Oligoelectrolyte Multilayers: Internal Structure and Hydration Properties at Early Growth Stages from Atomistic Simulations" Molecules 25, no. 8: 1848. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25081848