
Spirostanol Sapogenins and Saponins from Convallaria majalis L. Structural Characterization by 2D NMR, Theoretical GIAO DFT Calculations and Molecular Modeling

 and
and
Abstract
:1. Introduction
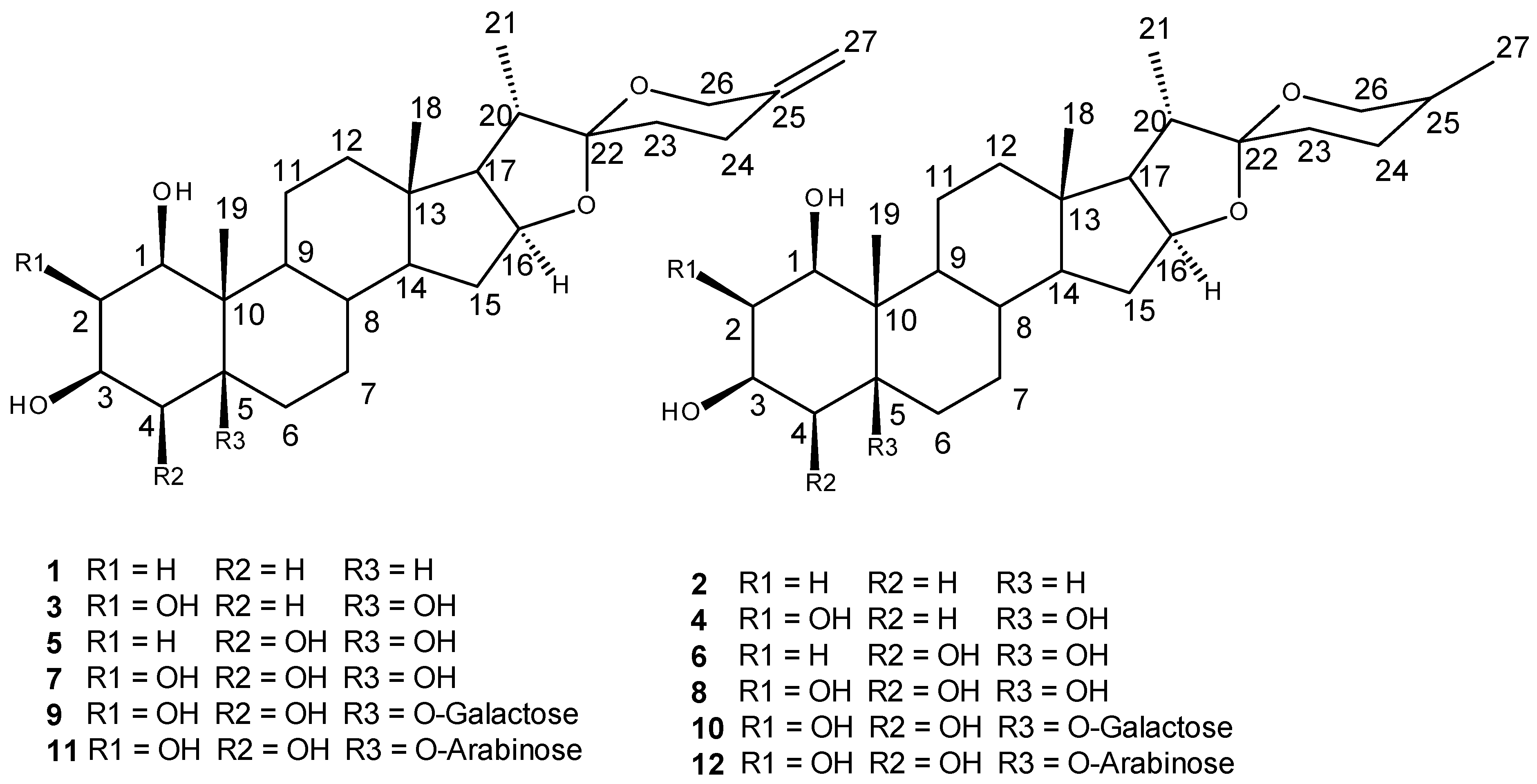
2. Results
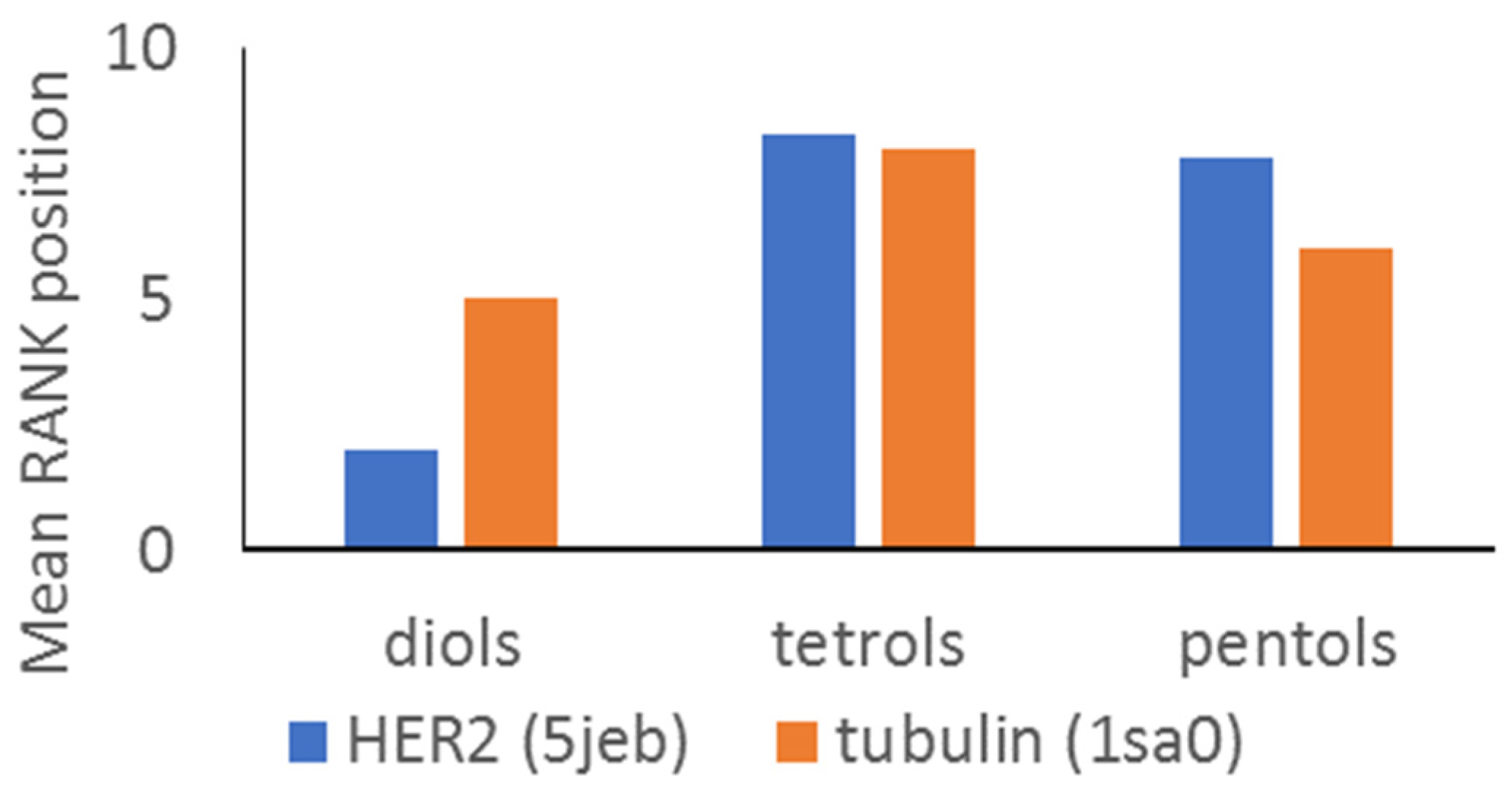
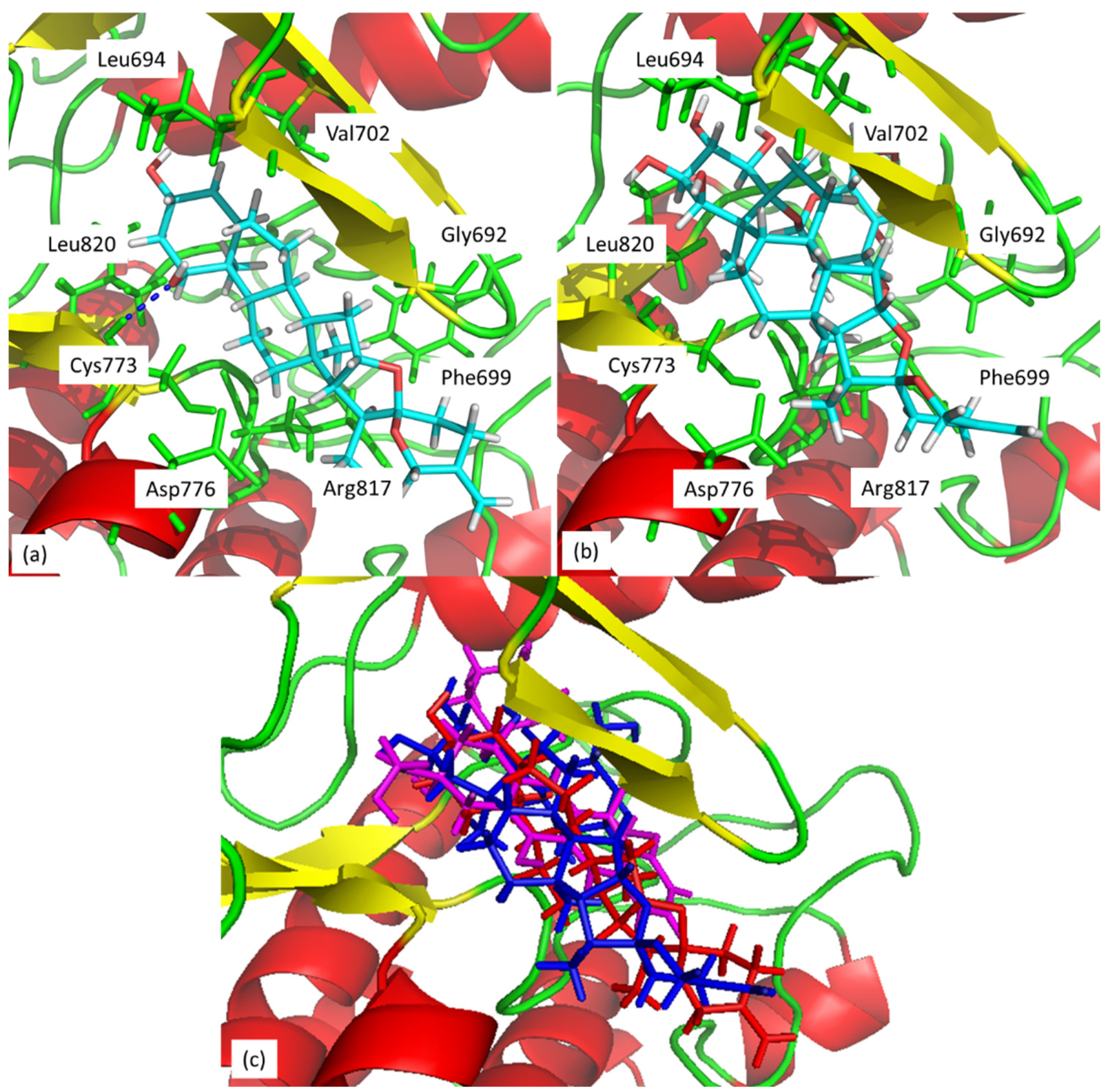
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Plant Material
4.3. Acid Hydrolysis of the Butanolic Fraction
4.4. Isolation of the Sapogenins
4.5. Isolation of Saponins
4.6. Catalytic Hydrogenation
4.7. Theoretical Calculations
4.8. Molecular Docking
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xiang, L.; Yi, X.; Wang, Y.; He, X.J. Antiproliferative and anti-inflammatory polyhydroxylated spirostanol saponins from Tupistra chinensis. Sci. Rep. 2016, 6, 31633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparg, S.; Light, M.; Van Staden, J. Biological activities and distribution of plant saponins. J. Ethnopharmacol. 2004, 94, 219–243. [Google Scholar] [CrossRef]
- Munafo, J.P., Jr.; Gianfagna, T.J. Chemistry and biological activity of steroidal glycosides from the Lilium genus. Nat. Prod. Rep. 2015, 32, 454–477. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Tohma, M.; Yoshizawa, I.J.C. Constituents of convallaria. IV. Isolation of convallasaponin-A,-B, and-C. Chem. Pharm. Bull. 1966, 14, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M.; Tohma, M.; Yoshizawa, I.J.C. Constituents of Convallaria. VIII. Structure of Convallagenin-B. Chem. Pharm. Bull. 1967, 15, 1713–1719. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M.; Tohma, M.; Yoshizawa, I.J.C. Constituents of convallaria. VII. Structure of convallagenin-A. Chem. Pharm. Bull. 1967, 15, 1204–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M.; Tohma, M.; Yoshizawa, I.J.C. Constituents of Convallaria. V. On the structure of Convallasaponin-C. Chem. Pharm. Bull. 1966, 14, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M.; Tohma, M.; Yoshizawa, I.; Akiyama, H.J.C. Constituents of Convallaria. X. Structures of convallasaponin-A,-B, and their glucosides. Chem. Pharm. Bull. 1968, 16, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M.; Tohma, M.; Yoshizawa, I.J.C. Constituents of Convallaria. XI. On the structure of convallasaponin-D. Chem. Pharm. Bull. 1968, 16, 1228–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschesche, R.; Wulff, G.J.T. Über saponine der spirostanolreihe—IX: Die konstitution des digitonins. Tetrahedron 1963, 19, 621–634. [Google Scholar] [CrossRef]
- Tschesche, R.; Hermann, K.H.; Langlais, R.; Tjoa, B.T.; Wulff, G. Steroidsaponine mit mehr als einer Zuckerkette, VII. Convallamarosid, ein trisdesmosidisches 22-Hydroxyfurostanol-Glycosid aus den Wurzeln von Convallaria majalis L. Chem. Ber. 1973, 106, 3010–3019. [Google Scholar] [CrossRef]
- Nartowska, J.; Strzelecka, H.J. Steroid saponins from the root and rhizome of Convallaria-Maialis L. 1. Isolation of saponosides. Acta Pol. Pharm. 1983, 40, 649–656. [Google Scholar]
- Tori, K.; Seo, S.; Terui, Y.; Nishikawa, J.; Yasuda, F.J. Carbon-13 NMR spectra of 5β-steroidal sapogenins. Reassignment of the F-ring carbon signals of (25S)-spirostans. Tetrahedron Lett. 1981, 22, 2405–2408. [Google Scholar] [CrossRef]
- Hirai, Y.; Konishi, T.; Sanada, S.; Ida, Y.; Shoji, J.J.C. Studies on the constituents of Aspidistra elatior BLUME. I. On the steroids of the underground part. Chem. Pharm. Bull. 1982, 30, 3476–3484. [Google Scholar] [CrossRef] [Green Version]
- Konishi, T.; Kiyosawa, S.; Shoji, J.J.C. Studies on the constituents of Aspidistra elatior Blume. II. On the steroidal glycosides of the leaves (1). Chem. Pharm. Bull. 1984, 32, 1451–1460. [Google Scholar] [CrossRef] [Green Version]
- Renzhou, Y.; Dezu, W.; Jian, F. Carbon-13 NMR spectra of ten steroidal sapogenins. Plant Divers. 1987, 3, 1–3. [Google Scholar]
- Higano, T.; Kuroda, M.; Sakagami, H.; Mimaki, Y.J.C. Convallasaponin A, a new 5β-spirostanol triglycoside from the rhizomes of Convallaria majalis. Chem. Pharm. Bull. 2007, 55, 337–339. [Google Scholar] [CrossRef] [Green Version]
- Nartowska, J.; Sommer, E.; Pastewka, K.; Sommer, S.; Skopińska-Rózewska, E. Anti-Angiogenic Activity Of Convallamaroside. Acta Pol. Pharm. 2004, 61, 279–282. [Google Scholar]
- Nartowska, J.; Sommer, E.; Siwicki, A.; Bany, J.; Skurzak, H.; Augustynowicz, J.; Skopińska-Różewska, E. The anti-angiogenic and immunotropic properties of convallamaroside, a steroidal saponin isolated from rhizomes and roots of Convallaria majalis L. Pol. J. Environ. Stud. 2005, 14, 296–299. [Google Scholar]
- Olayioye, M.A. Intracellular signaling pathways of ErbB2/HER-2 and family members. Breast Cancer Res. 2001, 3, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Rampogu, S.; Son, M.; Baek, A.; Park, C.; Rana, R.M.; Zeb, A.; Parameswaran, S.; Lee, K.W. Targeting natural compounds against HER2 kinase domain as potential anticancer drugs applying pharmacophore based molecular modelling approaches. Comput. Biol. Chem. 2018, 74, 327–338. [Google Scholar] [CrossRef]
- Zhou, J.; Giannakakou, P. Targeting microtubules for cancer chemotherapy. Curr. Med. Chem. Anti-Cancer Agents 2005, 5, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabrowska-Balcerzak, K.; Wawer, I.; Nartowska, J. Identification of Polyhydroxylated Spirostanes and their Glycosides from C. Majalis Using NMR Techniques. In Proceedings of the XXXVIII Polish Seminar on Nuclear Magnetic Resonance and Its Applications, Kralow, Poland, 1–2 December 2005. [Google Scholar]
- Matsumoto, N.J.C. Systematic Analysis of Steroids. II. Analysis of Steroid Sapogenins by Thin Layer Chromatography. Chem. Pharm. Bull. 1963, 11, 1189–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschesche, R.; Schwarz, H.; Snatzke, G. Über Saponine der Spirostanolreihe, VI. Die Konstitution des Convallamarogenins. Chem. Ber. 1961, 94, 1699–1705. [Google Scholar] [CrossRef]
- Blunt, J.; Stothers, J.B. 13C NMR spectra of steroids—A survey and commentary. Org. Matter Reson. 1977, 9, 439–464. [Google Scholar] [CrossRef]
- Kirk, D.N.; Harold, C.T.; Douglas, C.; White, K.A.; Smith, K.E.; Latif, S.; Robert, W.P. A survey of the high-field 1 H NMR spectra of the steroid hormones, their hydroxylated derivatives, and related compounds. Perkin Trans. 2 1990, 2, 1567–1594. [Google Scholar] [CrossRef]
- Eggert, H.; Djerassi, C. 13C NMR spectra of sapogenins. Tetrahedron Lett. 1975, 16, 3635–3638. [Google Scholar] [CrossRef]
- Roslund, M.U.; Klika, K.D.; Lehtilä, R.L.; Tähtinen, P.; Sillanpää, R.; Leino, R. Conformation of the galactose ring adopted in solution and in crystalline form as determined by experimental and DFT 1H NMR and single-crystal X-ray analysis. J. Org. Chem. 2004, 69, 18–25. [Google Scholar] [CrossRef]
- Moffitt, W.; Woodward, R.; Moscowitz, A.; Klyne, W.; Djerassi, C. Structure and the optical rotatory dispersion of saturated ketones. J. Am. Chem. Soc. 1961, 83, 4013–4018. [Google Scholar] [CrossRef]
- Takeda, K.I.; Kubota, T.; Shimaoka, A.J.T. Studies on the steroidal components of domestic plants—XIX: The structure of kogagenin, a sapogenin from Dioscorea tokoro, Makino. Tetrahedron 1959, 7, 62–69. [Google Scholar] [CrossRef]
- Matsuo, Y.; Shinoda, D.; Nakamaru, A.; Kamohara, K.; Sakagami, H.; Mimaki, Y.J. Steroidal glycosides from Convallaria majalis whole plants and their cytotoxic activity. Int. J. Mol. Sci. 2017, 18, 2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd El-kader, A.M.; Mahmoud, B.K.; Hajjar, D.; Mohamed, M.F.; Hayallah, A.M.; Abdelmohsen, U. Antiproliferative activity of new pentacyclic triterpene and a saponin from Gladiolus segetum Ker-Gawl corms supported by molecular docking study. RSC Adv. 2020, 10, 22730–22741. [Google Scholar] [CrossRef]
- Frisch, A.J.W. Gaussian 09w Reference; Gaussian Inc.: Wallingford, CT, USA, 2009; 25p. [Google Scholar]
- Siudem, P.; Bukowicki, J.; Wawer, I.; Paradowska, K. Structural studies of two capsaicinoids: Dihydrocapsaicin and nonivamide. 13 C and 15 N MAS NMR supported by genetic algorithm and GIAO DFT calculations. RSC Adv. 2020, 10, 18082–18092. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| H | 1 | 2 | 3 | 4 | 5 | 6 | 9/10 a | 11/12 b |
|---|---|---|---|---|---|---|---|---|
| 1 | 3.83 br s | 3.83 br s | 3.86, br s | 3.86, br s | 3.85 | 3.85 | 4.25, br s | 4.25, br s |
| 2 | 1.96; 1.74 | 1.96; 1.74 | 3.65, dd (3.3, 3.4), | 3.65, dd (3.3,3.4), | 2.16; 1.75 | 2.16; 1.75 | 4.10, br,s | 4.07 |
| 3 | 4.18 br s | 4.18 br s | 4.16, br s, | 4.16, br s | 4.14 | 4.14 | 4.68, br | 4.69, br s |
| 4 | 2.05, 1.48 | 2.05, 1.48 | 2.39 dd (15.6, 3.7); 1.85, d (15.6) | 2.39 dd (15.6, 3.7); 1.85, d (15.6) | 3.91 | 3.91 | 4.35 | 4.37 |
| 5 | 1.66 | 1.66 | - | - | - | - | - | |
| 6 | 1.46; 1.30 | 1.46; 1.30 | 1.74; 1.44 | 1.74; 1.44 | 1.95; 1.44 | 1.95; 1.44 | 2.72, d, (9.8); 1.85 | 2.75 d (10.1) 1.95 |
| 7 | 1.95, 1.12 | 1.95, 1.12 | 1.41, 1.91 | 1.41, 1.91 | 1.58, 1.04 | 1.58, 1.04 | 1.33, 1.15 | 1.35, 1.13 |
| 8 | 2.05 | 2.05 | 1.71 | 1.71 | 1.70 | 1.70 | 1.72 | 1.70 |
| 9 | 1.23 | 1.23 | 1.08 | 1.08 | 1.08 | 1.08 | 1.26, m | 1.21 |
| 11 | 1.30; 1.22 | 1.30, 1.22 | 1.45 | 1.45 | 1.45 | |||
| 12 | 1.72; 1.10 | 1.72; 1.10 | 1.75; 1.15 | 1.75; 1.15 | 1.72; 1.11 | 1.72; 1.11 | 1.59; 1.12 | 1.60; 1.05 |
| 14 | 1.14 | 1.14 | 1.13 | 1.13 | 1.11 | 1.11 | 1.12 | 1.00 |
| 15 | 2.05; 2.00 | 2.05 m | 1.99 m (16.5); 1.30 | 1.99 m (16.5); 1.30 | 1.99; 1.29 | 1.99; 1.29 | 2.02; 1.37 | 1.99, 1.40 |
| 16 | 4.44 dt (16.1, 7.5) | 4.43 | 4.45 | 4.44 | 4.45 | 4.46 | 4.58, m | 4.57m |
| 17 | 1.77 | 1.77 | 1.76 | 1.76 | 1.77 | 1.77 | 1.80 | 1.79 |
| 18 | 3H, s, 0.77 | 3H, s, 0.78 | 3H, s, 0.81 | 3H, s, 0.79 | 0.79 | 0.79 | 3H, s, 0.83 | 3H, s, 0.82 |
| 19 | 3H, s, 1.13 | 3H, s, 1.12 | 3H, s, 1.26 | 3H, s, 1.25 | 1.32 | 1.32 | 3H, s, 1.66 | 3H, s, 1.72 |
| 20 | 1.89m | 1.82m | 1.92 m | 1.84 m | 1.91 m | 1.85 m | 1.96 | 1.90, m |
| 21 | 3H, d (7.0) 0.95 | 3H, d (7.0) 0.95 | 3H, d (7.0), 0.96 | 3H, d (6.9) 0.99 | 3H, d 0.97 | 3H, d 0.98 | 3H, 1.05 | 3H, d (6.7) 1.07 |
| 23 | 1.27 | - | 1.74 | 1.02 d (3.0) 1.63 dt (15.8, 3.0) | 1.75; 1.28 | 1.09 | 1.80, m | 1.80 |
| 24 | 2.56 td (13.5, 5.5) 2.24 d (13.5, 2.2) | - | 2.56 td (13.1, 5.5) 2.27 dt (13.5, 2.2) | 2.27 (12.6, 2.2) | 2.54 | 1.94; 1.49 | 2.24 d (12.7) | 2.24 d; 2.69 |
| 25 | - | ca. 1.7 | - | 1.73 m | - | 1.74 | - | - |
| 26 | 4.30 d (12.2) 3.87 d (12.2) | 3.94 dd (11.0, 2.6) 3.30 d (11.0) | 4.29 d (12.0) 3.85 d (12.0) | 3.31 d (11.0) 3.94 dd (11.0, 2.7) | 4.25; 3.81 | 3.91; 3.26 | 4.47 d 3.36 4.2510 | 4.49 d; 4.04 4.0812 |
| 27 | 4.77, 4.74 | 3H, d (7.0) 1.07 | 4.79 d (0,9); 4.76 | 3H, d (7.0) 1.09 | 4.77 | 3H, d (7.0) 1.05 | 4.82, 4.79 4.73, 4.7610 | 4.81, 4.78 1.0912 |
| 1 | 3 | ||
|---|---|---|---|
| Long-range correlations in the HMBC spectra | |||
| H-18 (Me) H-19 (Me) H-21 (Me) H-27 H-1 C-3 C-14 C-16 C-17 C-22 C-25 | C-12, C-13, C-14, C-17 C-1, C-5, C-9, C-10 C-17, C-20, C-22 C-24, C-25, C-26 C-2, C-3 H-4ax H-15eq, ax, H-9, H-12 H-15eq, H-17, H-23 H-15eq, H-20 H-26ax, eq H-26ax, eq, H-23 | H-18 (Me) H-19 (Me) H-21 (Me) H-27 H-1 H-2 H-4eq H-4ax H-15eq H-17 H-26 | C-12, C-13, C-14, C-17, C-20 C-1, C-5, C-9, C-10 C-17, C-20, C-22 C-24, C-25, C-26 C-2, C-3, C-5, C-10, C-19 C-1, C-3 C-2, C-3, C-5, C-10 C-5, C-6 C-13, C-14, C-17 C-18 (Me), C-21 (Me) C-22, C25 (δ142.9), C-27 |
| Correlations assigned from COSY spectra | |||
| H-18 (Me) H-21 (Me) H-1 H-3 H-16 H-24eq H-24ax H-27 | H-14 H-20 H-2eq, ax H-2eq, ax; H-4 eq,ax H-15 eq,ax H-23, H-24ax, H-27 H-23, H-24eq, H-26ax H-26ax, eq, H-24eq | H-21 (Me) H-27 H-1 H-2 H-3 H-15eq H-16 H-27 | H-20, H-23 H26a, b H-2, H-3 H-1, H-3 H-1, H-2, H-4ax, eq H-15ax, H-14 H-15eq, ax, H-17 H-26eq |
| The “through-space” correlations in the NOESY spectra * | |||
| H-18 (Me) H-19 (Me) H-1 H-9 | H-8, H-11 H-8, H-11 H-11, H-19 (Me) H-2, H-4 | H-18 (Me) H-19(Me) H-21 (Me) H-1 H-3 H-9 | H-8, H-11, H15ax, H-17, H-20 H-1, H-8, H-11ax H-17, H-20, H-23 H-1, H-3, H-11, H-19 H-2, H-4ax, eq H-2, H-4ax (δ2.05) |
| 1 | 2 | 1 and 2 | 3 | 4 | 3 and 4 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| C Atom No. | δC sol. [ppm] | Calculated δC [ppm] | δC sol. [ppm] | Calculated δC [ppm] | δ C s. s. [ppm] | δC sol. [ppm] | Calculated δC [ppm] | δC sol. [ppm] | Calculated δC [ppm] | δ C s. s. [ppm] |
| 1 | 73.8 | 69.7 | 73.8 | 69.7 | 73.5 | 76.7 | 76.9 | 76.7 | 76.9 | 79.1 |
| 2 | 32.1 | 32.4 | 32.1 | 32.4 | 32.2 | 66.8 | 67.1 | 66.8 | 67.1 | 66.8 |
| 3 | 68.4 | 65.5 | 68.4 | 65.5 | 68.3 | 70.3 | 71.7 | 70.3 | 71.7 | 71.3 |
| 4 | 33.7 | 31.8 | 33.7 | 31.8 | 33.8 | 37.5 | 40.7 | 37.5 | 40.7 | 36.6 |
| 5 | 30.4 | 28.0 | 30.4 | 28.0 | 30.5 | 74.7 | 75.3 | 74.7 | 75.3 | 76.7 |
| 6 | 26.1 | 28.0 | 25.9 | 28.0 | 25.9–26.0 | 34.3 | 36.8 | 34.3 | 36.8 | 35.6 |
| 7 | 26.0 | 26.4 | 25.8 | 26.4 | 28.0 | 31.3 | 28.0 | 31.3 | 28.7 | |
| 8 | 35.5 | 37.3 | 35.5 | 37.3 | 36.4 | 34.0 | 36.5 | 34.0 | 36.5 | 34.0 |
| 9 | 41.9 | 43.7 | 41.9 | 43.7 | 42.2 | 45.0 | 46.8 | 45.0 | 46.8 | 46.0 |
| 10 | 39.7 | 38.5 | 39.7 | 38.5 | 40.0 | 44.5 | 43.9 | 44.5 | 43.9 | 46.7 |
| 11 | 20.7 | 24.9 | 20.7 | 24.9 | 21.3 | 20.8 | 24.9 | 20.8 | 24.9 | 23.6 |
| 12 | 40.1 | 37.6 | 40.0 | 37.6 | 40.8 | 39.3 | 36.9 | 39.3 | 36.9 | 41.6 |
| 13 | 40.4 | 41.0 | 40.3 | 41.0 | 40.8 | 40.0 | 41.3 | 40.0 | 41.3 | 42.4 |
| 14 | 56.3 | 55.8 | 56.3 | 55.8 | 56.5 | 55.6 | 55.5 | 55.6 | 55.5 | 56.9 |
| 15 | 31.7 | 35.0 | 31.7 | 35.0 | 32.2 | 31.2 | 35.2 | 31.1 | 34.5 | 31.4 |
| 16 | 81.0 | 83.3 | 81.1 | 83.3 | 80.9 | 80.7 | 83.0 | 80.5 | 85.2 | 82.2 |
| 17 | 62.4 | 63.1 | 62.6 | 63.1 | 63.6 | 61.8 | 63.2 | 61.7 | 53.8 | 58.9 |
| 18 | 16.5 | 17.4 | 16.5 | 17.8 | 17.2 | 16.3 | 17.3 | 15.8 | 17.6 | 19.2 |
| 19 | 18.9 | 12.8 | 18.9 | 15.1 | 19.5 | 12.3 | 12.9 | 12.3 | 12.9 | 15.7 |
| 20 | 41.5 | 45.1 | 41.5 | 45.6 | 41.9 | 41.2 | 45.0 | 41.7 | 47.2 | 41.2 |
| 21 | 14.5 | 16.3 | 14.7 | 16.3 | 14.5 | 13.7 | 16.2 | 13.5 | 15.7 | 16.5 |
| 22 | 109.4 | 108.9 | 109.4 | 109.3 | 109.6 | 109.2 | 109.2 | 109.7 | 107.3 | 110.6 |
| 23 | 32.8 | 35.3 | 25.9 | 30.0 | 32.0–34.0(1) | 32.3 | 35.1 | 25.4 | 29.5 | 32.8(3) |
| 24 | 28.5 | 27.7 | 25.7 | 24.7 | 25–29 | 27.9 | 27.9 | 25.1 | 25.0 | 27.6(3) |
| 25 | 143.6 | 134.4 | 27.1 | 26.3 | 141.0(1)/26.1(2) | 142.9 | 134.2 | 26.6 | 26.2 | 145.9(3) |
| 26 | 64.9 | 63.0 | 64.7 | 63.2 | 65.2 | 64.5 | 63.1 | 64.8 | 63.0 | 66.6 |
| 27 | 108.6 | 103.2 | 16.0 | 15.9 | 108.9(1) | 108.2 | 103.3 | 15.3 | 15.6 | 109.5(3) |
| R2 | 0.996 | 0.995 | 0.999 *(1) | 0.998 | 0.994 | 0.999 *(3) | ||||
| MAE | 2.43 | 1.91 | 0.49 * | 2.13 | 2.20 | 1.58 * | ||||
| 5 | 6 | 5 and 6 | 7 | 8 | |||||
|---|---|---|---|---|---|---|---|---|---|
| C Atom No. | δC sol. [ppm] | Calculated δC [ppm] | δC sol. [ppm] | Calculated δC [ppm] | δ C s. s. [ppm] | δC sol. [ppm] | Calculated δC [ppm] | δC sol. [ppm] | Calculated δC [ppm] |
| 1 | 73.9 | 70.6 | 73.9 | 70.6 | 75.6 | 78.8 | 77.6 | 78.8 | 77.6 |
| 2 | 32.3 | 32.2 | 32.3 | 32.2 | 30.0–34.0 | 67.6 | 67.0 | 67.6 | 67.0 |
| 3 | 70.7 | 70.1 | 70.7 | 70.1 | 73.8 | 75.7 | 76.3 | 75.7 | 76.3 |
| 4 | 67.7 | 67.5 | 67.7 | 67.5 | 69.8 | 68.5 | 69.6 | 68.5 | 69.6 |
| 5 | 78.5 | 78.6 | 78.5 | 78.6 | 79.4 | 78.7 | 77.4 | 78.7 | 77.4 |
| 6 | 29.9 | 31.4 | 29.9 | 31.4 | 30.7 28.1 | 30.4 | 32.1 | 30.4 | 32.1 |
| 7 | 28.2 | 30.1 | 28.2 | 30.1 | 28.9 | 29.5 | 28.9 | 29.5 | |
| 8 | 35.0 | 36.7 | 35.0 | 36.7 | 34.0 | 35.8 | 36.2 | 35.8 | 36.2 |
| 9 | 45.9 | 46.6 | 45.9 | 46.6 | 46.8 | 46.1 | 46.2 | 46.1 | 46.2 |
| 10 | 45.1 | 43.9 | 45.1 | 43.9 | 46.8 | 45.6 | 43.3 | 45.6 | 43.3 |
| 11 | 21.3 | 24.8 | 21.3 | 24.8 | 23.5 | 22.3 | 24.8 | 22.3 | 24.8 |
| 12 | 40.1 | 37.1 | 40.1 | 37.1 | 42.7 | 40.5 | 37.1 | 40.5 | 37.1 |
| 13 | 41.0 | 41.2 | 41.0 | 41.2 | 42.7 | 41.5 | 41.2 | 41.5 | 41.2 |
| 14 | 56.4 | 55.6 | 56.4 | 55.6 | 57.1 | 56.9 | 55.6 | 56.9 | 55.6 |
| 15 | 31.9 | 35.2 | 31.9 | 35.2 | 30.0–34.0 | 32.6 | 35.2 | 32.6 | 35.2 |
| 16 | 81.5 | 83.2 | 81.3 | 83.2 | 82.2 | 82.2 | 83.2 | 82.2 | 83.2 |
| 17 | 62.6 | 63.5 | 62.3 | 63.0 | 64.0 | 63.6 | 63.5 | 63.5 | 63.0 |
| 18 | 16.5 | 17.4 | 16.5 | 17.6 | 19.4 | 17.0 | 17.4 | 17.0 | 17.6 |
| 19 | 13.1 | 12.8 | 13.1 | 12.7 | 16.1 | 13.2 | 12.7 | 13.2 | 12.7 |
| 20 | 41.9 | 45.1 | 42.4 | 45.6 | 42.7 | 42.8 | 45.1 | 43.2 | 45.6 |
| 21 | 14.5 | 16.3 | 14.3 | 16.3 | 16.1 | 14.6 | 16.3 | 14.5 | 16.3 |
| 22 | 109.9 | 108.9 | 110.4 | 109.3 | 110.3 | 110.5 | 108.9 | 111 | 109.3 |
| 23 | 33.1 | 35.3 | 26.1 | 30.2 | 25.0–34.0 | 33.5 | 35.3 | 26.8 | 30.2 |
| 24 | 28.6 | 27.7 | 25.8 | 24.6 | 29.0 | 27.7 | 26.6 | 24.6 | |
| 25 | 143.6 | 134.4 | 27.4 | 26.4 | 145.1(5) | 144.2 | 134.4 | 28.2 | 26.4 |
| 26 | 65.2 | 63.0 | 65.2 | 63.2 | 66.3 | 65.6 | 63.0 | 66.0 | 63.2 |
| 27 | 109.0 | 103.2 | 16.0 | 15.9 | 109.7(5)/18.0(6) | 108.9 | 103.2 | 16.5 | 15.9 |
| R2 | 0.997 | 0.997 | 0.999 *(5) | 0.998 | 0.997 | ||||
| MAE | 1.93 | 1.51 | 1.35 * | 1.81 | 1.46 | ||||
| 9 | 10 | 9 and 10 | 11 | 12 | |||
|---|---|---|---|---|---|---|---|
| C Atom No. | δC sol. [ppm] | Calculated δC [ppm] | δC sol. [ppm] | Calculated δC [ppm] | δ C s. s. [ppm] | δC sol. [ppm] | δC sol. [ppm] |
| 1 | 78.1 | 77.5 | 78.1 | 77.5 | 79.8 | 77.2 | 77.2 |
| 2 | 67.3 | 67.2 | 67.3 | 67.2 | 67.0 | 67.1 | 67.1 |
| 3 | 75.9 | 76.9 | 75.9 | 76.9 | 75.8 | 75.6 | 75.6 |
| 4 | 68.2 | 69.2 | 68.2 | 69.2 | 68.7 | 67.6 | 67.6 |
| 5 | 87.9 | 83.7 | 87.9 | 83.7 | 93.6 | 86.9 | 86.9 |
| 6 | 25.0 | 30.2 | 25.0 | 30.2 | 27.0 28.7 | 24.4 | 24.4 |
| 7 | 28.8 | 30.4 | 28.8 | 30.4 | 28.1 | 28.1 | |
| 8 | 35.3 | 36.6 | 35.3 | 36.6 | 34.7 | 34.2 | 34.2 |
| 9 | 47.0 | 47.1 | 47.0 | 47.1 | 46.7 | 46.2 | 46.2 |
| 10 | 46.9 | 44.4 | 46.9 | 44.4 | 46.7 | 45.8 | 45.8 |
| 11 | 22.1 | 25.2 | 22.1 | 25.2 | 23.2 | 21.2 | 21.2 |
| 12 | 40.5 | 37.1 | 40.5 | 37.1 | 40.2 | 39.5 | 39.5 |
| 13 | 41.1 | 41.2 | 41.1 | 41.2 | 42.7 | 41.0 | 41.0 |
| 14 | 56.5 | 55.5 | 56.5 | 55.5 | 57.4 | 55.6 | 55.6 |
| 15 | 32.4 | 35.1 | 32.4 | 35.1 | 32.1 | 31.7 | 31.7 |
| 16 | 81.9 | 83.0 | 81.9 | 83.0 | 83.1 | 81 | 80.8 |
| 17 | 63.0 | 63.1 | 63.0 | 63.0 | 66.4 | 62.6 | 62.4 |
| 18 | 16.9 | 17.5 | 16.9 | 17.6 | 17.1 | 16.1 | 16.1 |
| 19 | 13.5 | 13.1 | 13.5 | 12.7 | 14.0 | 13.5 | 13.5 |
| 20 | 42.5 | 45.1 | 43.0 | 45.6 | 42.8–44.8 | 42.1 | 41.5 |
| 21 | 14.6 | 16.2 | 14.5 | 16.3 | 16.0 | 14.6 | 14.4 |
| 22 | 110.5 | 109.3 | 110.9 | 109.3 | 110.2 | 109.0 | 109.3 |
| 23 | 33.5 | 34.9 | 26.6 | 30.2 | 32.9(9) | 32.8 | 26.0 |
| 24 | 29.0 | 28.0 | 26.4 | 24.6 | 29.5(9) | 28.6 | 25.8 |
| 25 | 144.8 | 134.1 | 28.0 | 26.4 | 146.2(9) | 144.0 | 27.1 |
| 26 | 65.5 | 63.0 | 65.9 | 63.2 | 66.4 | 64.7 | 64.6 |
| 27 | 109.1 | 103.2 | 16.4 | 15.9 | 108.8(9) | 108.4 | 15.9 |
| 1′ | 97.3 | 97.8 | 97.3 | 97.8 | 96.1 | 97.2 | 97.2 |
| 2′ | 73.0 | 76.0 | 73.0 | 76.0 | 73.7 | 72.8 | 72.8 |
| 3′ | 74.7 | 75.1 | 74.7 | 75.1 | 74.2 | 73.8 | 73.8 |
| 4′ | 69.8 | 68.0 | 69.8 | 68.0 | 70.7 | 68.1 | 68.1 |
| 5′ | 76.5 | 75.2 | 76.5 | 75.2 | 78.9 | 65.8 | 65.8 |
| 6′ | 61.8 | 59.3 | 61.8 | 59.3 | 62.5 | - | - |
| R2 | 0.997 | 0.997 | 0.999* | - | |||
| MAE | 2.01 | 1.70 | 1.01* | - | |||
| Compound | HER2 | Tubulin |
|---|---|---|
| Ranking Position (RANK) | ||
| 1 | 1 | 3 |
| 2 | 3 | 7 |
| 3 | 11 | 4 |
| 4 | 7 | 8 |
| 5 | 5 | 9 |
| 6 | 10 | 11 |
| 7 | 8 | 10 |
| 8 | 9 | 12 |
| 9 | 2 | 1 |
| 10 | 12 | 5 |
| 11 | 6 | 2 |
| 12 | 4 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dąbrowska-Balcerzak, K.; Nartowska, J.; Wawer, I.; Siudem, P.; Paradowska, K. Spirostanol Sapogenins and Saponins from Convallaria majalis L. Structural Characterization by 2D NMR, Theoretical GIAO DFT Calculations and Molecular Modeling. Molecules 2021, 26, 2999. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26102999
Dąbrowska-Balcerzak K, Nartowska J, Wawer I, Siudem P, Paradowska K. Spirostanol Sapogenins and Saponins from Convallaria majalis L. Structural Characterization by 2D NMR, Theoretical GIAO DFT Calculations and Molecular Modeling. Molecules. 2021; 26(10):2999. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26102999
Chicago/Turabian StyleDąbrowska-Balcerzak, Karolina, Jadwiga Nartowska, Iwona Wawer, Paweł Siudem, and Katarzyna Paradowska. 2021. "Spirostanol Sapogenins and Saponins from Convallaria majalis L. Structural Characterization by 2D NMR, Theoretical GIAO DFT Calculations and Molecular Modeling" Molecules 26, no. 10: 2999. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26102999