
Computational Insights on the Potential of Some NSAIDs for Treating COVID-19: Priority Set and Lead Optimization

 , ,
, ,  ,
,  and
and 
Abstract
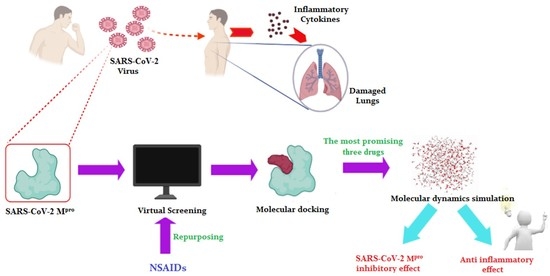
:
1. Introduction
2. Materials and Methods
2.1. Molecular Docking
2.1.1. NSAIDs Preparation
2.1.2. Target (SARS-CoV-2 Mpro) Preparation
2.1.3. Docking of the Tested NSAIDs to the Viral Mpro Binding Site
2.2. Molecular Dynamics (MD) Simulations
2.3. Quantum Mechanical Studies
3. Results and Discussion
3.1. Docking Studies
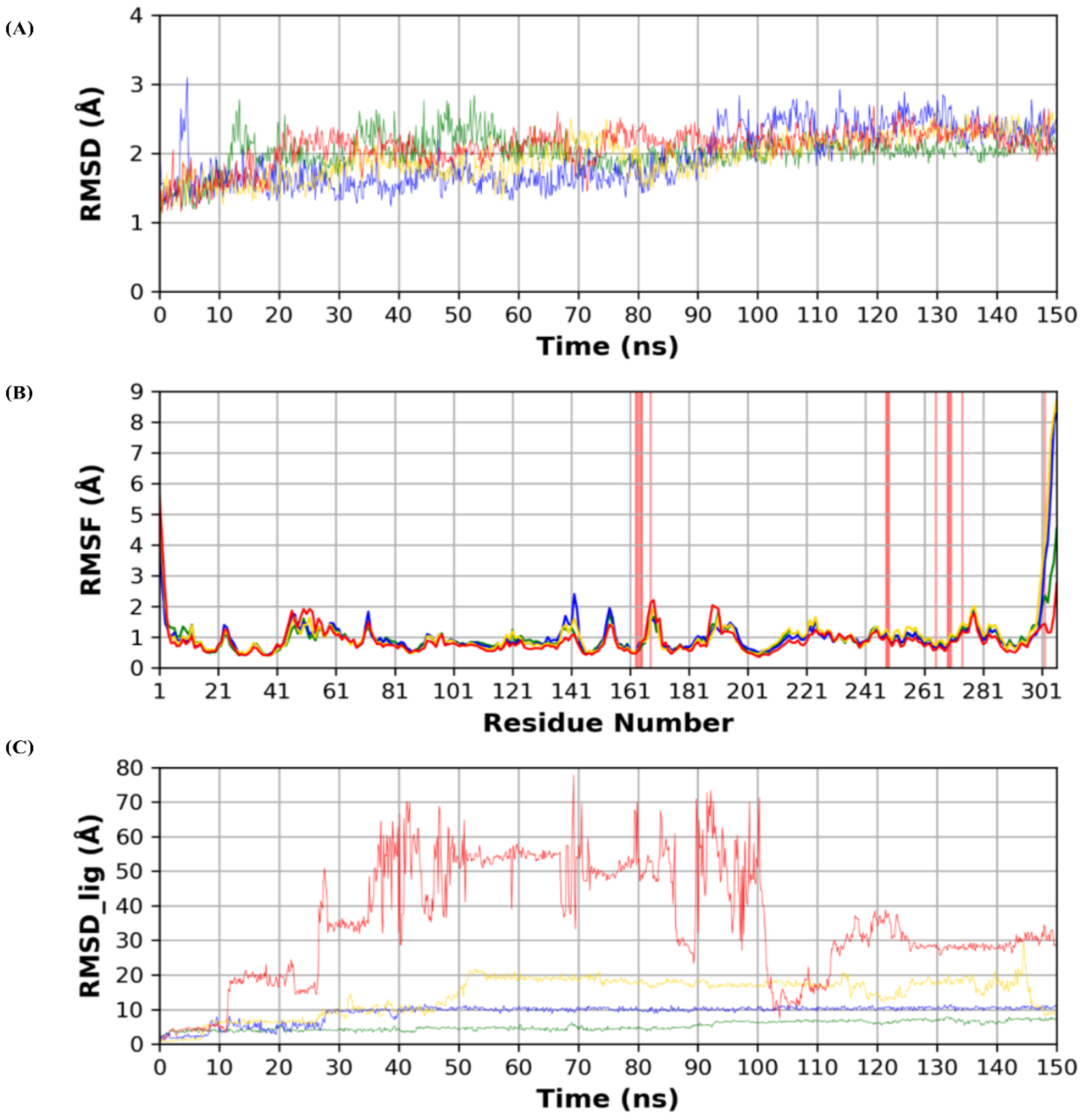
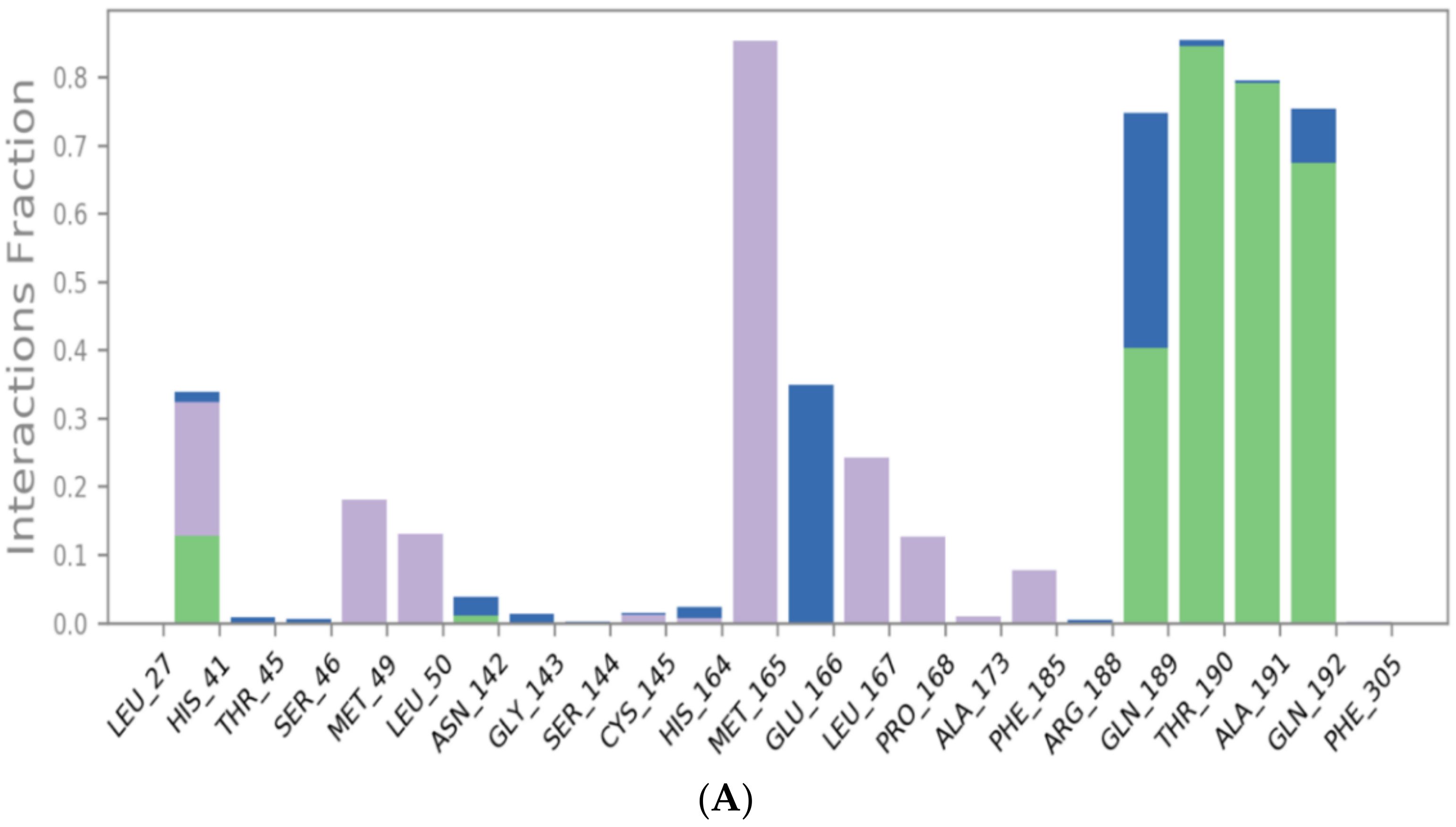
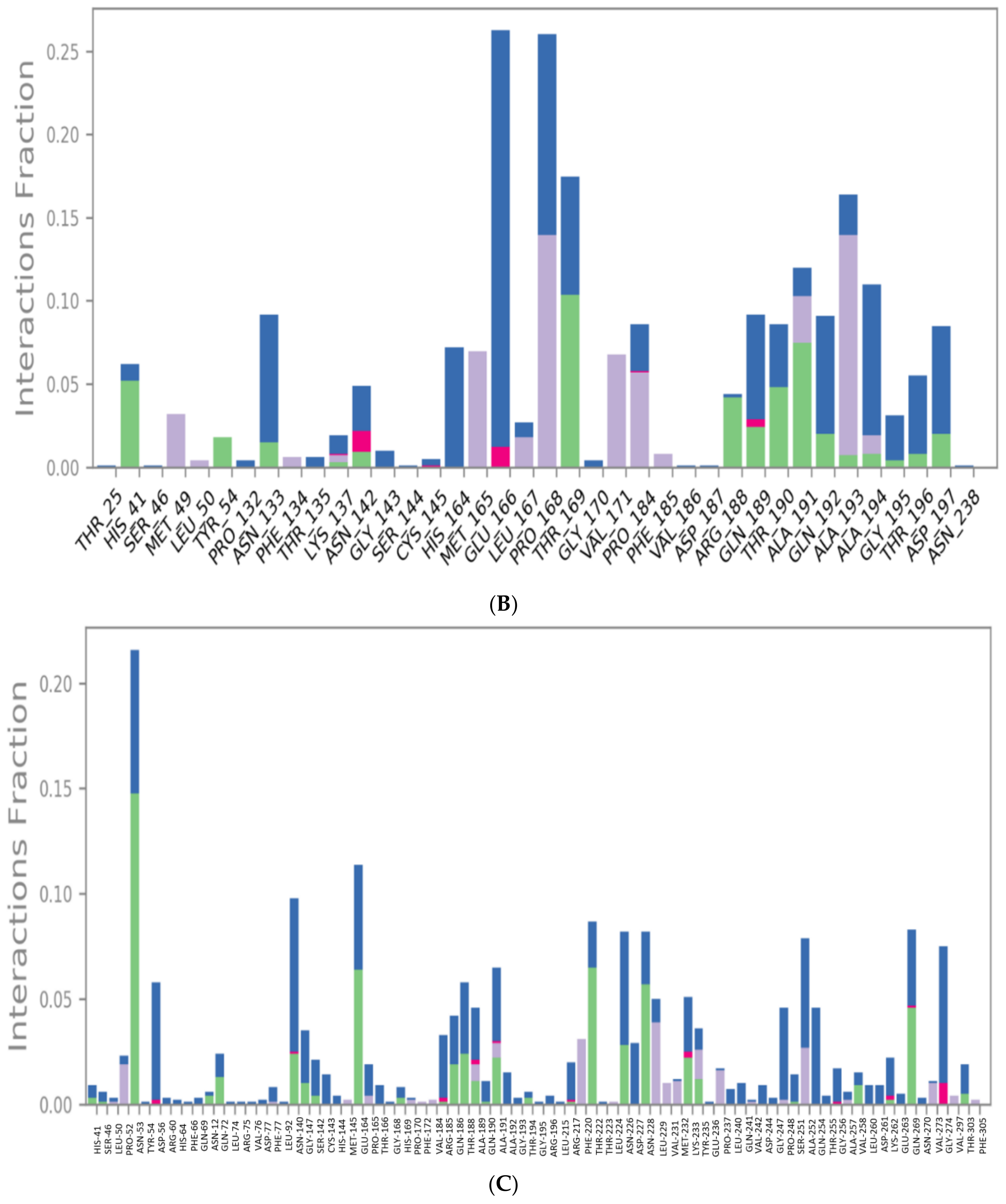
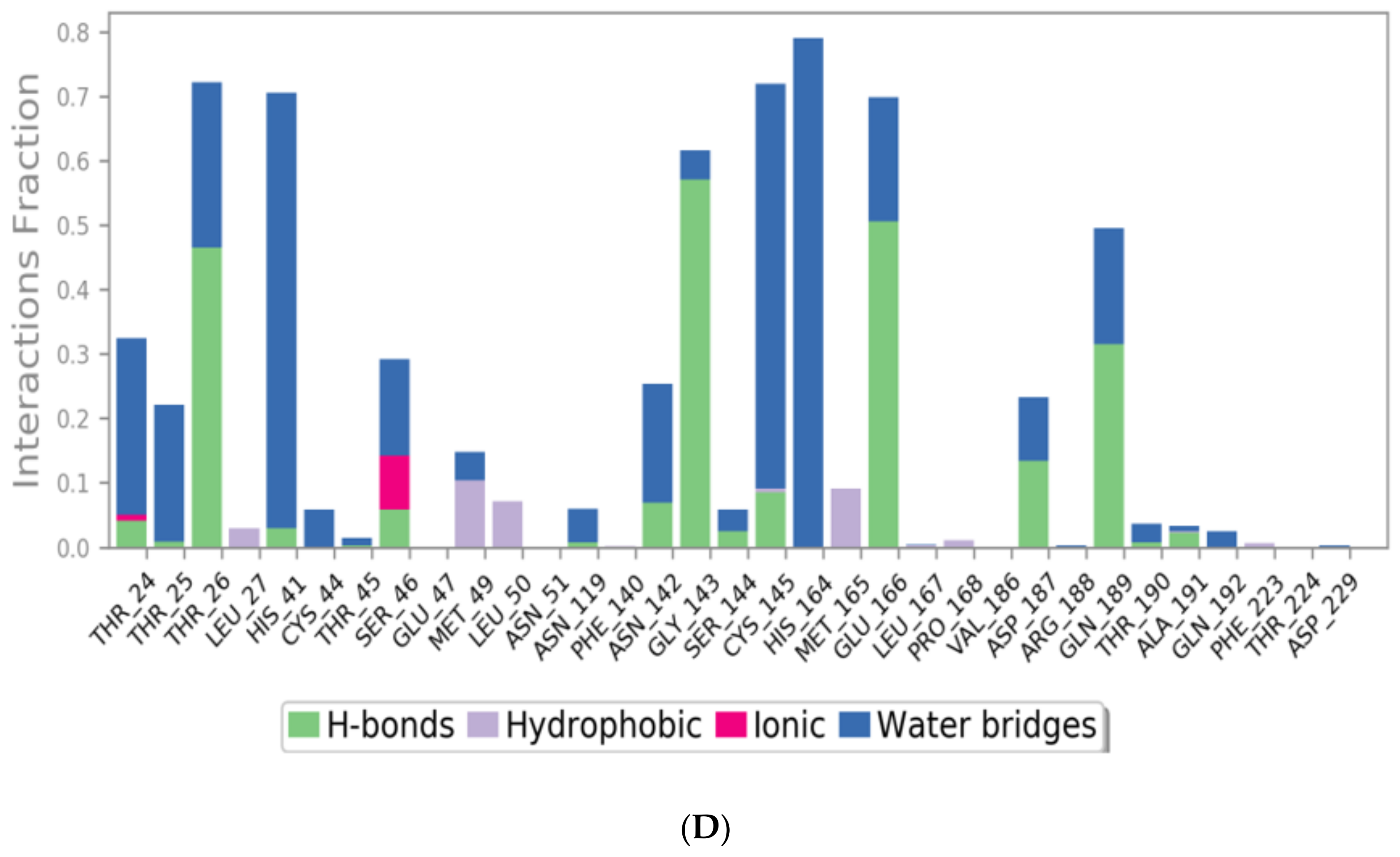
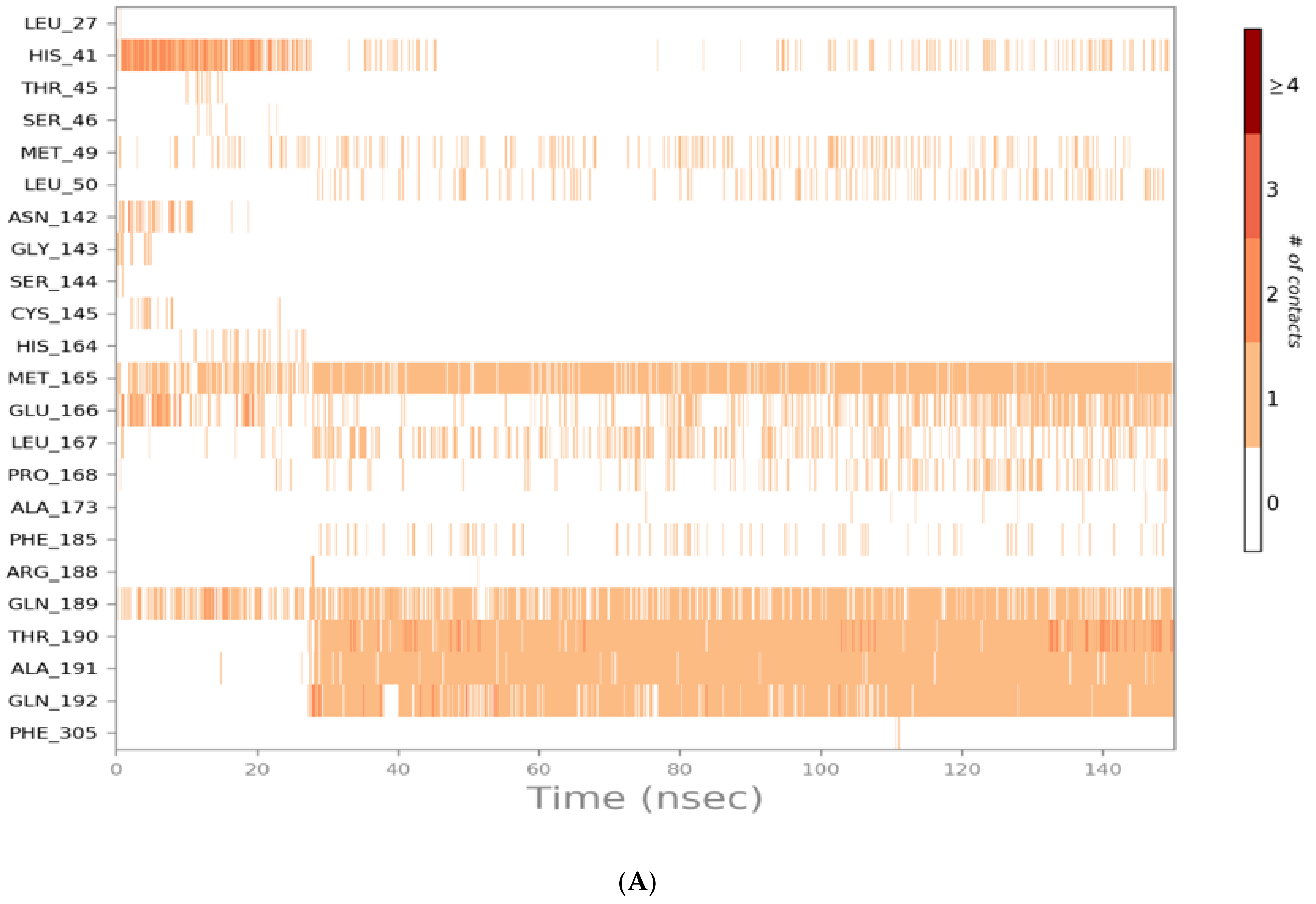
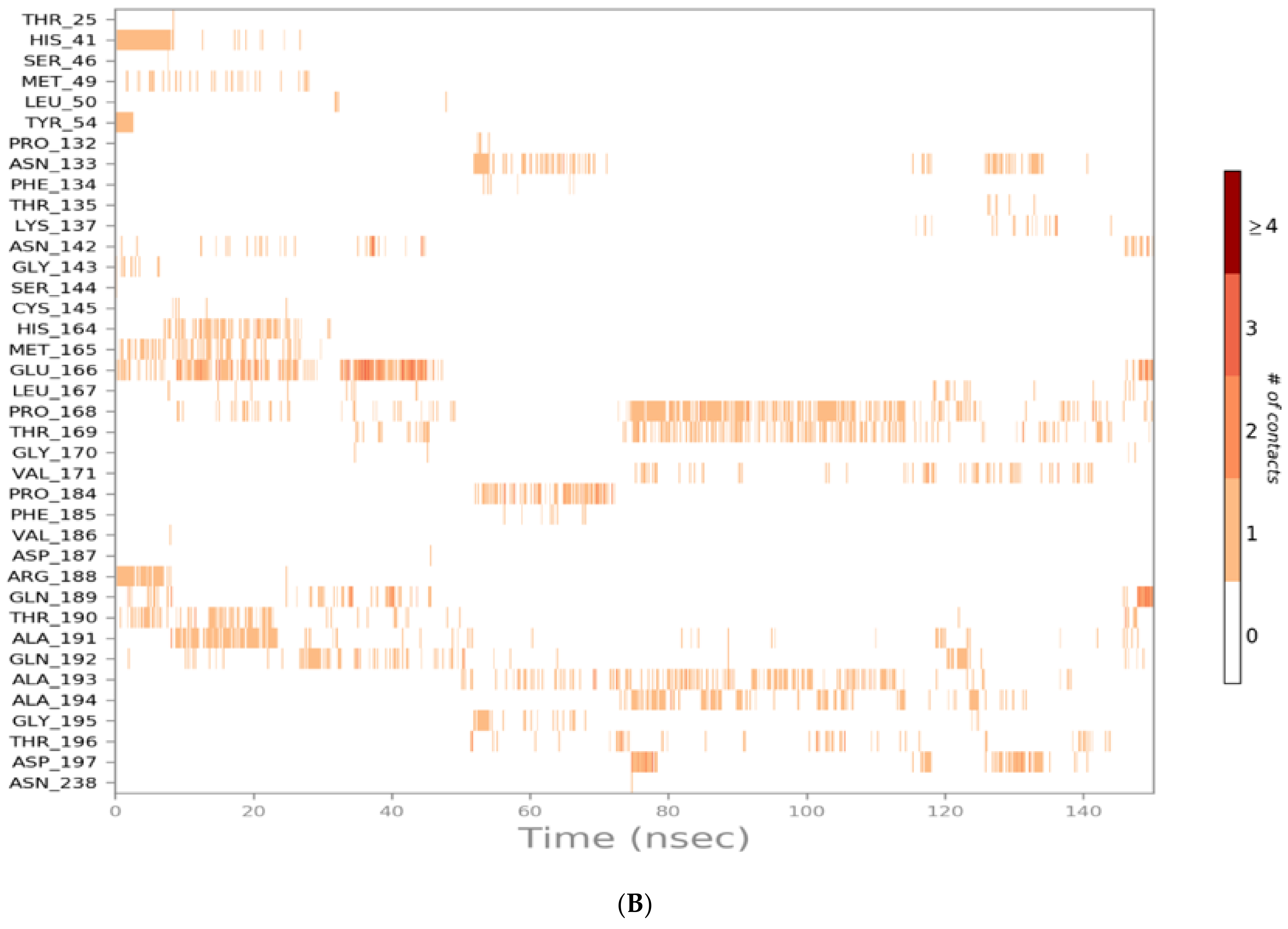
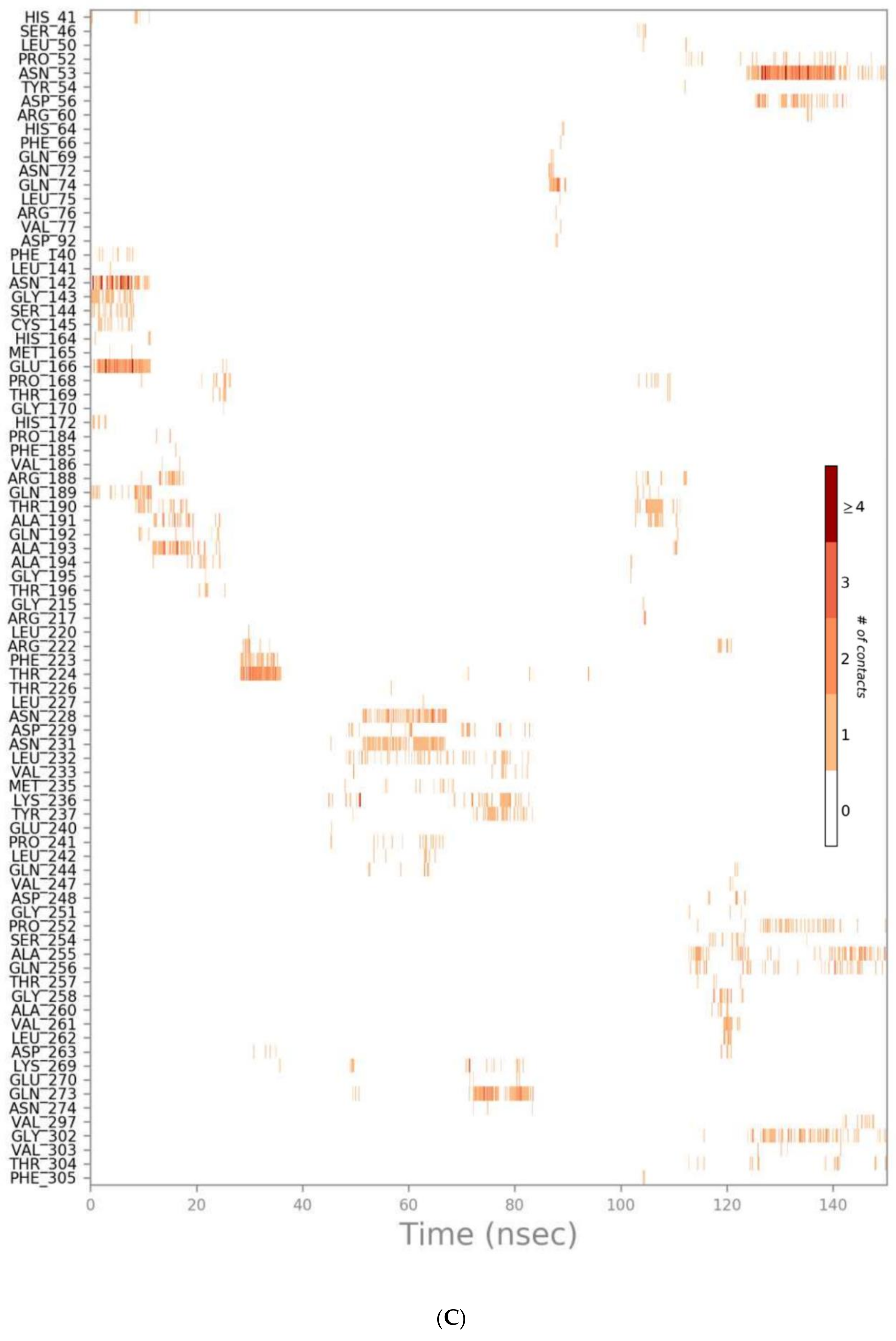
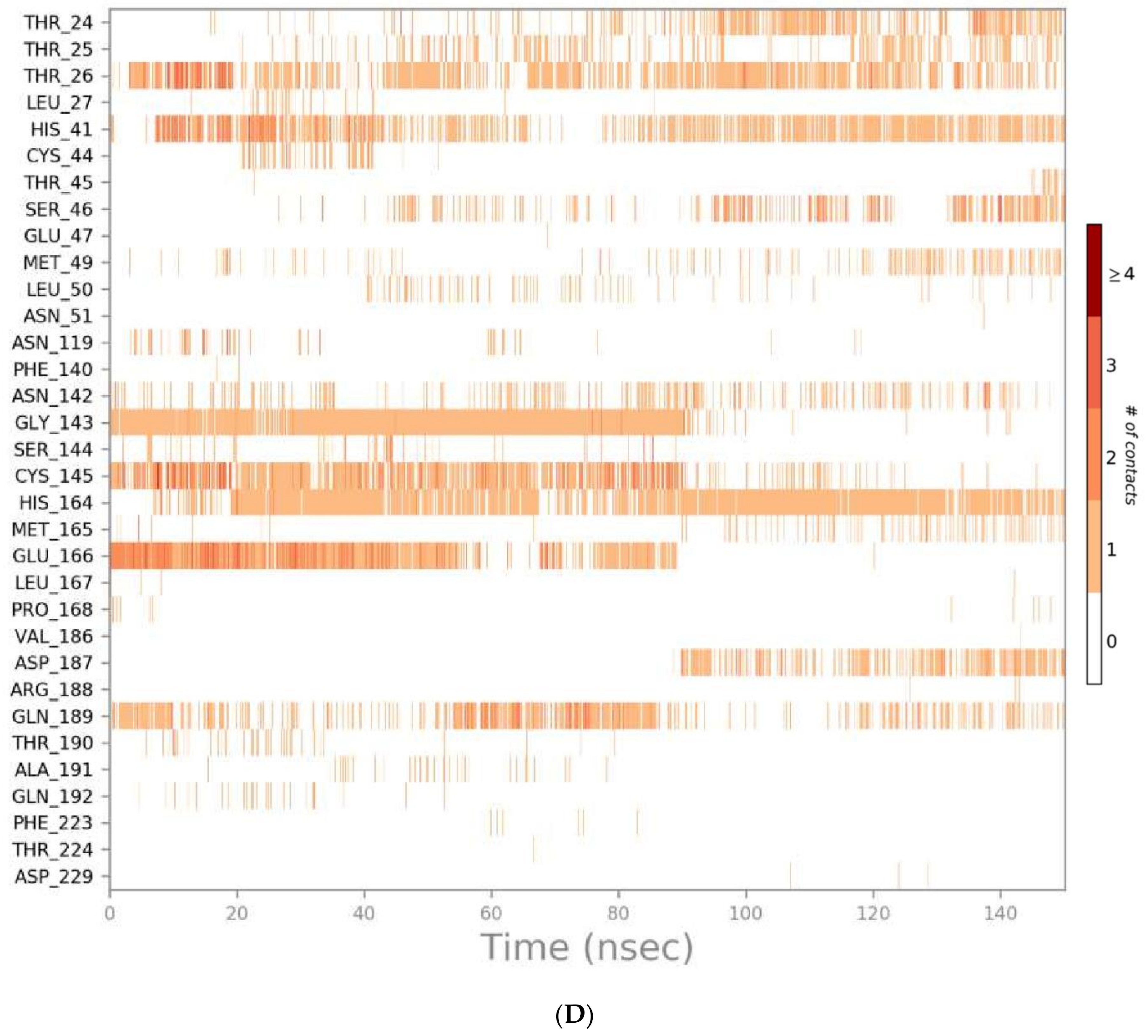
3.2. Molecular Dynamics (MD) Simulations
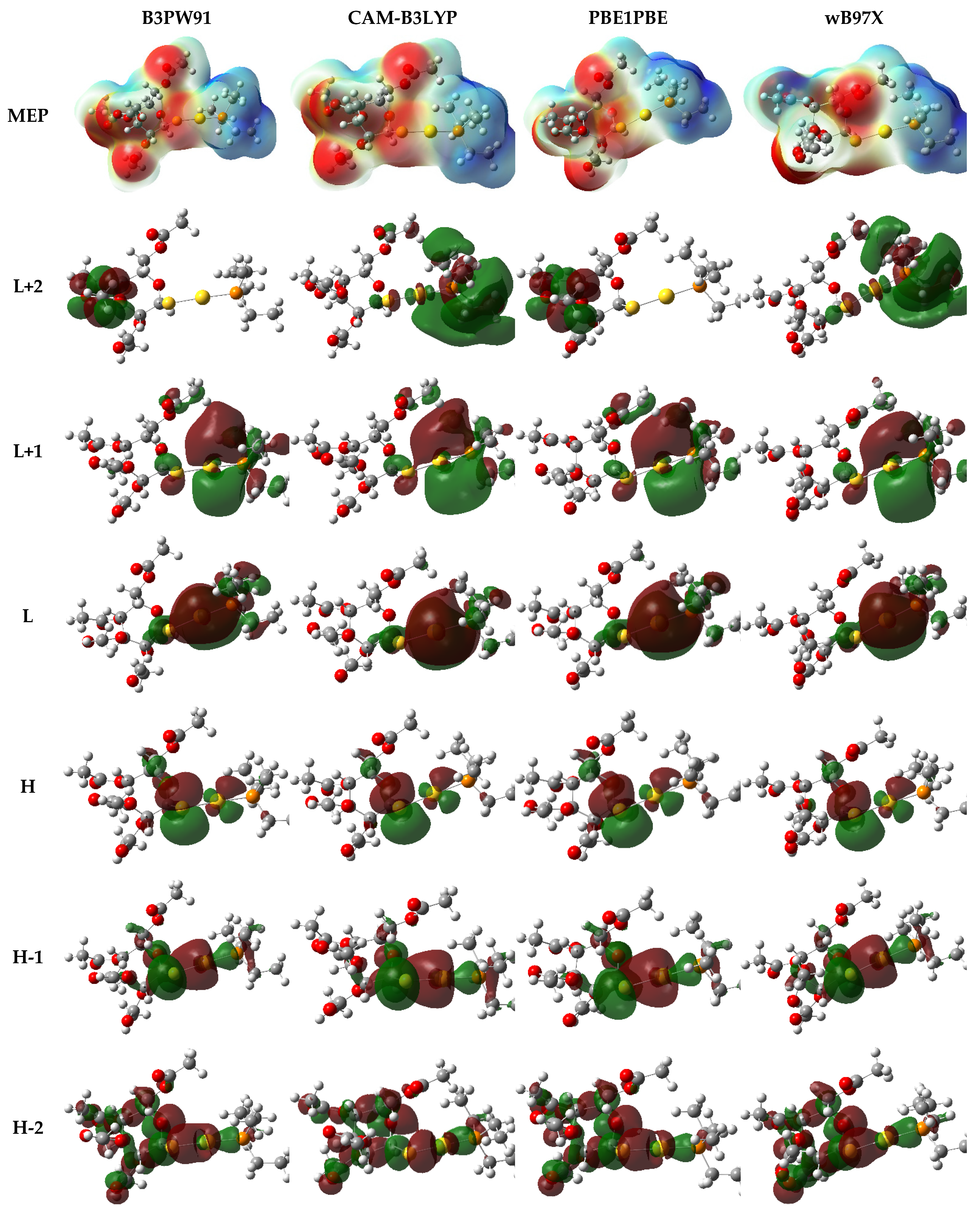
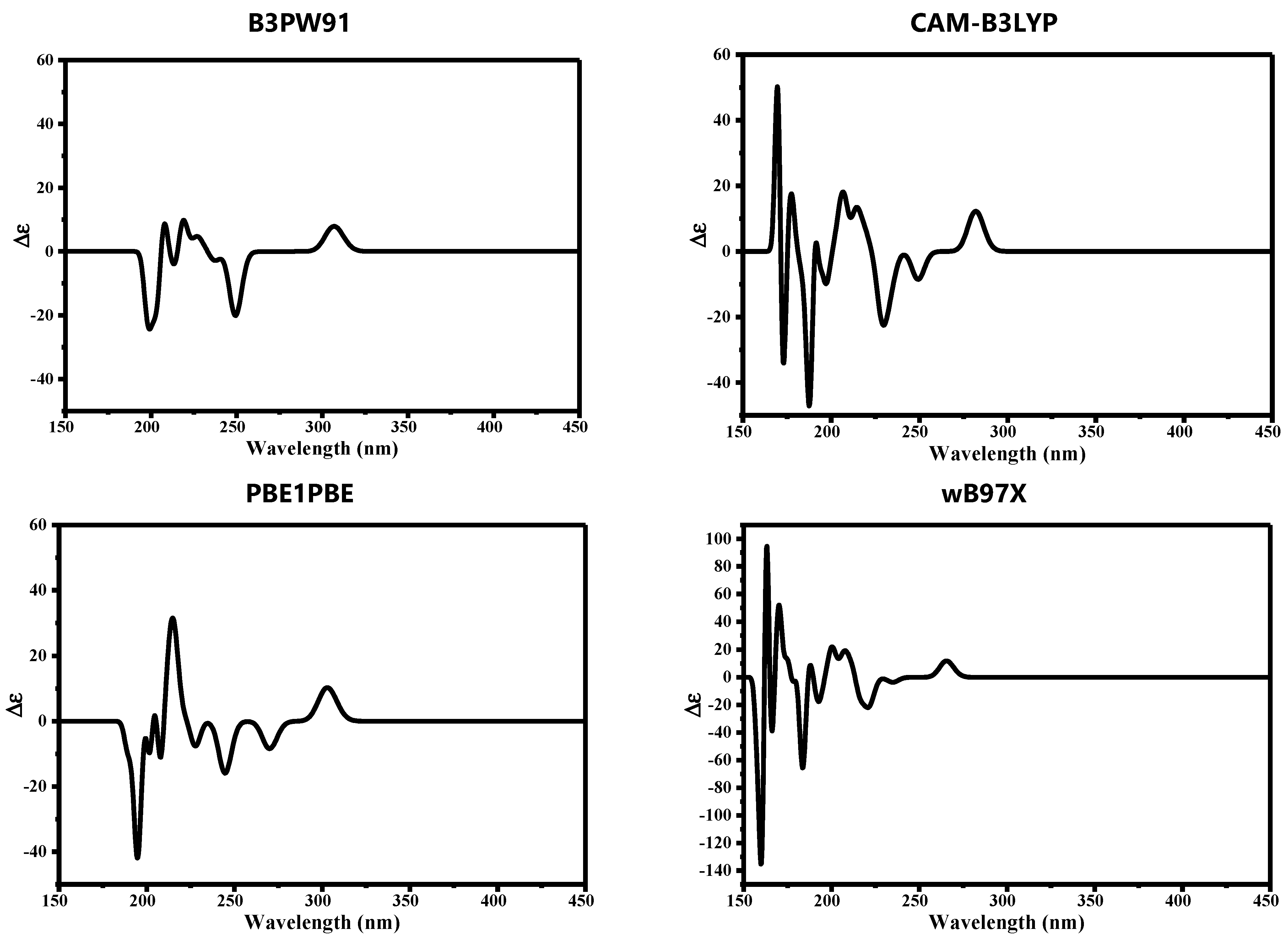
3.3. Quantum Mechanical Studies
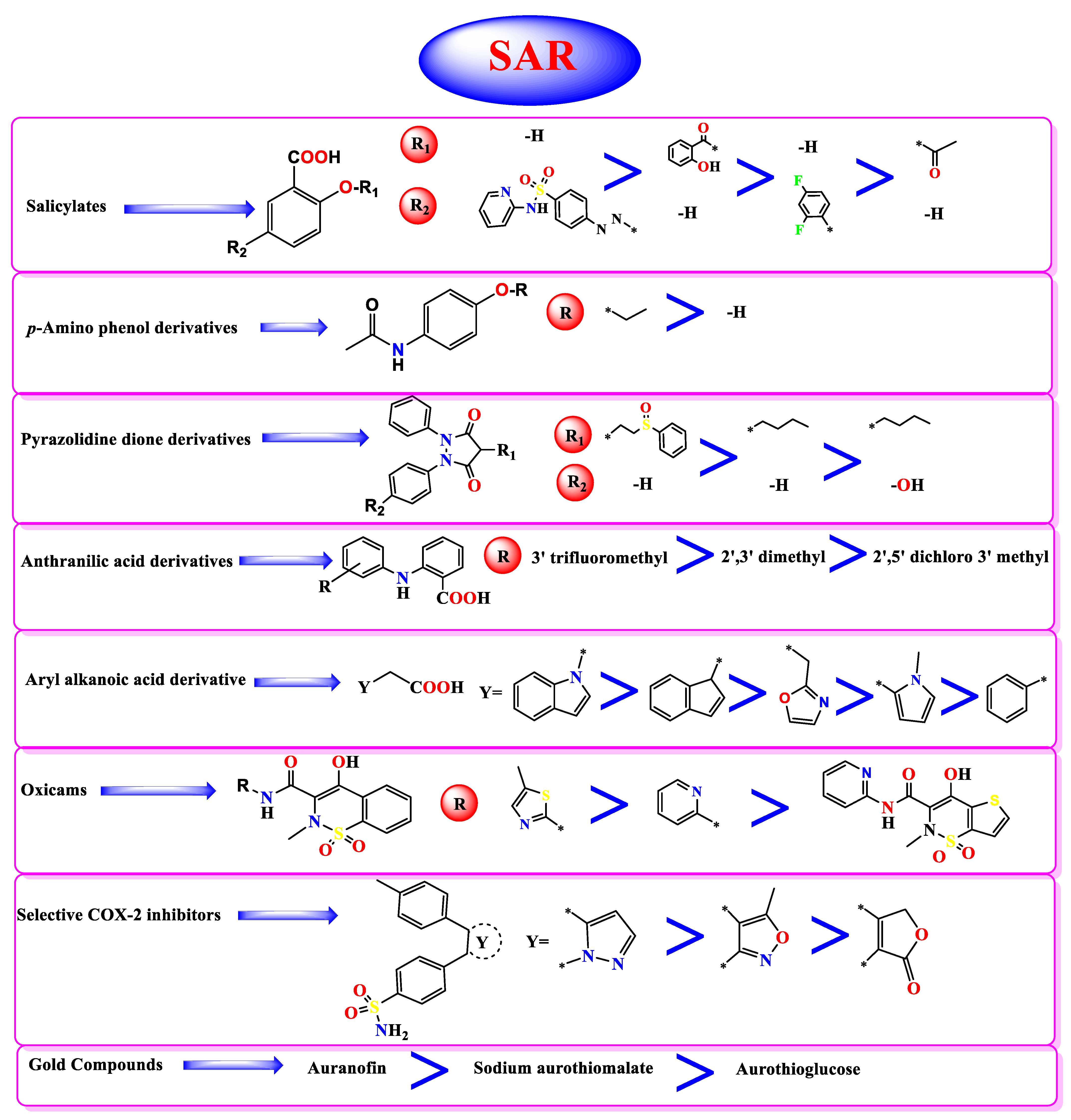
3.4. Structure–Activity Relationship Studies
- I.
- Salicylic acid derivatives: (Sulfasalazine 7, Salsalate 29, Diflunisal 36, and Aspirin 38).
- II.
- p-Amino phenol derivatives: (Phenacetin 35 and Paracetamol 40).
- III.
- Pyrazolidine dione derivatives: (Sulfinpyrazone 2, Phenylbutazone 5, and Oxyphenbutazone 8).
- IV.
- Anthranilic acid derivatives: (Flufenamic acid 31, Mefenamic acid 32, and Meclofenamic acid 34).
- V.
- Aryl alkanoic acid derivatives:
- Indole acetic acid: (Indomethacin 3).
- Indene acetic acid: (Sulindac 9).
- Pyrrole acetic acid: (Zomepirac 16 and Tolmetin 22).
- Phenyl acetic (propionic) acid: (Oxaprozin 12, Etodolac 18, Carprofen 20, Ketoprofen 21, Ketorolac 25, Ibuprofen 26, Fenoprofen 27, Flurbiprofen 28, Naproxen 30, and Diclofenac 33).
- VI.
- Oxicams: (Meloxicam 11, Piroxicam 14, and Tenoxicam 19).
- VII.
- Selective COX-2 inhibitors: (Celecoxib 6, Valdecoxib 15, and Rofecoxib 17).
- VIII.
- Gold compounds: (Auranofin 4, Aurothioglucose 37, and Aurothiomalate sodium 39).
- IX.
- Miscellaneous: (Metamizole 10, Nimesulide 13, Nabumetone 23, Probenecid 24, and Allopurinol 41).
- (a)
- Concerning salicylic acid derivatives, the best activity was attained by maintaining a salicylic acid scaffold without –OH or –COOH substitution, yet it was preferable to substitute a phenyl ring at the para position to –OH of the salicylic scaffold to ensure the best activity (compound 7).
- (b)
- In addition, for p-Amino phenol derivatives, better activity was achieved when phenolic –OH was substituted by ethyl group (compound 35) than unsubstituted one (compound 40).
- (c)
- For pyrazolidine dione NSAIDs, the best activity was accomplished by substitution of a pyrazolidine ring at position 4 by [2-(phenylsulfinyl)ethyl] moiety (compound 2).
- (d)
- Moreover, studying the structure–activity relationship for anthranilic acid derivatives revealed that substitution of a phenyl ring attached to the anthranilic acid scaffold by trifluoromethyl group at position 3 attained the best activity (compound 31).
- (e)
- Furthermore, concerning aryl acetic/propionic acid derivatives, the best activity was attained when the indole acetic acid drug was used (compound 3).
- (f)
- (g)
- On the other hand, with regards to selective cox-2 inhibitors, it worth noting that substitution of a benzenesulfonamide scaffold at position 4 with 3-trifluoromethyl pyrazole moiety (compound 6) showed better activity than 5-methyl isoxazole moiety (compound 15) and 5H-furan-2-one (compound 17).
- (h)
- Additionally, for gold anti-inflammatory compounds, the best activity was attained when gold was attached to 3,4,5-triacetyloxy-6-(acetyloxymethyl) oxane-2-thiolate moiety (compound 4).
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- WHO. Coronavirus Disease (COVID-19) Pandemic. 2021. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019?gclid=Cj0KCQjw38-DBhDpARIsADJ3kjmxpTjUS_7o4K1orvnoq-MfSNKEhLJl_TeOoN6DlYz3RBAC8hfU_rQaAsNwEALw_wcB (accessed on 11 April 2021).
- Worldometer. Coronavirus. 2021. Available online: https://www.worldometers.info/coronavirus/?utm_campaign=homeAdvegas1? (accessed on 11 April 2021).
- Chen, J.; Wang, Y.K.; Gao, Y.; Hu, L.S.; Yang, J.W.; Wang, J.R.; Sun, W.J.; Liang, Z.Q.; Cao, Y.M.; Cao, Y.B. Protection against COVID-19 injury by qingfei paidu decoction via anti-viral, anti-inflammatory activity and metabolic programming. Biomed. Pharmacother. 2020, 129, 110281. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, A.A.; Ashour, N.A.; Al-Karmalawy, A.A. The journey of antimalarial drugs against SARS-CoV-2: Review article. Inform. Med. Unlocked 2021, 24, 100604. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Dis. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Khattab, M.; Al-Karmalawy, A.A. Revisiting Activity of Some Nocodazole Analogues as a Potential Anticancer Drugs Using Molecular Docking and DFT Calculations. Front. Chem. 2021, 9, 92. [Google Scholar] [CrossRef]
- Talevi, A.; Bellera, C.L. Challenges and opportunities with drug repurposing: Finding strategies to find alternative uses of therapeutics. Expert Opin. Drug Dis. 2020, 15, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Masoudi-Sobhanzadeh, Y.; Omidi, Y.; Amanlou, M.; Masoudi-Nejad, A. Drug databases and their contributions to drug repurposing. Genomics 2020, 112, 1087–1095. [Google Scholar] [CrossRef]
- Brogi, S. Computational approaches for drug discovery. Molecules 2019, 24, 3061. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Li, X.; Lin, X. A review on applications of computational methods in drug screening and design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef] [Green Version]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef] [PubMed]
- Kosuge, M.; Furusawa-Nishii, E.; Ito, K.; Saito, Y.; Ogasawara, K. Point mutation bias in SARS-CoV-2 variants results in increased ability to stimulate inflammatory responses. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Russell, B.; Moss, C.; Rigg, A.; Van Hemelrijck, M. COVID-19 and treatment with NSAIDs and corticosteroids: Should we be limiting their use in the clinical setting? Ecancermedicalscience 2020, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guererero, C.A.; Murillo, A.; Acosta, O. Inhibition of rotavirus infection in cultured cells by N-acetyl-cysteine, PPARγ agonists and NSAIDs. Antivir. Res. 2012, 96, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Madeira, J.M.; Gibson, D.L.; Kean, W.F.; Klegeris, A. The biological activity of auranofin: Implications for novel treatment of diseases. Inflammopharmacology 2012, 20, 297–306. [Google Scholar] [CrossRef]
- Mostafa, A.; Kandeil, A.; Elshaier, Y.A.M.M.; Kutkat, O.; Moatasim, Y.; Rashad, A.A.; Shehata, M.; Gomaa, M.R.; Mahrous, N.; Mahmoud, S.H.; et al. FDA-Approved Drugs with Potent In Vitro Antiviral Activity against Severe Acute Respiratory Syndrome Coronavirus 2. Pharmaceuticals 2020, 13, 443. [Google Scholar] [CrossRef] [PubMed]
- Alnajjar, R.; Mostafa, A.; Kandeil, A.; Al-Karmalawy, A.A. Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease. Heliyon 2020, 6, e05641. [Google Scholar] [CrossRef]
- Zaki, A.A.; Al-Karmalawy, A.A.; El-Amier, Y.A.; Ashour, A. Molecular docking reveals the potential of Cleome amblyocarpa isolated compounds to inhibit COVID-19 virus main protease. New J. Chem. 2020, 44, 16752–16758. [Google Scholar] [CrossRef]
- Elmaaty, A.A.; Alnajjar, R.; Hamed, M.I.; Khattab, M.; Khalifa, M.M.; Al-Karmalawy, A.A. Revisiting activity of some glucocorticoids as a potential inhibitor of SARS-CoV-2 main protease: Theoretical study. RSC Adv. 2021, 11, 10027–10042. [Google Scholar] [CrossRef]
- Eissa, I.; Al-Karmalawy, A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Darwish, K.M. Molecular docking and dynamics simulation revealed the potential inhibitory activity of ACEIs against SARS-CoV-2 targeting hACE2 receptor. Front. Chem. 2021, 9, 227. [Google Scholar]
- Al-Karmalawy, A.A.; Alnajjar, R.; Dahab, M.; Metwaly, A.; Eissa, I. Molecular docking and dynamics simulations reveal the potential of anti-HCV drugs to inhibit COVID-19 main protease. Pharm. Sci. 2021, 9, 661230. [Google Scholar] [CrossRef]
- Zaki, A.A.; Ashour, A.; Elhady, S.S.; Darwish, K.M.; Al-Karmalawy, A.A. Calendulaglycoside A Showing Potential Activity Against SARS-CoV-2 Main Protease: Molecular Docking, Molecular Dynamics, and SAR Studies. J. Tradit. Complement. Med. 2021, in press. [Google Scholar] [CrossRef]
- Soltane, R.; Chrouda, A.; Mostafa, A.; Al-Karmalawy, A.; Chouaïb, K.; Dhahri, A.; Pashameah, R.; Alasiri, A.; Kutkat, O.; Shehata, M.; et al. Strong Inhibitory Activity and Action Modes of Synthetic Maslinic Acid Derivative on Highly Pathogenic Coronaviruses: COVID-19 Drug Candidate. Pathogens 2021, 10, 623. [Google Scholar] [CrossRef]
- Elmaaty, A.A.; Darwish, K.M.; Khattab, M.; Elhady, S.S.; Salah, M.; Hamed, M.I.; Al-Karmalawy, A.A.; Saleh, M.M. In a search for potential drug candidates for combating COVID-19: Computational study revealed salvianolic acid B as a potential therapeutic targeting 3CLpro and spike proteins. J. Biomol. Struct. Dyn. 2021, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Borges, R.S.; Palheta, I.C.; Ota, S.S.B.; Morais, R.B.; Barros, V.A.; Ramos, R.S.; Silva, R.C.; Costa, J.D.S.; Silva, C.H.T.P.; Campos, J.M.; et al. Toward of Safer Phenylbutazone Derivatives by Exploration of Toxicity Mechanism. Molecules 2019, 24, 143. [Google Scholar] [CrossRef] [Green Version]
- Aceves-Hernandez, J.; Nicolás-Vázquez, I.; Aceves, F.; Hinojosa-Torres, J.; Paz, M.; Castaño, V. Indomethacin polymorphs: Experimental and conformational analysis. J. Pharm. Sci. 2009, 98, 2448–2463. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, Y.; Jing, P.; Xu, G.; Zhou, Q.; Cai, Y.; Deng, X. Terahertz spectroscopic characterizations and DFT calculations of indomethacin cocrystals with nicotinamide and saccharin. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2021, 249, 119309. [Google Scholar] [CrossRef]
- Chemical Computing Group. Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2021. [Google Scholar]
- Release, S. 3: Desmond Molecular Dynamics System, DE Shaw Research; Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2017. [Google Scholar]
- Al-Karmalawy, A.A.; Khattab, M. Molecular modelling of mebendazole polymorphs as a potential colchicine binding site inhibitor. New J. Chem. 2020, 44, 13990–13996. [Google Scholar] [CrossRef]
- Ghanem, A.A.; Emara, H.A.; Muawia, S.; El Maksoud, A.I.A.; Al-Karmalawy, A.A.; Elshal, M.F. Tanshinone IIA synergistically enhances the antitumor activity of doxorubicin by interfering with the PI3K/AKT/mTOR pathway and inhibition of topoisomerase II: In vitro and molecular docking studies. New J. Chem. 2020, 44, 17374–17381. [Google Scholar] [CrossRef]
- Samra, R.M.; Soliman, A.F.; Zaki, A.A.; Ashour, A.; Al-Karmalawy, A.A.; Hassan, M.A.; Zaghloul, A.M. Bioassay-guided isolation of a new cytotoxic ceramide from Cyperus rotundus L. S. Afr. J. Bot. 2021, 139, 210–216. [Google Scholar] [CrossRef]
- Davis, I.W.; Baker, D. RosettaLigand Docking with Full Ligand and Receptor Flexibility. J. Mol. Biol. 2009, 385, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Eliaa, S.G.; Al-Karmalawy, A.A.; Saleh, R.M.; ElShal, M.F. Empagliflozin and Doxorubicin Synergistically Inhibit the Survival of Triple-Negative Breast Cancer Cells via Interfering with the mTOR Pathway and Inhibition of Calmodulin: In Vitro and Molecular Docking Studies. ACS Pharmacol. Transl. Sci. 2020, 3, 1330–1338. [Google Scholar] [CrossRef]
- Neria, E.; Fischer, S.; Karplus, M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Schrödinger. Desmond Molecular Dynamics System, DE Shaw Research: New York, 2015; Schrödinger: New York, NY, USA, 2015. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.R.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Ropo, M.; Kokko, K.; Vitos, L. Proving the Perdew-Burke-Ernzerhof density functional designed for metallic bulk and surface systems. Mater. Sci. 2007, 77, 195445. [Google Scholar] [CrossRef] [Green Version]
- Austin, A.J.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density Functional with Spherical Atom Dispersion Terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar]
- Hill, D.; Sutton, B. Gold. C20H34AuO9P5. Cryst. Struct. Commun. 1980, 9, 679–686. [Google Scholar]
- Alagarsamy, V. Textbook of Medicinal Chemistry; CBS Publishers and Distributors: New Delhi, India, 2018; Volume 2. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | NSAID | S a kcal/mol | RMSD_Refine b | Amino Acid Bond | Distance Å |
|---|---|---|---|---|---|
| 1 | N3 | −9.39 | 1.78 | Glu166/H-donor | 2.94 |
| Gln189/H-acceptor | 3.05 | ||||
| Ser46/H-acceptor | 3.12 | ||||
| Met49/H-acceptor | 3.51 | ||||
| His41/pi-H | 4.19 | ||||
| 2 | Sulfinpyrazone | −7.12 | 1.66 | Glu166/H-donor | 2.98 |
| His41/H-donor | 3.21 | ||||
| Gly143/H-pi | 3.58 | ||||
| 3 | Indomethacin | −7.07 | 1.51 | His163/H-donor | 3.49 |
| Met165/H-acceptor | 3.89 | ||||
| Met165/H-acceptor | 4.11 | ||||
| His41/pi-H | 3.87 | ||||
| Glu166/H-pi | 4.30 | ||||
| 4 | Auranofin | −6.91 | 0.84 | His41/H-donor | 2.90 |
| His163/H-donor | 3.10 | ||||
| Leu141/H-acceptor | 3.39 | ||||
| Asn142/H-donor | 3.41 | ||||
| Gln189/H-donor | 3.49 | ||||
| His41/pi-H | 4.20 | ||||
| 5 | Phenylbutazone | −6.88 | 1.07 | Glu166/H-donor | 3.43 |
| 6 | Celecoxib | −6.79 | 1.17 | Ser144/H-donor | 3.01 |
| His163/H-donor | 3.04 | ||||
| Asn142/H-acceptor | 3.88 | ||||
| Gln189/H-pi | 4.38 | ||||
| 7 | Sulfasalazine | −6.76 | 1.77 | Thr190/H-acceptor | 2.82 |
| Glu166/H-acceptor | 3.03 | ||||
| Gly143/H-donor | 3.14 | ||||
| His41/H-donor | 3.16 | ||||
| 8 | Oxyphenbutazone | −6.75 | 2.00 | His164/H-acceptor | 3.12 |
| Asn142/H-donor | 3.47 | ||||
| Gly143/H-donor | 3.56 | ||||
| 9 | Sulindac | −6.67 | 1.25 | Gly143/H-donor | 2.99 |
| Cys145/H-donor | 3.14 | ||||
| Glu166/H-pi | 4.49 | ||||
| 10 | Metamizole | −6.56 | 1.49 | Gln189/H-acceptor | 3.49 |
| Met165/H-acceptor | 3.55 | ||||
| Met165/H-acceptor | 3.85 | ||||
| Met165/H-pi | 3.50 | ||||
| His41/pi-H | 4.18 | ||||
| Glu166/H-pi | 4.24 | ||||
| 11 | Meloxicam | −6.47 | 1.35 | His163/H-donor | 2.84 |
| His164/H-acceptor | 3.15 | ||||
| His164/H-acceptor | 3.28 | ||||
| 12 | Oxaprozin | −6.43 | 1.20 | Ser144/H-donor | 2.97 |
| Glu166/H-pi | 3.83 | ||||
| Gln189/H-pi | 4.03 | ||||
| 13 | Nimesulide | −6.35 | 1.35 | His41/H-donor | 2.86 |
| His163/H-donor | 2.91 | ||||
| Cys145/H-donor | 3.46 | ||||
| 14 | Piroxicam | −6.30 | 1.32 | His41/H-donor | 3.30 |
| Cys145/H-acceptor | 3.81 | ||||
| Met165/H-pi | 3.46 | ||||
| His41/pi-H | 3.54 | ||||
| Glu166/H-pi | 4.54 | ||||
| 15 | Valdecoxib | −6.30 | 1.12 | Glu166/H-acceptor | 2.89 |
| Met165/H-acceptor | 3.40 | ||||
| Gln189/H-donor | 3.41 | ||||
| 16 | Zomepirac | −6.25 | 1.40 | His163/H-donor | 3.14 |
| Met165/H-pi | 4.42 | ||||
| 17 | Rofecoxib | −6.24 | 1.02 | Cys145/H-donor | 2.99 |
| Met165/H-acceptor | 3.48 | ||||
| Asn142/H-pi | 4.15 | ||||
| 18 | Etodolac | −6.19 | 0.68 | Arg188/H-donor | 3.28 |
| Glu166/H-pi | 3.74 | ||||
| 19 | Tenoxicam | −6.18 | 1.47 | Gly143/H-donor | 2.92 |
| His164/H-acceptor | 3.14 | ||||
| Asn142/H-donor | 3.18 | ||||
| Gly143/H-donor | 3.29 | ||||
| 20 | Carprofen | −6.15 | 0.90 | His164/H-acceptor | 2.95 |
| Gln192/H-acceptor | 3.77 | ||||
| Gln189/H-pi | 4.53 | ||||
| 21 | Ketoprofen | −6.15 | 1.57 | Glu166/H-donor | 2.99 |
| 22 | Tolmetin | −6.08 | 1.64 | Gly143/H-donor | 3.01 |
| His164/H-acceptor | 3.08 | ||||
| Cys145/H-donor | 3.36 | ||||
| Met49/H-acceptor | 3.93 | ||||
| 23 | Nabumetone | −6.02 | 1.14 | His163/H-donor | 3.16 |
| Met165/H-pi | 3.74 | ||||
| Glu166/H-pi | 4.16 | ||||
| 24 | Probenecid | −5.96 | 2.19 | Glu166/H-donor | 3.17 |
| Gln189/H-acceptor | 3.44 | ||||
| 25 | Ketorolac | −5.89 | 1.57 | Glu166/H-donor | 3.05 |
| Glu166/H-acceptor | 3.27 | ||||
| 26 | Ibuprofen | −5.88 | 0.87 | Leu141/H-acceptor | 2.99 |
| His163/H-donor | 3.03 | ||||
| 27 | Fenoprofen | −5.84 | 1.14 | His163/H-donor | 3.01 |
| His163/H-donor | 3.14 | ||||
| Glu166/H-pi | 4.04 | ||||
| Met165/H-pi | 4.22 | ||||
| 28 | Flurbiprofen | −5.74 | 1.03 | Phe140/H-acceptor | 2.91 |
| His163/H-donor | 3.08 | ||||
| Asn142/H-pi | 3.82 | ||||
| 29 | Salsalate | −5.72 | 1.78 | Gln189/H-acceptor | 3.08 |
| Glu166/H-donor His41/pi-H | 3.17 | ||||
| 3.90 | |||||
| 30 | Naproxen | −5.72 | 1.61 | Gly143/H-donor | 3.08 |
| Cys145/H-donor | 3.31 | ||||
| 31 | Flufenamic acid | −5.70 | 1.26 | His164/H-acceptor | 2.95 |
| Ser144/H-donor | 2.98 | ||||
| Ser144/H-donor | 3.09 | ||||
| His164/H-acceptor | 3.13 | ||||
| Cys145/H-acceptor | 3.21 | ||||
| 32 | Mefenamic acid | −5.68 | 2.08 | Glu166/H-donor | 3.06 |
| Gln189/H-acceptor | 3.21 | ||||
| Met165/H-acceptor | 3.68 | ||||
| Gln189/H-pi | 4.05 | ||||
| 33 | Diclofenac | −5.54 | 1.66 | Gln189/H-acceptor | 2.89 |
| Glu166/H-donor | 2.94 | ||||
| Gly143/H-donor | 3.25 | ||||
| Leu141/H-acceptor | 3.73 | ||||
| 34 | Meclofenamic acid | −5.48 | 1.18 | Glu166/H-acceptor | 2.84 |
| Gln192/H-donor | 3.09 | ||||
| Glu166/H-acceptor | 3.17 | ||||
| 35 | Phenacetin | −5.43 | 1.27 | Glu166/H-acceptor | 3.03 |
| Gln189/H-donor | 3.37 | ||||
| His41/pi-H | 4.18 | ||||
| 36 | Diflunisal | −5.26 | 1.52 | Leu141/H-acceptor | 2.80 |
| His163/H-donor | 2.97 | ||||
| His41/pi-H | 3.83 | ||||
| 37 | Aurothioglucose | −4.90 | 1.45 | His163/H-donor | 3.17 |
| Glu166/H-acceptor | 3.22 | ||||
| Glu166/H-donor | 3.76 | ||||
| Met165/H-donor | 4.08 | ||||
| 38 | Aspirin | −4.81 | 1.31 | Gln189/H-acceptor | 2.82 |
| Glu 166/H-donor | 3.53 | ||||
| 39 | Sodium aurothiomalate | −4.67 | 1.42 | His164/H-acceptor | 2.83 |
| Arg188/H-donor | 3.55 | ||||
| Met49/H-acceptor | 3.87 | ||||
| 40 | Paracetamol | −4.53 | 0.44 | Glu166/H-acceptor | 3.11 |
| Glu166/H-pi | 4.25 | ||||
| 41 | Allopurinol | −4.33 | 1.13 | Asp187/H-acceptor | 3.24 |
| Gln189/H-pi | 3.52 |
| Drug | 3 D Interaction | 3 D Pocket Positioning |
|---|---|---|
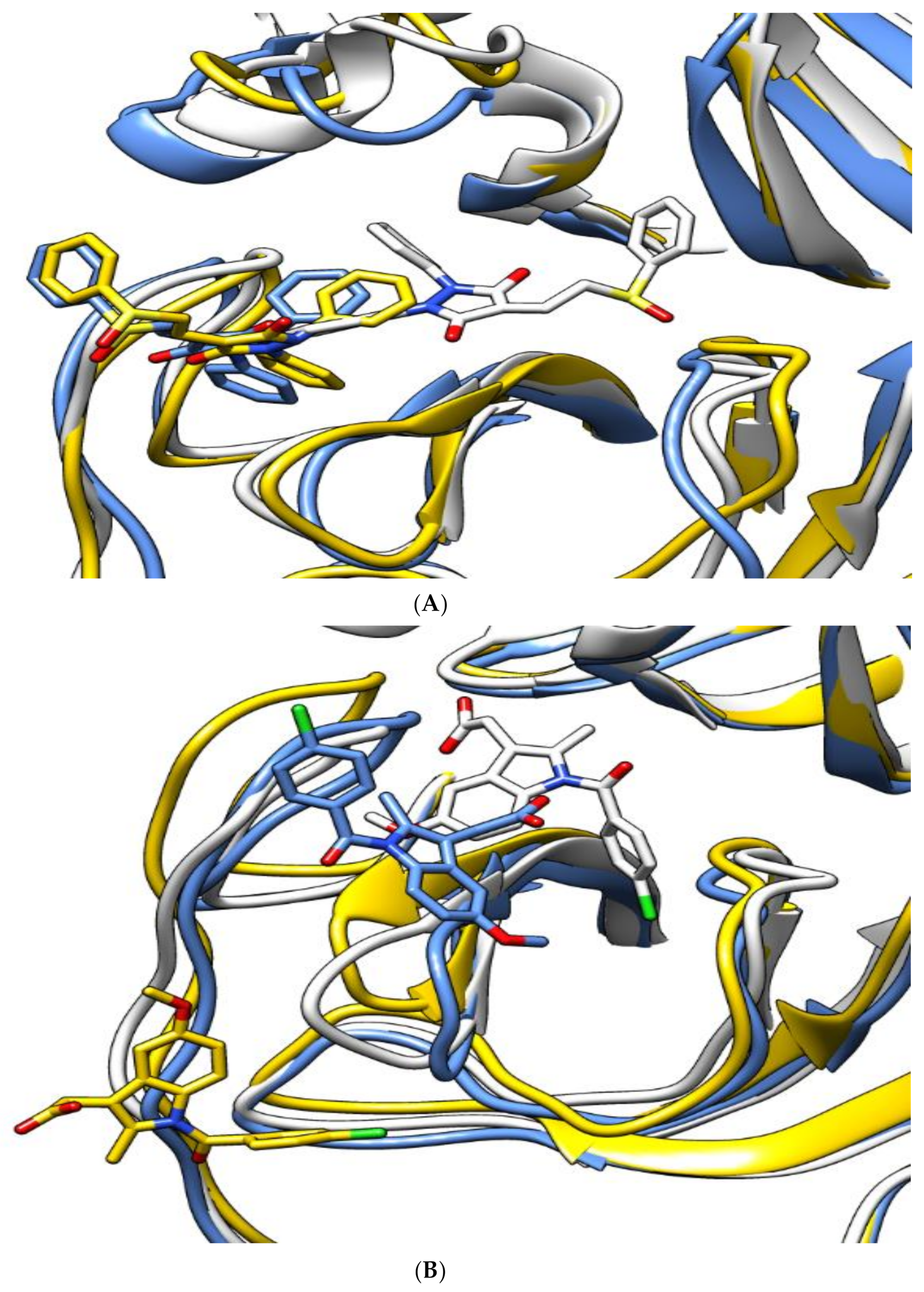
| Sulfinpyrazone 2 |  |  |
| Indomethacin 3 |  |  |
| Auranofin 2 |  |  |
| N3 1 |  |  |
| B3PW91 | CAM-B3LYP | PBE1PBE | wB97X | Exp | |
|---|---|---|---|---|---|
| Au-P (Å) | 2.28 | 2.29 | 2.28 | 2.29 | 2.26 |
| Au-S (Å) | 2.31 | 2.32 | 2.31 | 2.32 | 2.29 |
| ∠ S-Au-P | 177.4 | 179.2 | 178.9 | 177.4 | 173.6 |
| ∠ Au-S-C | 102.7 | 101.6 | 101.3 | 102.2 | 105.6 |
| Eh (a.u.) | −2335.103739 | −2334.956583 | −2333.714375 | −2335.316967 | - |
| ZPE (a.u.) | 0.534394 | 0.540545 | 0.537098 | 0.542842 | - |
| Eh + ZPE (a.u.) | −2334.569345 | −2334.416038 | −2333.177278 | −2334.774126 | - |
| Polarizability (a.u.) | 348.400064 | 337.552355 | 344.065746 | 334.158185 | - |
| μ (D) | 11.5681 | 11.6597 | 11.4449 | 11.8367 | - |
| HOMO-LUMO gap (eV) | 4.94 | 7.30 | 5.21 | 9.21 | - |
| Entropy (cal/mol-kelvin) | 261.262 | 256.099 | 256.603 | 250.821 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abo Elmaaty, A.; Hamed, M.I.A.; Ismail, M.I.; B. Elkaeed, E.; S. Abulkhair, H.; Khattab, M.; Al-Karmalawy, A.A. Computational Insights on the Potential of Some NSAIDs for Treating COVID-19: Priority Set and Lead Optimization. Molecules 2021, 26, 3772. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26123772
Abo Elmaaty A, Hamed MIA, Ismail MI, B. Elkaeed E, S. Abulkhair H, Khattab M, Al-Karmalawy AA. Computational Insights on the Potential of Some NSAIDs for Treating COVID-19: Priority Set and Lead Optimization. Molecules. 2021; 26(12):3772. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26123772
Chicago/Turabian StyleAbo Elmaaty, Ayman, Mohammed I. A. Hamed, Muhammad I. Ismail, Eslam B. Elkaeed, Hamada S. Abulkhair, Muhammad Khattab, and Ahmed A. Al-Karmalawy. 2021. "Computational Insights on the Potential of Some NSAIDs for Treating COVID-19: Priority Set and Lead Optimization" Molecules 26, no. 12: 3772. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26123772