
Design, Sustainable Synthesis and Biological Evaluation of a Novel Dual α2A/5-HT7 Receptor Antagonist with Antidepressant-Like Properties

 , , , , , ,
, , , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
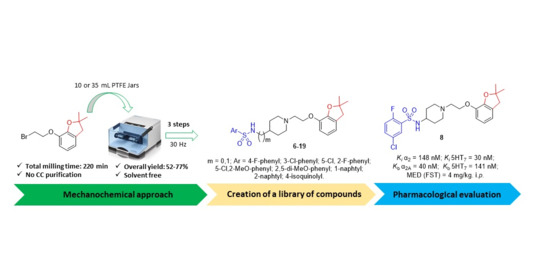
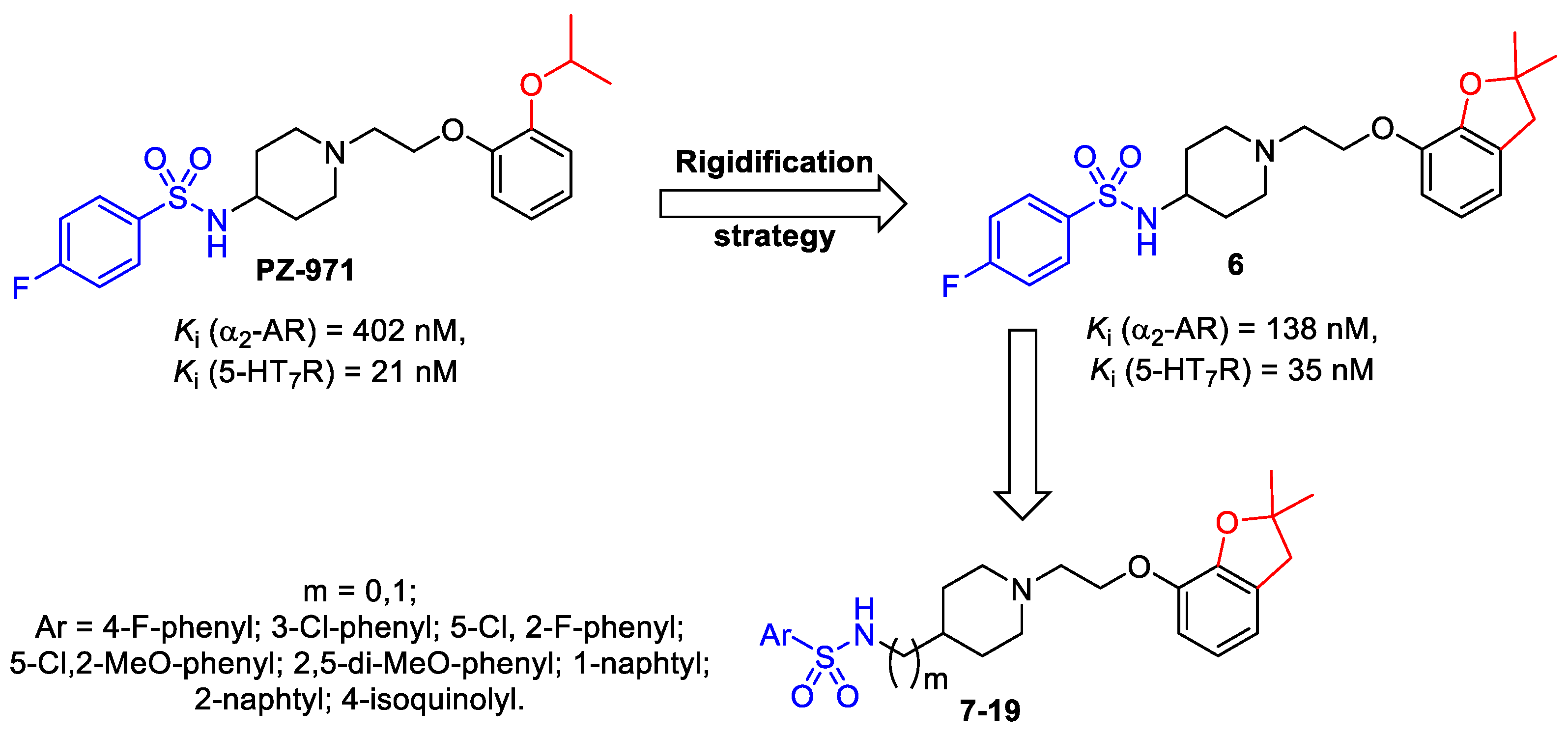
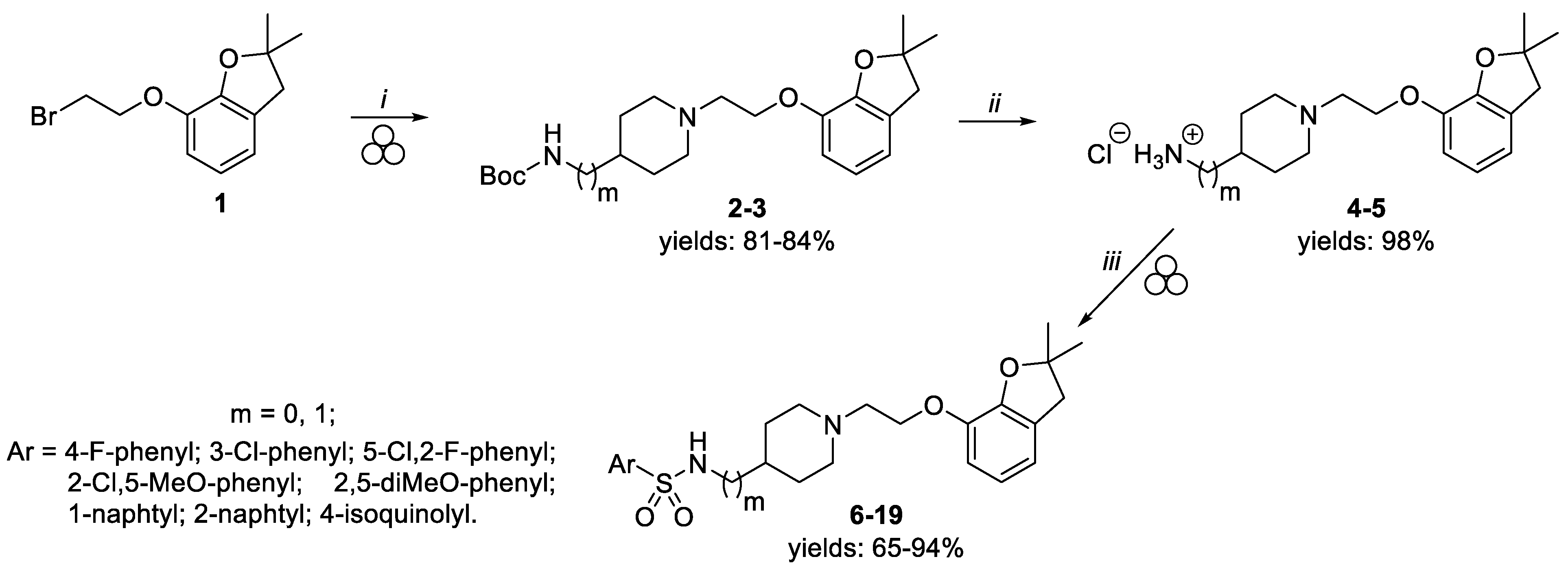
2.1. Chemistry
2.2. In Vitro Pharmacology
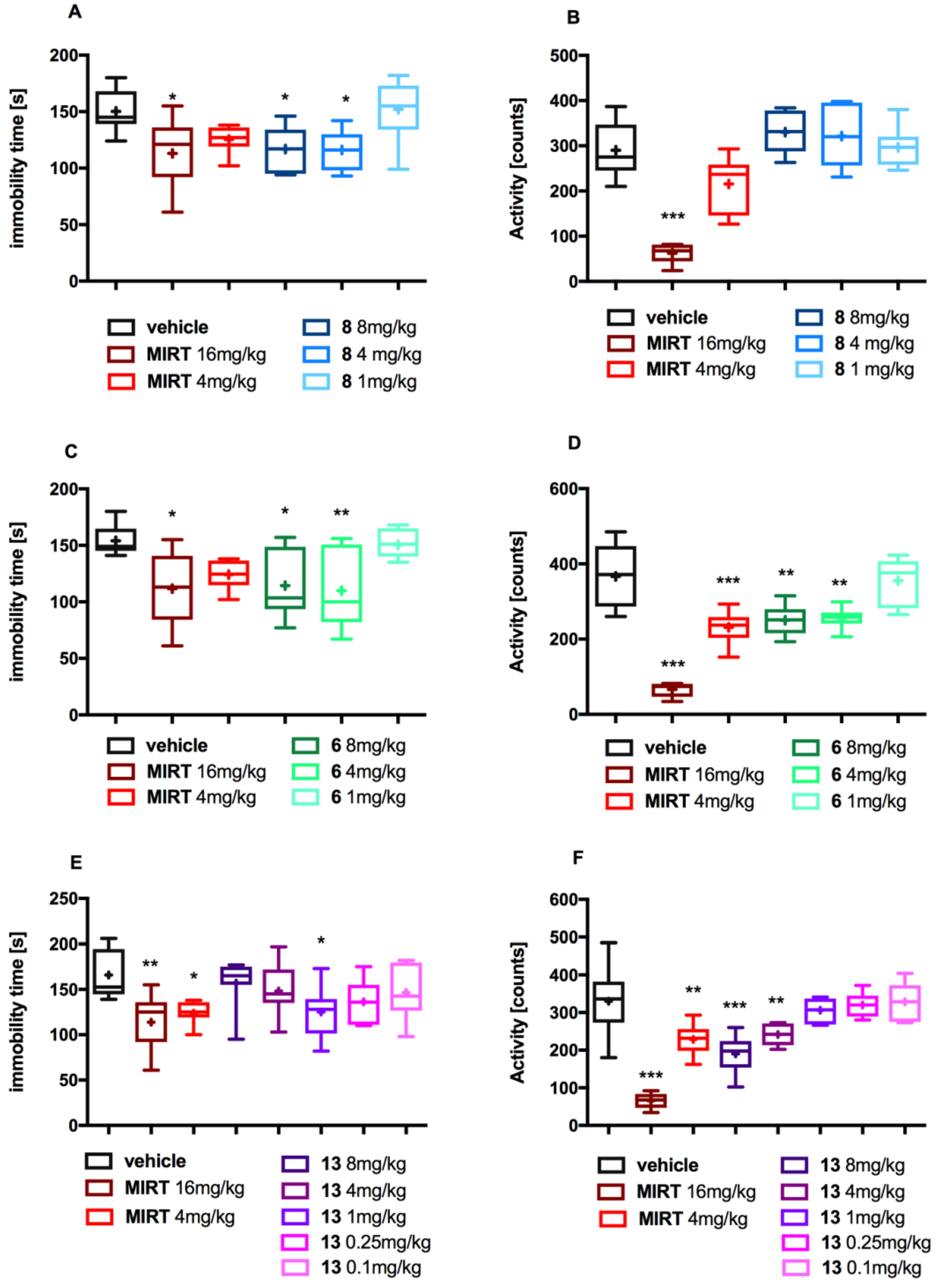
2.3. In Vivo Pharmacology
3. Materials and Methods
3.1. Chemistry
3.1.1. General Chemical Methods
3.1.2. Alkylation of Boc-Protected 4-Aminopiperidine in Ball Mill (Procedure A)
3.1.3. Alkylation of Boc-Protected 4-Aminopiperidine and 4-Aminomethylpiperidine in Ball Mill (Procedure B)
Tert-butyl {1-[2-(2,2-Dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}carbamate (2)
Tert-butyl ({1-[2-(2,2-Dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}methyl)carbamate (3)
3.1.4. General Procedure for the Deprotection of Boc Function in Solid State (Procedure C)
1-{2-[(2,2-Dimethyl-2,3-dihydrobenzofuran-7-yl)oxy]ethyl}piperidin-4-amine hydrochloride (4)
(1-{2-[(2,2-Dimethyl-2,3-dihydrobenzofuran-7-yl)oxy]ethyl}piperidin-4-yl)methanamine hydrochloride (5)
3.1.5. Sulfonylation of Primary Amine (Procedure D) for the Preparation of Final Compounds (6–19)
3.1.6. Sulfonylation of Primary Amine (Procedure E) for the High-Scale Preparation of Selected Final Compounds (6, 8 and 13)
3.1.7. Characterization of Final Compounds
4-Fluoro-N-{1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl} benzenesulfonamide (6)
3-Chloro-N-{1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}benzenesulfonamide (7)
5-Chloro-2-fluoro-N-{1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}benzenesulfonamide (8)
5-Chloro-2-methoxy-N-{1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}benzenesulfonamide (9)
2,5-Dimethoxy-N-{1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethy]piperidin-4-yl}benzenesulfonamide (10)
1-Naphthalene-N-{1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}sulfonamide (11)
2-Naphthalene-N-{1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}sulfonamide (12)
4-Isoquinoline-N-{1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}sulfonamide (13)
4-Fluoro-N-({1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}methyl)benzenesulfonamide (14)
3-Chloro-N-({1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}methyl)benzenesulfonamide (15)
5-Chloro-2-flluoro-N-({1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethylpiperidin-4-yl}methyl)benzenesulfonamide (16)
5-Chloro-2-methoxy-N-({1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}methyl)benzenesulfonamide (17)
1-Naphthalene-N-({1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}methyl)sulfonamide (18)
4-Isoquinoline-N-({1-[2-(2,2-dimethyl-2,3-dihydrobenzofuran-7-yloxy)ethyl]piperidin-4-yl}methyl)sulfonamide (19)
3.2. In Vitro Pharmacology
3.2.1. Determination of the Affinity of the Test Compounds at the α1- and α2-ARs
3.2.2. Determination of the Affinity of the Test Compounds at the Serotonin 5-HT1A, 5-HT2A, 5-HT6, 5-HT7 and Dopaminergic D2 Receptors
3.2.3. Determination of the Intrinsic Activity of the Test Compounds at the α2-AR Subtypes
3.2.4. Determination of the Intrinsic Activity of the Test Compounds at the 5-HT7R
3.3. In Vivo Pharmacology
3.3.1. Animals
3.3.2. Drug Administration
3.3.3. Forced Swim Test
3.3.4. Spontaneous Locomotor Activity
3.3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- World Health Organization. Depression and Other Common Mental Disorders: Global Health Estimates; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Conradi, H.J.; Ormel, J.; De Jonge, P. Presence of individual (residual) symptoms during depressive episodes and periods of remission: A 3-year prospective study. Psychol. Med. 2010, 41, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Philipp, M.; Brede, M.; Hein, L. Physiological significance of α2-adrenergic receptor subtype diversity: One receptor is not enough. Am. J. Physiol. Integr. Comp. Physiol. 2002, 283, R287–R295. [Google Scholar] [CrossRef] [Green Version]
- Bücheler, M.; Hadamek, K.; Hein, L. Two α2-adrenergic receptor subtypes, α2A and α2C, inhibit transmitter release in the brain of gene-targeted mice. Neuroscience 2002, 109, 819–826. [Google Scholar] [CrossRef]
- Scheinin, M.; Lomasney, J.W.; Hayden-Hixson, D.M.; Schambra, U.B.; Caron, M.G.; Lefkowitz, R.J.; Fremeau, R.T. Distribution of α2-adrenergic receptor subtype gene expression in rat brain. Mol. Brain Res. 1994, 21, 133–149. [Google Scholar] [CrossRef]
- Scheibner, J.; Trendelenburg, A.-U.; Hein, L.; Starke, K. α2 -Adrenoceptors modulating neuronal serotonin release: A study in α2 -adrenoceptor subtype-deficient mice. Br. J. Pharmacol. 2001, 132, 925–933. [Google Scholar] [CrossRef] [Green Version]
- Cottingham, C.; Wang, Q. α2 adrenergic receptor dysregulation in depressive disorders: Implications for the neurobiology of depression and antidepressant therapy. Neurosci. Biobehav. Rev. 2012, 36, 2214–2225. [Google Scholar] [CrossRef] [Green Version]
- Lovenberg, T.W.; Baron, B.M.; De Lecea, L.; Miller, J.D.; Prosser, R.; Rea, M.A.; Foye, P.E.; Racke, M.; Slone, A.L.; Siegel, B.W.; et al. A novel adenylyl cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythms. Neuron 1993, 11, 449–458. [Google Scholar] [CrossRef]
- Guseva, D.; Wirth, A.; Ponimaskin, E. Cellular mechanisms of the 5-HT7receptor-mediated signaling. Front. Behav. Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikiforuk, A. Targeting the Serotonin 5-HT7 Receptor in the Search for Treatments for CNS Disorders: Rationale and Progress to Date. CNS Drugs 2015, 29, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modica, M.N.; LaCivita, E.; Intagliata, S.; Salerno, L.; Romeo, G.; Pittalà, V.; Leopoldo, M. Structure–Activity Relationships and Therapeutic Potentials of 5-HT7 Receptor Ligands: An Update. J. Med. Chem. 2018, 61, 8475–8503. [Google Scholar] [CrossRef]
- Langer, S.Z. α2-Adrenoceptors in the treatment of major neuropsychiatric disorders. Trends Pharmacol. Sci. 2015, 36, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.M.; Platt, B.J.; Rizzo, S.J.S.; Pulicicchio, C.M.; Wantuch, C.; Zhang, M.-Y.; Cummons, T.; Leventhal, L.; Bender, C.N.; Zhang, J.; et al. Preclinical characterization of BRL 44408: Antidepressant—And analgesic-like activity through selective α2A-adrenoceptor antagonism. Int. J. Neuropsychopharmacol. 2010, 13, 1193–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guscott, M.; Bristow, L.; Hadingham, K.; Rosahl, T.; Beer, M.; Stanton, J.; Bromidge, F.; Owens, A.; Huscroft, I.; Myers, J.; et al. Genetic knockout and pharmacological blockade studies of the 5-HT7 receptor suggest therapeutic potential in depression. Neuropharmacology 2005, 48, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Wesołowska, A.; Tatarczynska, E.; Nikiforuk, A.; Chojnacka-Wójcik, E. Enhancement of the anti-immobility action of antidepressants by a selective 5-HT7 receptor antagonist in the forced swimming test in mice. Eur. J. Pharmacol. 2007, 555, 43–47. [Google Scholar] [CrossRef]
- Sanacora, G.; Berman, R.M.; Cappiello, A.; Oren, D.A.; Kugaya, A.; Liu, N.; Gueorguieva, R.; Fasula, D.; Charney, D.S. Addition of the α2-Antagonist Yohimbine to Fluoxetine: Effects on Rate of Antidepressant Response. Neuropsychopharmacology 2004, 29, 1166–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhir, A.; Kulkarni, S. Effect of Addition of Yohimbine (Alpha-2-Receptor Antagonist) to the Antidepressant Activity of Fluoxetine or Venlafaxine in the Mouse Forced Swim Test. Pharmacology 2007, 80, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Canale, V.; Partyka, A.; Kurczab, R.; Krawczyk, M.; Kos, T.; Satała, G.; Kubica, B.; Jastrzębska-Więsek, M.; Wesołowska, A.; Bojarski, A.; et al. Novel 5-HT 7 R antagonists, arylsulfonamide derivatives of (aryloxy)propyl piperidines: Add-on effect to the antidepressant activity of SSRI and DRI, and pro-cognitive profile. Bioorg. Med. Chem. 2017, 25, 2789–2799. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R. The pharmacology of mianserin-an update. Br. J. Clin. Pharmacol. 1983, 15, 263S–268S. [Google Scholar] [CrossRef]
- Anttila, S.A.; Leinonen, E. V A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001, 7, 249–264. [Google Scholar] [CrossRef]
- Canale, V.; Kurczab, R.; Partyka, A.; Satała, G.; Lenda, T.; Jastrzębska-Więsek, M.; Wesołowska, A.; Bojarski, A.; Zajdel, P. Towards new 5-HT 7 antagonists among arylsulfonamide derivatives of (aryloxy)ethyl-alkyl amines: Multiobjective based design, synthesis, and antidepressant and anxiolytic properties. Eur. J. Med. Chem. 2016, 108, 334–346. [Google Scholar] [CrossRef]
- Tan, D.; Loots, L.; Friščić, T. Towards medicinal mechanochemistry: Evolution of milling from pharmaceutical solid form screening to the synthesis of active pharmaceutical ingredients (APIs). Chem. Commun. 2016, 52, 7760–7781. [Google Scholar] [CrossRef]
- Colacino, E.; Porcheddu, A.; Charnay, C.; Delogu, F. From enabling technologies to medicinal mechanochemistry: An eco-friendly access to hydantoin-based active pharmaceutical ingredients. React. Chem. Eng. 2019, 4, 1179–1188. [Google Scholar] [CrossRef]
- Friščić, T.; Mottillo, C.; Titi, H.M. Mechanochemistry for Synthesis. Angew. Chem. Int. Ed. 2020, 59, 1018–1029. [Google Scholar] [CrossRef]
- Beillard, A.; Bantreil, X.; Métro, T.-X.; Martinez, J.; Lamaty, F. Alternative Technologies That Facilitate Access to Discrete Metal Complexes. Chem. Rev. 2019, 119, 7529–7609. [Google Scholar] [CrossRef] [PubMed]
- Milbeo, P.; Quintin, F.; Moulat, L.; Didierjean, C.; Martinez, J.; Bantreil, X.; Calmès, M.; Lamaty, F. Synthesis, characterisation and cytotoxic activity evaluation of new metal-salen complexes based on the 1,2-bicyclo[2.2.2]octane bridge. Tetrahedron Lett. 2021, 63, 152706. [Google Scholar] [CrossRef]
- Métro, T.-X.; Martinez, J.; Lamaty, F. 1,1′-Carbonyldiimidazole and Mechanochemistry: A Shining Green Combination. ACS Sustain. Chem. Eng. 2017, 5, 9599–9602. [Google Scholar] [CrossRef]
- Ardila-Fierro, K.J.; Hernández, J.G. Sustainability Assessment of Mechanochemistry by Using the Twelve Principles of Green Chemistry. ChemSusChem 2021, 14, 2145–2162. [Google Scholar] [CrossRef] [PubMed]
- Canale, V.; Frisi, V.; Bantreil, X.; Lamaty, F.; Zajdel, P. Sustainable Synthesis of a Potent and Selective 5-HT7 Receptor Antagonist Using a Mechanochemical Approach. J. Org. Chem. 2020, 85, 10958–10965. [Google Scholar] [CrossRef] [PubMed]
- Declerck, V.; Nun, P.; Martinez, J.; Lamaty, F. Solvent-Free Synthesis of Peptides. Angew. Chem. Int. Ed. 2009, 48, 9318–9321. [Google Scholar] [CrossRef]
- Zajdel, P.; Canale, V.; Partyka, A.; Marciniec, K.; Kurczab, R.; Satała, G.; Siwek, A.; Jastrzębska-Więsek, M.; Wesołowska, A.; Kos, T.; et al. Arylsulfonamide derivatives of (aryloxy)ethylpiperidines as selective 5-HT7 receptor antagonists and their psychotropic properties. MedChemComm 2015, 6, 1272–1277. [Google Scholar] [CrossRef]
- Rak, A.; Canale, V.; Marciniec, K.; Żmudzki, P.; Kotańska, M.; Knutelska, J.; Siwek, A.; Stachowicz, G.; Bednarski, M.; Nowiński, L.; et al. Arylsulfonamide derivatives of (aryloxy)ethyl pyrrolidines and piperidines as α 1 -adrenergic receptor antagonist with uro-selective activity. Bioorg. Med. Chem. 2016, 24, 5582–5591. [Google Scholar] [CrossRef]
- Canale, V.; Rak, A.; Kotańska, M.; Knutelska, J.; Siwek, A.; Bednarski, M.; Nowiński, L.; Zygmunt, M.; Koczurkiewicz, P.; Pękala, E.; et al. Synthesis and Pharmacological Evaluation of Novel Silodosin-Based Arylsulfonamide Derivatives as α1A/α1D-Adrenergic Receptor Antagonist with Potential Uroselective Profile. Molecules 2018, 23, 2175. [Google Scholar] [CrossRef] [Green Version]
- De Boer, T.; Maura, G.; Raiteri, M.; De Vos, C.; Wieringa, J.; Pinder, R. Neurochemical and autonomic pharmacological profiles of the 6-aza-analogue of mianserin, org 3770 and its enantiomers. Neuropharmacology 1988, 27, 399–408. [Google Scholar] [CrossRef]
- Fernandez, J.; Alonso, J.M.; Andres, J.I.; Cid, J.M.; Diaz, A.; Iturrino, L.; Gil, P.; Megens, A.; Sipido, V.K.; Trabanco, A.A. Discovery of New Tetracyclic Tetrahydrofuran Derivatives as Potential Broad-Spectrum Psychotropic Agents. J. Med. Chem. 2005, 48, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Zajdel, P.; Marciniec, K.; Satała, G.; Canale, V.; Kos, T.; Partyka, A.; Jastrzębska-Więsek, M.; Wesołowska, A.; Basińska-Ziobroń, A.; Wójcikowski, J.; et al. N1-Azinylsulfonyl-1H-indoles: 5-HT6 Receptor Antagonists with Procognitive and Antidepressant-Like Properties. ACS Med. Chem. Lett. 2016, 7, 618–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partyka, A.; Kurczab, R.; Canale, V.; Satała, G.; Marciniec, K.; Pasierb, A.; Jastrzębska-Więsek, M.; Pawłowski, M.; Wesołowska, A.; Bojarski, A.; et al. The impact of the halogen bonding on D 2 and 5-HT 1A /5-HT 7 receptor activity of azinesulfonamides of 4-[(2-ethyl)piperidinyl-1-yl]phenylpiperazines with antipsychotic and antidepressant properties. Bioorg. Med. Chem. 2017, 25, 3638–3648. [Google Scholar] [CrossRef] [PubMed]
- Zajdel, P.; Kos, T.; Marciniec, K.; Satała, G.; Canale, V.; Kamiński, K.; Hołuj, M.; Lenda, T.; Koralewski, R.; Bednarski, M.; et al. Novel multi-target azinesulfonamides of cyclic amine derivatives as potential antipsychotics with pro-social and pro-cognitive effects. Eur. J. Med. Chem. 2018, 145, 790–804. [Google Scholar] [CrossRef]
- Campbell, S.; MacQueen, G. The role of the hippocampus in the pathophysiology of major depression. J. Psychiatry Neurosci. 2004, 29, 417–426. [Google Scholar]
- Marcinkowska, M.; Kotańska, M.; Zagórska, A.; Śniecikowska, J.; Kubacka, M.; Siwek, A.; Bucki, A.; Pawłowski, M.; Bednarski, M.; Sapa, J.; et al. Synthesis and biological evaluation of N-arylpiperazine derivatives of 4,4-dimethylisoquinoline-1,3(2H,4H)-dione as potential antiplatelet agents. J. Enzym. Inhib. Med. Chem. 2018, 33, 536–545. [Google Scholar] [CrossRef] [Green Version]
- Bogdanova, O.V.; Kanekar, S.; D’Anci, K.E.; Renshaw, P.F. Factors influencing behavior in the forced swim test. Physiol. Behav. 2013, 118, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, N.; Omori, I.M.; Nakagawa, A.; Cipriani, A.; Barbui, C.; Churchill, R.; Furukawa, T.A. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst. Rev. 2011, CD006528. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.J.; Peterson, M.J. Sleep Disturbances in Depression. Sleep Med. Clin. 2015, 10, 17–23. [Google Scholar] [CrossRef]
- Maj, J.; Klimek, V.; Nowak, G. Antidepressant drugs given repeatedly increase binding to α1-adrenoceptors in the rat cortex. Eur. J. Pharmacol. 1985, 119, 113–116. [Google Scholar] [CrossRef]
- Kurczab, R.; Canale, V.; Satała, G.; Zajdel, P.; Bojarski, A.J. Amino Acid Hot Spots of Halogen Bonding: A Combined Theoretical and Experimental Case Study of the 5-HT7 Receptor. J. Med. Chem. 2018, 61, 8717–8733. [Google Scholar] [CrossRef]
- Hogendorf, A.S.; Hogendorf, A.; Kurczab, R.; Kalinowska-Tłuścik, J.; Popik, P.; Nikiforuk, A.; Krawczyk, M.; Satała, G.; Lenda, T.; Knutelska, J.; et al. 2-Aminoimidazole-based antagonists of the 5-HT6 receptor—A new concept in aminergic GPCR ligand design. Eur. J. Med. Chem. 2019, 179, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Porsolt, R.D.; Bertin, A.; Jalfre, M. Behavioral despair in mice: A primary screening test for antidepressants. Arch. Int. Pharmacodyn. Ther. 1977, 229, 327–336. [Google Scholar] [PubMed]
- Pytka, K.; Socala, K.; Rapacz, A.; Nieoczym, D.; Pieróg, M.; Gryboś, A.; Siwek, A.; Waszkielewicz, A.; Wlaź, P. HBK-14 and HBK-15, triple 5-HT1A, 5-HT7 and 5-HT3 antagonists with potent antidepressant- and anxiolytic-like properties, increase seizure threshold in various seizure tests in mice. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2017, 79, 378–385. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | Ar | m | Ki [nM] ± SEM | |
|---|---|---|---|---|
| α2 a | 5-HT7 b | |||
| 6 | 4-F-phenyl | 0 | 138 ± 44 | 35 ± 22 |
| 7 | 3-Cl-phenyl | 0 | 649 ± 62 | 64 ± 15 |
| 8 | 5-Cl,2-F-phenyl | 0 | 148 ± 23 | 30 ± 11 |
| 9 | 5-Cl,2-MeO-phenyl | 0 | 573 ± 56 | 86 ± 26 |
| 10 | 2,5-diMeO-phenyl | 0 | 244 ± 52 | 95 ± 18 |
| 11 | 1-naphthyl | 0 | 366 ± 46 | 50 ± 16 |
| 12 | 2-naphthyl | 0 | 200 ± 51 | 67 ± 21 |
| 13 | 4-isoquinolyl | 0 | 80 ± 42 | 91 ± 25 |
| 14 | 4-F-phenyl | 1 | 1194 ± 44 | 727 ± 65 |
| 15 | 3-Cl-phenyl | 1 | 743 ± 32 | 664 ± 82 |
| 16 | 5-Cl,2-F-phenyl | 1 | 154 ± 28 | 316 ± 73 |
| 17 | 5-Cl,2-MeO-phenyl | 1 | 1097 ± 78 | 488 ± 70 |
| 18 | 1-naphthyl | 1 | 907 ± 64 | 366 ± 69 |
| 19 | 4-isoquinolyl | 1 | 298 ± 24 | 387 ± 58 |
| Clonidine | 2.7 ± 0.3 | NT c | ||
| SB-267790 | NTc | 3 ± 0.5 | ||
| Mirtazapine | 112 d | 265 e | ||
| Compd | Ki [nM] | ||||
|---|---|---|---|---|---|
| α1 a | 5-HT1A b | 5-HT2A b | 5-HT6 b | D2 b | |
| 6 | 761 ± 86 | 221 ± 25 | 538 ± 38 | 839 ± 70 | 327 ± 29 |
| 8 | 1256 ± 101 | 260 ± 29 | 1420 ± 98 | 873 ± 91 | 388 ± 37 |
| 13 | 429 ± 66 | 260 ± 18 | 1422 ± 105 | 1123 ± 114 | 326 ± 25 |
| Compd | α2 | α2A | α2B | 5-HT7 | |
|---|---|---|---|---|---|
| Ki [nM] a | Kb [nM] b | Kb [nM] c | Ki [nM] d | Kb[nM] e | |
| 6 | 138 ± 44 | 12 ± 4 | 103 ± 25 | 35 ± 22 | 186 ± 37 |
| 8 | 148 ± 23 | 40 ± 11 | 142 ± 31 | 30 ± 11 | 141 ± 43 |
| 13 | 80 ± 42 | 31 ± 8 | 27 ± 13 | 91 ± 25 | 155 ± 30 |
| Yohimbine | 164 ± 55 | 0.81 ± 0.3 | 2.99 ± 0.7 | NT f | NT f |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canale, V.; Kotańska, M.; Dziubina, A.; Stefaniak, M.; Siwek, A.; Starowicz, G.; Marciniec, K.; Kasza, P.; Satała, G.; Duszyńska, B.; et al. Design, Sustainable Synthesis and Biological Evaluation of a Novel Dual α2A/5-HT7 Receptor Antagonist with Antidepressant-Like Properties. Molecules 2021, 26, 3828. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26133828
Canale V, Kotańska M, Dziubina A, Stefaniak M, Siwek A, Starowicz G, Marciniec K, Kasza P, Satała G, Duszyńska B, et al. Design, Sustainable Synthesis and Biological Evaluation of a Novel Dual α2A/5-HT7 Receptor Antagonist with Antidepressant-Like Properties. Molecules. 2021; 26(13):3828. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26133828
Chicago/Turabian StyleCanale, Vittorio, Magdalena Kotańska, Anna Dziubina, Matylda Stefaniak, Agata Siwek, Gabriela Starowicz, Krzysztof Marciniec, Patryk Kasza, Grzegorz Satała, Beata Duszyńska, and et al. 2021. "Design, Sustainable Synthesis and Biological Evaluation of a Novel Dual α2A/5-HT7 Receptor Antagonist with Antidepressant-Like Properties" Molecules 26, no. 13: 3828. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26133828