
Bioactive Abietane-Type Diterpenoid Glycosides from Leaves of Clerodendrum infortunatum (Lamiaceae)

 , , , , and
, , , , and 
Abstract
:
1. Introduction
2. Results and Discussion
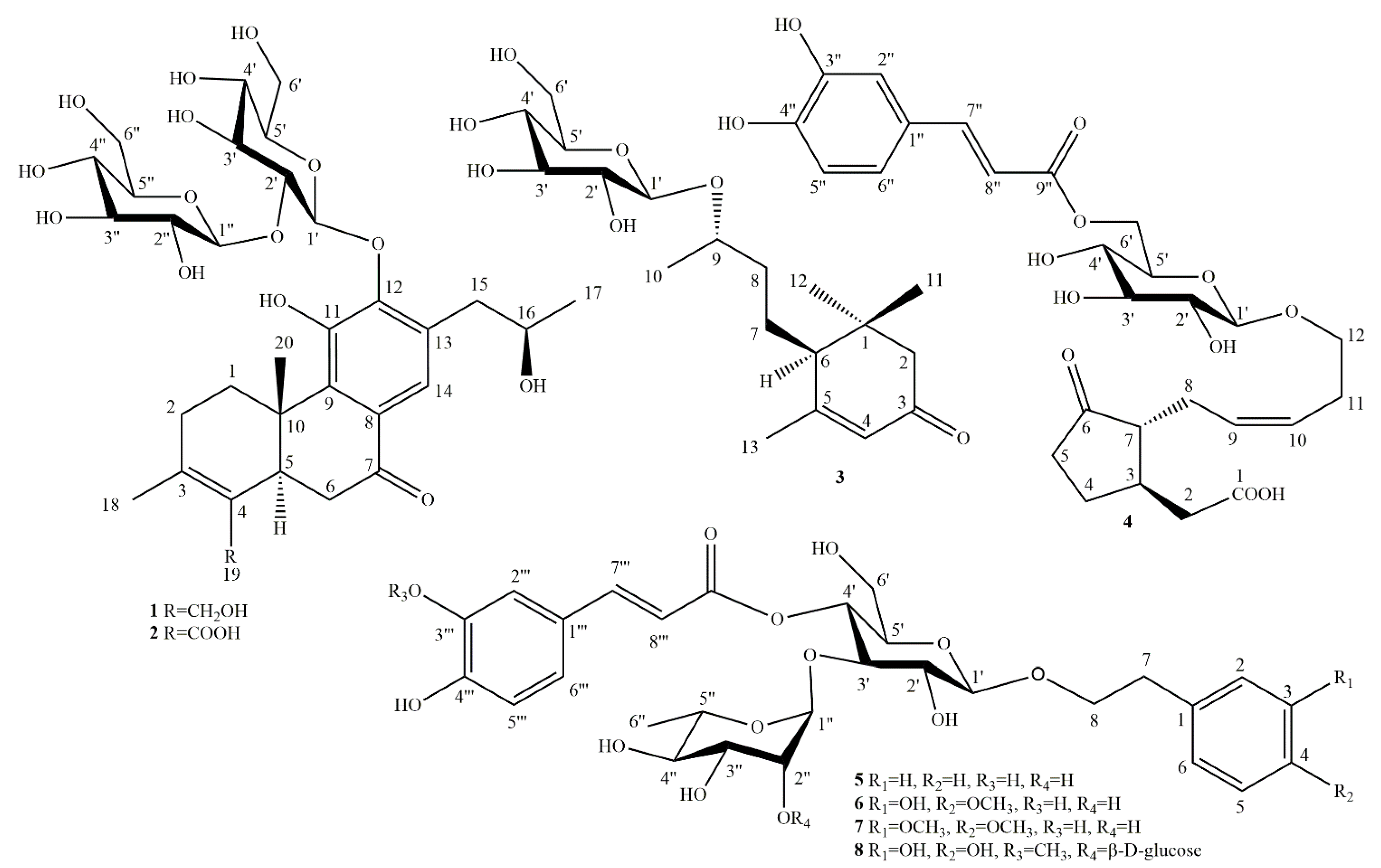
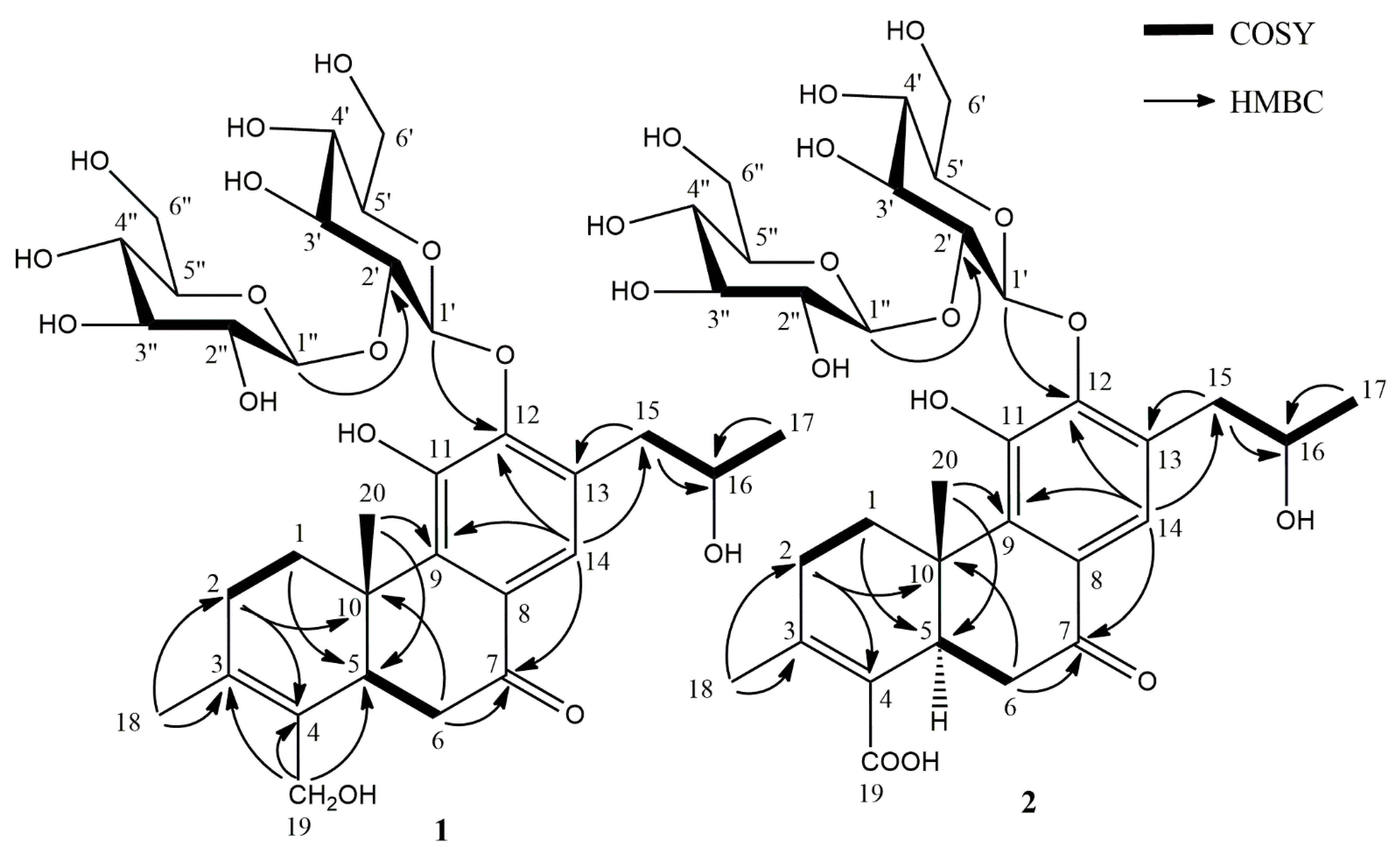
2.1. Phytochemical Investigation
2.2. α-Amylase and α-Glucosidase Inhibition
2.3. Cholinesterase Inhibitory Properties
2.4. Antiproliferative and Cytotoxic Activities
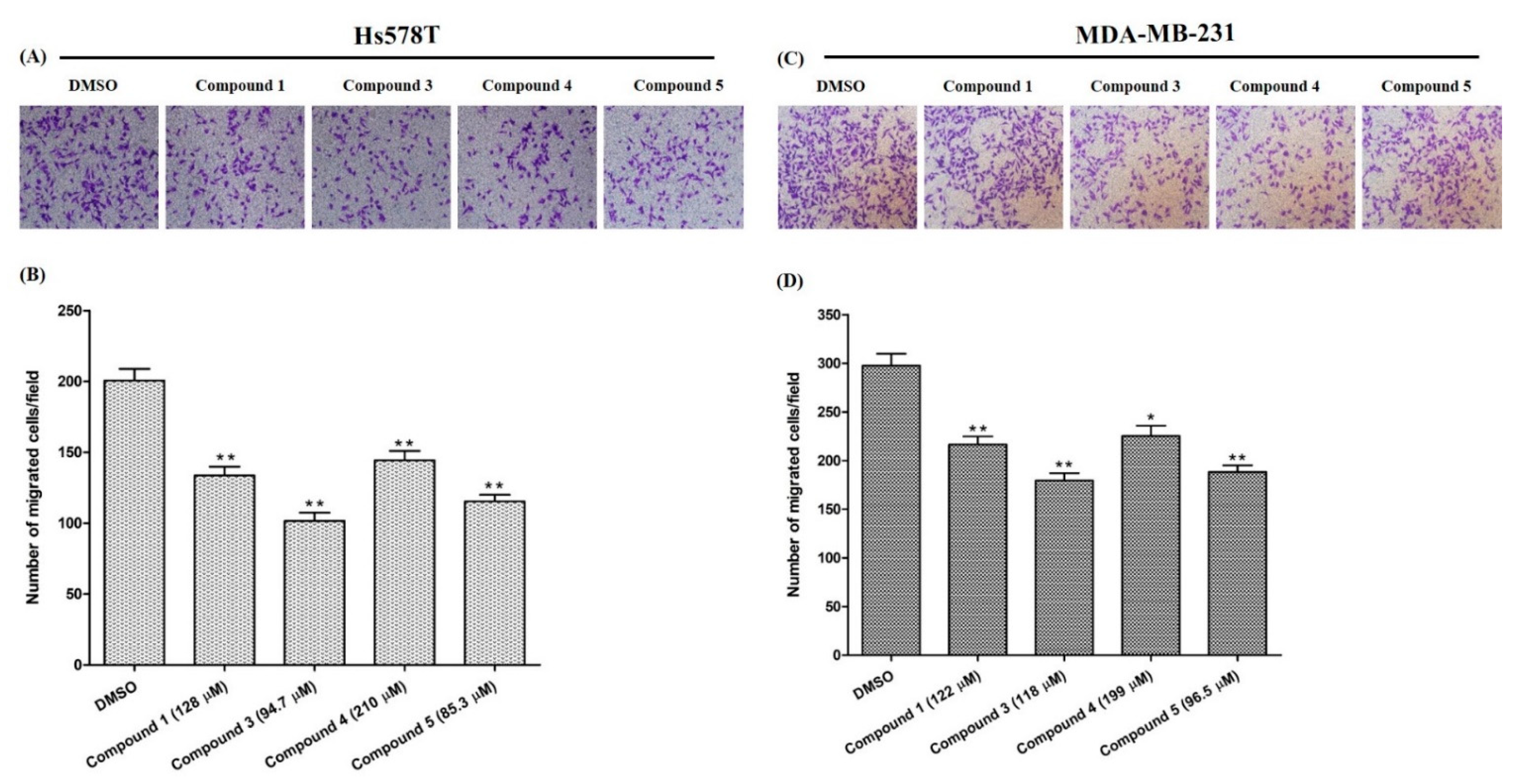
2.4.1. Effects on TNBC Cell Proliferation
2.4.2. Effects on Cell Migration
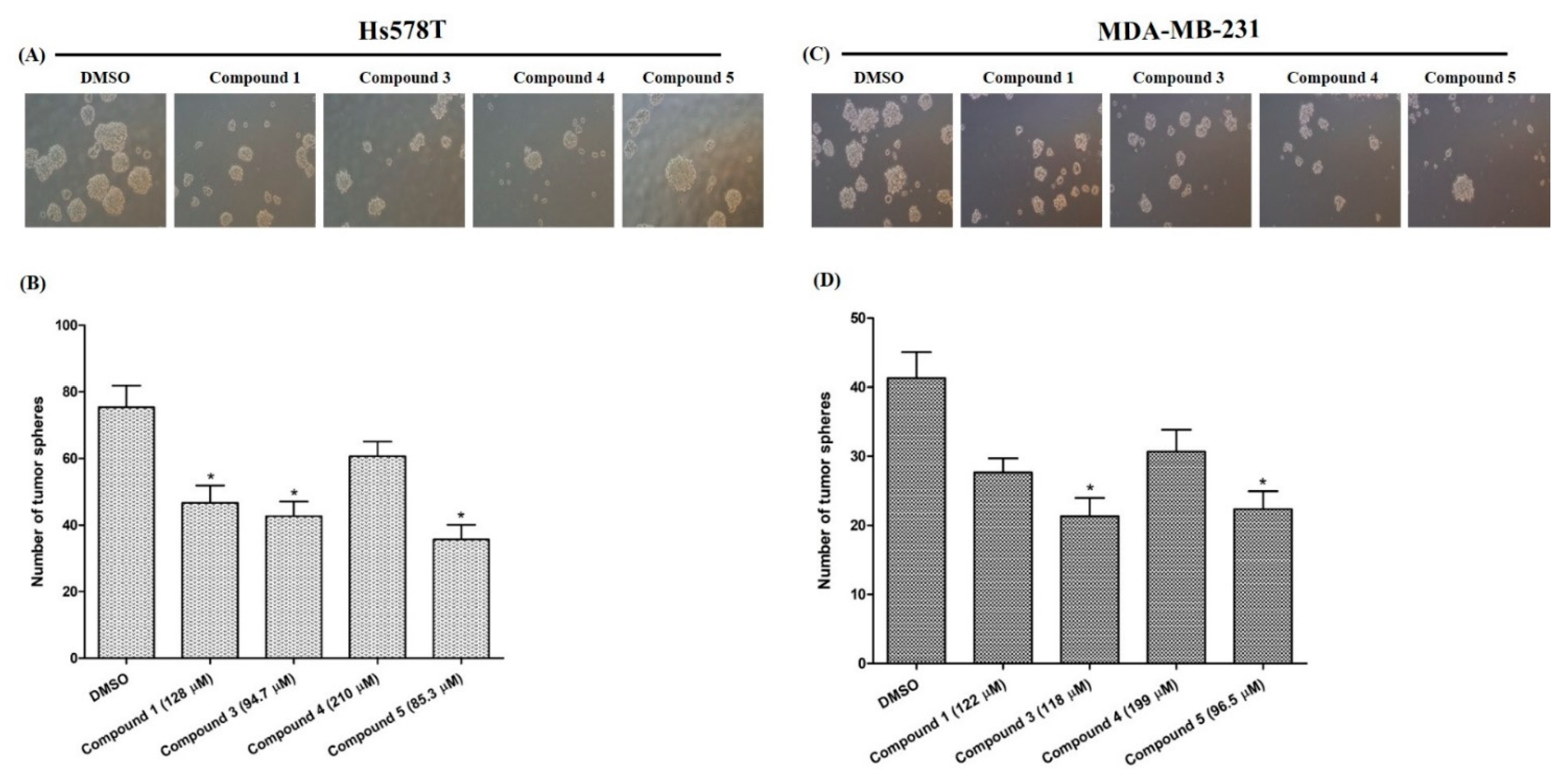
2.4.3. Effects on Tumor-Sphere Formation
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Chemicals
3.3. Plant Material
3.4. Extraction and Isolation
3.5. α-Amylase Inhibition Assay
3.6. α-Glucosidase Inhibition Assay
3.7. Determination of Cholinesterase Inhibitory Activities
3.8. Determination of Anticancer Activities
3.8.1. Cell Lines and Culture Condition
3.8.2. Colony Formation Assay
3.8.3. Cell Viability Assay
3.8.4. Transwell Cell Migration Assay
3.8.5. Tumor-Sphere Formation Assay
3.9. Determination of the Absolute Sugar Configuration
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Munir, A.A. A taxonomic revision of the genus Clerodendrum L. (Verbenaceae) in Australia. J. Adel. Bot. Gard. 1989, 11, 101–173. [Google Scholar]
- Verdcourt, B. Flora of Tropical East Africa—Verbenaceae; Polhill, R.M., Ed.; A.A. Balkema: Rotterdam, The Netherlands, 1992. [Google Scholar]
- Nandi, S.; Lyndem, M.L. Clerodendrum viscosum: Traditional uses, pharmacological activities and phytochemical constituents. Nat. Prod. Res. 2016, 30, 497–506. [Google Scholar] [CrossRef]
- Ghani, A. Medicinal Plants of Bangladesh, 2nd ed.; The Asiatic Society of Bangladesh: Dhaka, Bangladesh, 2003; pp. 1–398. [Google Scholar]
- Uddin, M.S. Traditional Knowledge of Medicinal Plants in Bangladesh. Nature Info. Electronic Database. Available online: https://www.natureinfo.com.bd/category/flora/medicinal-palnts (accessed on 12 January 2021).
- Lobo, R.; Punitha, I.S.R.; Rajendran, K.; Shirwaikar, A. Prelimenary study on the antisnake venom activity of alcoholic root extract of Clerodendrun viscosum in Naja naja venom. J. Nat. Prod. Sci. 2006, 129, 153–156. [Google Scholar]
- Pal, D.K.; Sannigrahi, S.; Mazumder, U.K. Analgesic and anticonvulsant effect of saponin isolated from the leaves of Clerodendrum infortunatum Linn in mice. Ind. J. Exp. Biol. 2009, 47, 743–747. [Google Scholar]
- Gupta, R.; Singh, H.K. Nootropic potential of Alternanthera sessilis and Clerodendrum infortunatum leaves on mice. Asian Pac. J. Trop. Dis. 2012, 2, 465–470. [Google Scholar] [CrossRef]
- Ghosh, G.; Sahoo, S.; Das, D.; Dubey, D.; Padhy, R.N. Antibacterial and antioxidant activities of methanol extract and fractions of Clerodendrum viscosum Vent. leaves. Ind. J. Nat. Prod. Resour. 2014, 5, 134–142. [Google Scholar]
- Swargiary, A.; Daimari, A.; Daimari, M.; Basumatary, N.; Narzary, E. Phytochemicals, antioxidant, and anthelmintic activity of selected traditional wild edible plants of lower Assam. Indian J. Pharmacol. 2016, 48, 418–423. [Google Scholar] [CrossRef]
- Shendge, A.K.; Basu, T.; Panja, S.; Chaudhuri, D.; Mandal, N. An ellagic acid isolated from Clerodendrum viscosum leaves ameliorates iron-overload induced hepatotoxicity in Swiss albino mice through inhibition of oxidative stress and the apoptotic pathway. Biomed. Pharmacother. 2018, 106, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.J.; Çiçek, S.S.; Willer, J.; Shulha, O.; Abdalla, M.A.; Sönnichsen, F.; Girreser, U.; Zidorn, C. Phenylpropanoid and flavonoid glycosides from the leaves of Clerodendrum infortunatum (Lamiaceae). Biochem. Syst. Ecol. 2020, 92, 104131. [Google Scholar] [CrossRef]
- Buford, T.W. Hypertension and aging. Ageing Res. Rev. 2016, 26, 96–111. [Google Scholar] [CrossRef] [Green Version]
- Belikov, A.V. Age-related diseases as vicious cycles. Ageing Res. Rev. 2019, 49, 11–26. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Yang, W.; Chen, X.; Li, Y.; Guo, S.; Wang, Z.; Yu, X. Advances in pharmacological activities of terpenoids. Nat. Prod. Commun. 2020, 15, 1934578X20903555. [Google Scholar] [CrossRef] [Green Version]
- Panda, P.; Appalashetti, M.; Judeh, Z.M.A. Phenylpropanoid sucrose esters: Plant derived natural products as potential leads for new therapeutics. Curr. Med. Chem. 2011, 18, 3234–3251. [Google Scholar] [CrossRef]
- Neelam; Khatkar, A.; Sharma, K.K. Phenylpropanoids and its derivatives: Biological activities and its role in food, pharmaceutical and cosmetic industries. Crit. Rev. Food Sci. Nutr. 2020, 60, 2655–2675. [Google Scholar] [CrossRef]
- González, M.A. Aromatic abietane diterpenoids: Their biological activity and synthesis. Nat. Prod. Rep. 2015, 32, 684–704. [Google Scholar] [CrossRef] [PubMed]
- Pu, D.B.; Wang, T.; Zhang, X.J.; Gao, J.B.; Zhang, R.H.; Li, X.N.; Wang, Y.M.; Li, X.L.; Wang, H.Y.; Xiao, W.L. Isolation, identification and bioactivities of abietane diterpenoids from Premna szemaoensis. RSC Adv. 2018, 8, 6425–6435. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Ishikawa, Y.; Saruul, E.; Selenge, E.; Sasaki, K.; Umehara, K.; Yoshizaki, F.; Batkhuu, J. Abietane-type diterpenoids from the roots of Caryopteris mongolica and their cholinesterase inhibitory activities. Phytochemistry 2016, 130, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Matsunami, K.; Otsuka, H.; Takeda, Y. Structural revisions of blumenol C glucoside and byzantionoside B. Chem. Pharm. Bull. 2010, 58, 438–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, U.; Ryu, S.M.; Lee, D.; Seo, E.K. Chemical constituents of the leaves of Brassica oleracea var. acephala. Chem. Nat. Compd. 2018, 54, 1023–1026. [Google Scholar] [CrossRef]
- Devaraj, S.; Yip, Y.M.; Panda, P.; Ong, L.L.; Wong, P.W.K.; Zhang, D.; Judeh, Z. Cinnamoyl sucrose esters as alpha glucosidase inhibitors for the treatment of diabetes. Molecules 2021, 26, 469. [Google Scholar] [CrossRef] [PubMed]
- Etsassala, N.G.; Cupido, C.N.; Iwuoha, E.I.; Hussein, A.A. Abietane diterpenes as potential candidates for the management of type 2 diabetes. Curr. Pharm. Des. 2020, 26, 2885–2891. [Google Scholar] [CrossRef] [PubMed]
- Güvenalp, Z.; Özbek, H.; Dursunoğlu, B.; Yuca, H.; Gözcü, S.; Çil, Y.M.; Demirezer, Ö.L. α-Amylase and α-glucosidase inhibitory activities of the herbs of Artemisia dracunculus L. and its active constituents. Med. Chem. Res. 2017, 26, 3209–3215. [Google Scholar] [CrossRef]
- Santoro, V.; Parisi, V.; D’Ambola, M.; Sinisgalli, C.; Monné, M.; Milella, L.; Tommasi, N.D. Chemical profiling of Astragalus membranaceus roots (Fish.) bunge herbal preparation and evaluation of its bioactivity. Nat. Prod. Commun. 2020, 15, 1934578X20924152. [Google Scholar] [CrossRef]
- Pinho, B.R.; Ferreres, F.; Valentão, P.; Andrade, P.B. Nature as a source of metabolites with cholinesterase-inhibitory activity: An approach to Alzheimer’s disease treatment. J. Pharm. Pharmacol. 2013, 65, 1681–1700. [Google Scholar] [CrossRef]
- Robles, A.J.; Du, L.; Cichewicz, R.H.; Mooberry, S.L. Maximiscin induces DNA damage, activates DNA damage response pathways, and has selective cytotoxic activity against a subtype of triple-negative breast cancer. J. Nat. Prod. 2016, 79, 1822–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirshfield, K.M.; Ganesan, S. Triple-negative breast cancer: Molecular subtypes and targeted therapy. Curr. Opin. Obstet. Gynecol. 2014, 26, 34–40. [Google Scholar] [CrossRef]
- Chudzik, M.; Korzonek-Szlacheta, I.; Król, W. Triterpenes as potentially cytotoxic compounds. Molecules 2015, 20, 1610–1625. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Lu, J.J.; Huang, M.Q.; Bao, J.L.; Chen, X.P.; Wang, Y.T. Terpenoids: Natural products for cancer therapy. Expert Opin. Invest. Drugs 2012, 21, 1801–1818. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T. Protection against cancer by plant phenylpropenoids: Induction of mammalian anticarcinogenic enzymes. Mini-Rev. Med. Chem. 2002, 2, 595–610. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Shah, M.A. Targeting the cell cycle: A new approach to cancer therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef]
- Geiger, T.R.; Peeper, D.S. Metastasis mechanisms. Biochim. Biophys. Acta 2009, 1796, 293–308. [Google Scholar] [CrossRef]
- Aznavoorian, S.; Murphy, A.N.; Stetler-Stevenson, W.G.; Liotta, L.A. Molecular aspects of tumor cell invasion and metastasis. Cancer 1993, 71, 1368–1383. [Google Scholar] [CrossRef]
- Zhu, Z.W.; Chen, L.; Liu, J.X.; Huang, J.W.; Wu, G.; Zheng, Y.F.; Yao, K.T. A novel three-dimensional tumorsphere culture system for the efficient and low-cost enrichment of cancer stem cells with natural polymers. Exp. Ther. Med. 2018, 15, 85–92. [Google Scholar] [CrossRef]
- Hooker, J.D. The Flora of British India; L. Reeve and Company: London, UK, 1954. [Google Scholar]
- Faraone, I.; Rai, D.K.; Russo, D.; Chiummiento, L.; Fernandez, E.; Choudhary, A.; Milella, L. Antioxidant, antidiabetic, and anticholinesterase activities and phytochemical profile of Azorella glabra Wedd. Plants 2019, 8, 265. [Google Scholar] [CrossRef] [Green Version]
- Lelario, F.; De Maria, S.; Rivelli, A.R.; Russo, D.; Milella, L.; Bufo, S.A.; Scrano, L. A complete survey of glycoalkaloids using LC-FTICR-MS and IRMPD in a commercial variety and a local landrace of eggplant (Solanum melongena L.) and their anticholinesterase and antioxidant activities. Toxins 2019, 11, 230. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.A.; Islam, M.A.U. Pleurotus highking mushroom induces apoptosis by altering the balance of proapoptotic and antiapoptotic genes in breast cancer cells and inhibits tumor sphere formation. Medicina 2019, 55, 716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, M.A.; Reza, A.S.M.A.; Nasrin, M.S.; Rahman, M.A. Pleurotus highking mushrooms potentiate antiproliferative and antimigratory activity against triple-negative breast cancer cells by suppressing Akt signaling. Integr. Cancer Ther. 2020, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Yu, C.C.; Wang, B.Y.; Chang, W.W. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget 2016, 7, 1215–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakeney, A.B.; Harris, P.J.; Henry, R.J.; Stone, B.A. A simple and rapid preparation of alditol acetates for monosaccharide analysis. Carbohydr. Res. 1983, 113, 291–299. [Google Scholar] [CrossRef]
- Leontein, K.; Lindberg, B.; Lōnngren, J. Assignment of absolute configuration of sugars by GLC of their acetylated glycosides formed from chiral alcohols. Carbohydr. Res. 1978, 62, 359–362. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 1 b | 2 a | 2 b | 2 c |
|---|---|---|---|---|---|
| δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | |
| 1 | 1.54, td (12.5, 6.5) | 1.43, td (12.5, 6.5) | 1.58, td (12.5, 6.5) | 1.45, m | 1.40, m |
| 3.46, m | 3.31, dd (13.0, 6.5) | 3.48, m d | 3.34, m | 3.16, m | |
| 2 | 2.11, dd (18.5, 6.0) | 2.00, dd (18, 6.0) | 2.06, m | 1.23, m | 1.98, m |
| 2.32, m | 2.20, t (10.0) | 2.29, m | 2.14, m | ||
| 5 | 2.94, br d (15.5) | 2.83, br d (15.5) | 3.08, m | 2.53, m | 2.90, m |
| 6 | 2.59, dd (16.5, 15.5) | 2.94, m d | 2.39, m | ||
| 3.01, dd (17.0, 3.0) | 2.50, m d | 2.50, m | |||
| 14 | 7.47, s | 7.42, s | 7.41, s | 7.41, s | 7.35, s |
| 15 | 2.67, dd (13.5, 7.0) | 2.66, dd (13.5, 6.0) | 2.67, m d | 2.67, dd (13.5, 6.0) | 2.62, dd (13.5, 6.0) |
| 3.25, dd (13.5, 6.0) | 2.92, m d | 3.27, dd (13.5, 6.5) | 2.93, dd (13.5, 6.5) | 2.93, dd (13.5, 6.5) | |
| 16 | 4.17, dd (13.0, 6.5) | 3.97, dd (12.5, 6.0) | 4.16, dd (13.0, 6.5) | 3.97, dd (12.5, 6.0) | 3.99, dd (12.5, 6.0) |
| 17 | 1.10, d (6.0) | 1.00, d (6.0) | 1.00, d (6.0) | 1.00, d (6.0) | 0.97, d (6.0) |
| 18 | 1.78, s | 1.70, s | 1.75, s | 1.70, s | 1.60, s |
| 19 | 4.08, d (12.0) | 3.85, d (12.0) | |||
| 4.28, d (12.0) | 4.13, d (12.0) | ||||
| 20 | 1.28, s | 1.18, s | 1.32, s | 1.21, s | 1.10, s |
| 1′ | 4.72, d (8.0) | 4.64, d (8.0) | 4.87, d (8.0) | 4.74, d (8.0) | 4.74, m d |
| 2′ | 3.88, dd (9.0, 8.0) | 3.70, m d | 3.88, dd (17.0, 8.0) | 3.71, m d | 3.79, m d |
| 3′ | 3.41, m d | 3.50, m d | 3.42, m d | 3.51, m d | 3.35, m d |
| 4′ | 3.47, m d | 3.23, m d | 3.48, m d | 3.23, m d | 3.39, m d |
| 5′ | 3.37, m d | 3.10, m d | 3.39, m d | 3.11, m d | 3.26, m d |
| 6′ | 3.67, m d | 3.42, dd (12.0, 6.0) | 3.68, dd (12.0, 6.0) | 3.44, dd (12.0, 6.0) | 3.49, dd (12.0, 6.0) |
| 3.83, m d | 3.66, m d | 3.82, dd (6.0, 1.5) | 3.65, m d | 3.62, m d | |
| 1″ | 4.87, d (8.0) | 4.75, d (8.0) | 4.73, d (8.0) | 4.64, d (8.0) | 4.71, m d |
| 2″ | 3.38, m d | 3.12, m d | 3.40, m d | 3.12, m d | 3.25, m d |
| 3″ | 3.66, m d | 3.20, m d | 3.67, m d | 3.21, m d | 3.58, m d |
| 4″ | 3.30, m d | 3.24, m d | 3.36, m d | 3.23, m d | 3.28, m d |
| 5″ | 3.36, m d | 3.18, m d | 3.31, m d | 3.19, m d | 3.21, m d |
| 6″ | 3.72, dd (12.0, 5.5) | 3.47, m d | 3.72, dd (12.0, 5.0) | 3.48, m d | 3.59, m d |
| 3.83, m d | 3.68, m d | 3.84, dd (6.0, 2.0) | 3.68, m d | 3.64, m d | |
| Position | 1 a | 1 b | 2 a | 2 b | 2 c |
|---|---|---|---|---|---|
| δC, Type | δC, Type | δC, Type | δC, Type | δC, Type | |
| 1 | 32.7, CH2 | 31.1, CH2 | 32.5, CH2 | 30.7, CH2 | 32.3, CH2 |
| 2 | 31.1, CH2 | 29.7, CH2 | 29.7, CH2 | 29.0, CH2 | 29.6, CH2 |
| 3 | 130.2, C | 129.2, C | 130.5, C | 129.4, C | 131.2, C |
| 4 | 132.5, C | 129.9, C | 132.5, C | 129.6, C | 133.3, C |
| 5 | 44.4, CH | 42.7, CH | 43.2, CH | 40.4, CH | 42.5, CH |
| 6 | 38.1, CH2 | 36.6, CH2 | 38.6, CH2 | 36.9, CH2 | 38.9, CH2 |
| 7 | 200.9, C | 197.8, C | 200.3, C | 197.0, C | 202.9, C |
| 8 | 129.5, C | 128.3, C | 128.4, C | 128.4, C | 130.4, C |
| 9 | 139.7, C | 137.2, C | 139.6, C | 136.8, C | 140.5, C |
| 10 | 39.0, C | 37.3, C | 39.1, C | 37.5, C | 38.7, C |
| 11 | 149.4, C | 147.8, C | 149.3, C | 147.8, C | 149.1, C |
| 12 | 150.2, C | 148.4, C | 150.1, C | 148.4, C | 150.4, C |
| 13 | 133.8, C | 131.4, C | 134.6, C | 131.6, C | 133.0, C |
| 14 | 122.5, CH | 120.7, CH | 122.7, CH | 121.0, CH | 123.4, CH |
| 15 | 41.1, CH2 | 39.2, CH2 | 41.1, CH2 | 39.2, CH2 | 40.0, CH2 |
| 16 | 68.1, CH | 65.5, CH | 68.1, CH | 65.5, CH | 68.6, CH |
| 17 | 22.7, CH3 | 23.3, CH3 | 22.6, CH3 | 23.3, CH3 | 23.4, CH3 |
| 18 | 19.0, CH3 | 18.8, CH3 | 20.5, CH3 | 20.1, CH3 | 21.2, CH3 |
| 19 | 59.2, CH2 | 57.5, CH2 | ---- | 166.2, COOH | 170.7, COOH |
| 20 | 15.7, CH3 | 15.2, CH3 | 16.1, CH3 | 15.3, CH3 | 16.8, CH3 |
| 1′ | 105.5, CH | 103.6, CH | 105.3, CH | 103.6, CH | 104.8, CH |
| 2′ | 82.7, CH | 80.8, CH | 82.7, CH | 80.7, CH | 82.0, CH |
| 3′ | 77.8, CH | 76.1, CH | 77.8, CH | 76.1, CH | 77.1, CH |
| 4′ | 70.8, CH | 69.5, CH | 70.8, CH | 69.5, CH | 70.4, CH |
| 5′ | 71.4, CH | 69.9, CH | 71.4, CH | 69.9, CH | 71.0, CH |
| 6′ | 62.6, CH2 | 61.1, CH2 | 62.6, CH2 | 61.1, CH2 | 62.1, CH2 |
| 1″ | 105.4, CH | 103.6, CH | 105.5, CH | 103.7, CH | 104.9, CH |
| 2″ | 75.7, CH | 74.1, CH | 75.7, CH | 74.1, CH | 75.2, CH |
| 3″ | 77.9, CH | 76.2, CH | 77.9, CH | 76.2, CH | 77.2, CH |
| 4″ | 78.6, CH | 77.5, CH | 78.6, CH | 77.5, CH | 77.9, CH |
| 5″ | 78.5, CH | 77.4, CH | 78.6, CH | 77.4, CH | 77.8, CH |
| 6″ | 62.3, CH2 | 60.9, CH2 | 62.2, CH2 | 60.8, CH2 | 61.7, CH2 |
| Position | Szemaoenoid A | Szemaoenoid C | E | Compound 1 | Compound 2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | δC | δH | δC | δH | |
| 15 | 40.9 | 2.71, 3.20 | 33.6 | 2.87, 3.17 | 40.4 | 2.70, 2.82 | 41.1 | 2.67, 3.25 | 41.1 | 2.67, 3.27 |
| 16 | 68.3 | 4.10 | 68.3 | 4.15 | 68.7 | 4.04 | 68.1 | 4.17 | 68.1 | 4.16 |
| 17 | 22.8 | 1.12 | 22.9 | 1.12 | 23.3 | 1.15 | 22.7 | 1.10 | 22.6 | 1.00 |
| Compound | IC50 (µM) | |||
|---|---|---|---|---|
| α-Amylase | α-Glucosidase | AChE | BChE | |
| 1 | 18.5 ± 0.6 b | 24.6 ± 0.2 a | 191 ± 10.2 a | >1000 |
| 2 | 64.6 ± 7.1 c | 78.3 ± 3 a | 139 ± 7.2 a | >1000 |
| 3 | 284 ± 13.2 d | >1000 | >1000 | >1000 |
| 4 | 13.0 ± 1.3 a,b | >1000 | >1000 | >1000 |
| 5 | 24.9 ± 0.4 b | 96 ± 10.5 a | 160 ± 12.2 a | >1000 |
| 6 | 3.4 ± 0.2 a | 55.8 ± 0.2 a | >1000 | >1000 |
| 7 | 221 ± 24.5 d | >1000 | >1000 | >1000 |
| 8 | 19.8 ± 0.50 b | >1000 | 178 ± 11.3 a | >1000 |
| acarbose | 5.9 ± 0.1 | 665 ± 42 | ||
| galanthamine | 2.9 ± 0.4 | 22.5 ± 1.9 | ||
| Compounds | IC50 (μM) | |
|---|---|---|
| Hs578T | MDA-MB-231 | |
| 1 | 128 ± 2.2 | 122 ± 1.2 |
| 3 | 94.7 ± 1.3 | 118 ± 3.3 |
| 4 | 210 ± 5.1 | 199 ± 3.1 |
| 5 | 85.3 ± 2.4 | 96.5 ± 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uddin, M.J.; Russo, D.; Haque, M.A.; Çiçek, S.S.; Sönnichsen, F.D.; Milella, L.; Zidorn, C. Bioactive Abietane-Type Diterpenoid Glycosides from Leaves of Clerodendrum infortunatum (Lamiaceae). Molecules 2021, 26, 4121. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144121
Uddin MJ, Russo D, Haque MA, Çiçek SS, Sönnichsen FD, Milella L, Zidorn C. Bioactive Abietane-Type Diterpenoid Glycosides from Leaves of Clerodendrum infortunatum (Lamiaceae). Molecules. 2021; 26(14):4121. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144121
Chicago/Turabian StyleUddin, Md. Josim, Daniela Russo, Md. Anwarul Haque, Serhat Sezai Çiçek, Frank D. Sönnichsen, Luigi Milella, and Christian Zidorn. 2021. "Bioactive Abietane-Type Diterpenoid Glycosides from Leaves of Clerodendrum infortunatum (Lamiaceae)" Molecules 26, no. 14: 4121. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144121