
The HDAC/HSP90 Inhibitor G570 Attenuated Blue Light-Induced Cell Migration in RPE Cells and Neovascularization in Mice through Decreased VEGF Production

 , , ,
, , ,
Abstract
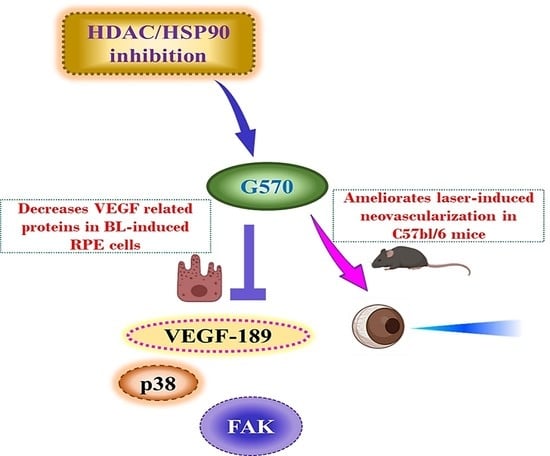
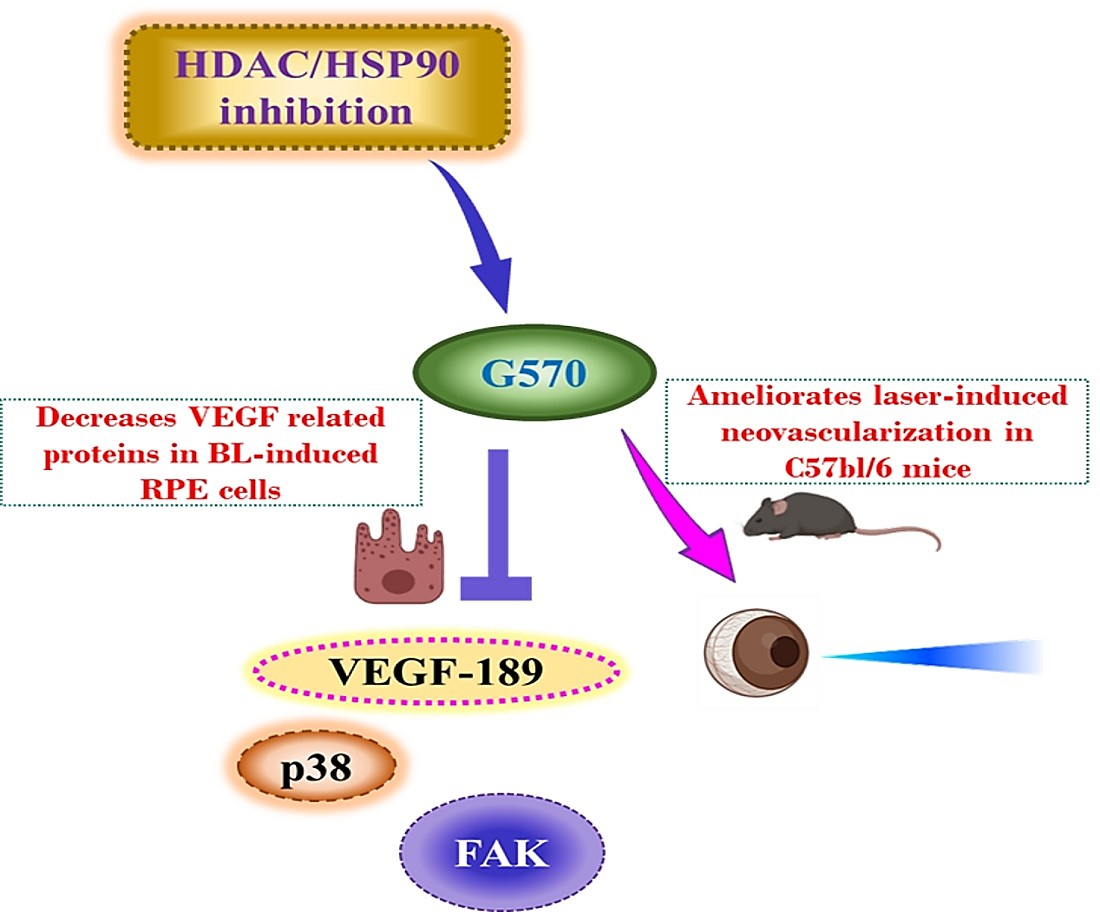
:
1. Introduction
2. Results
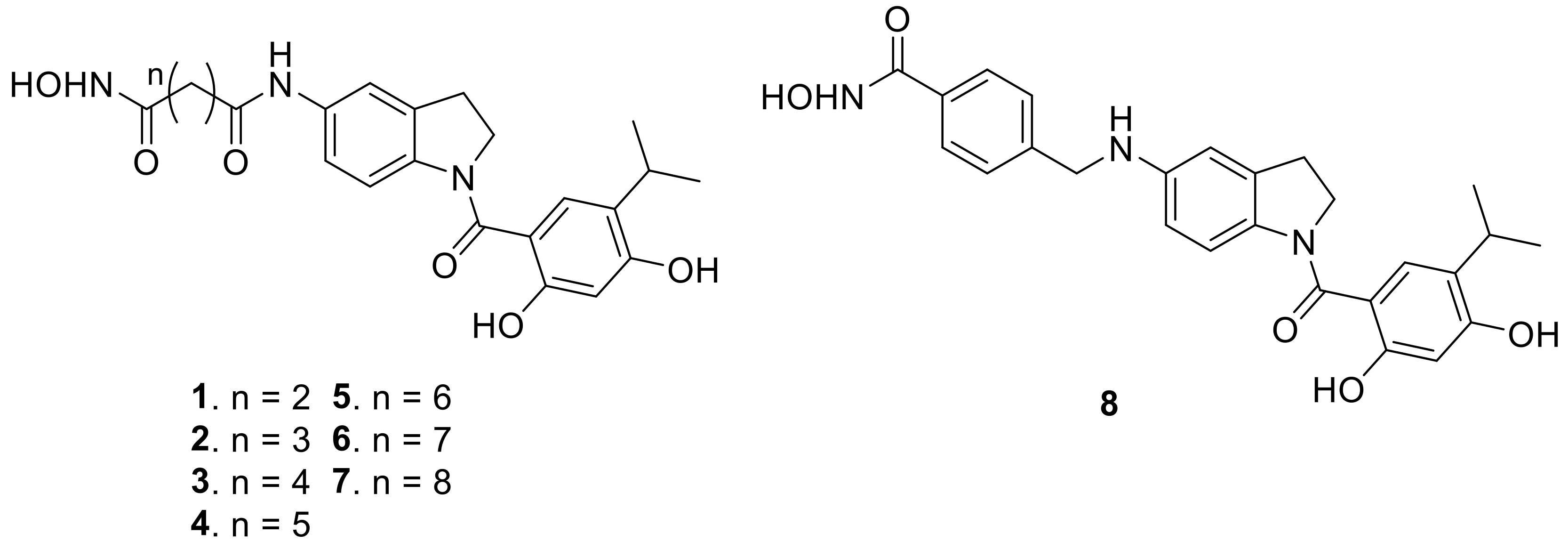
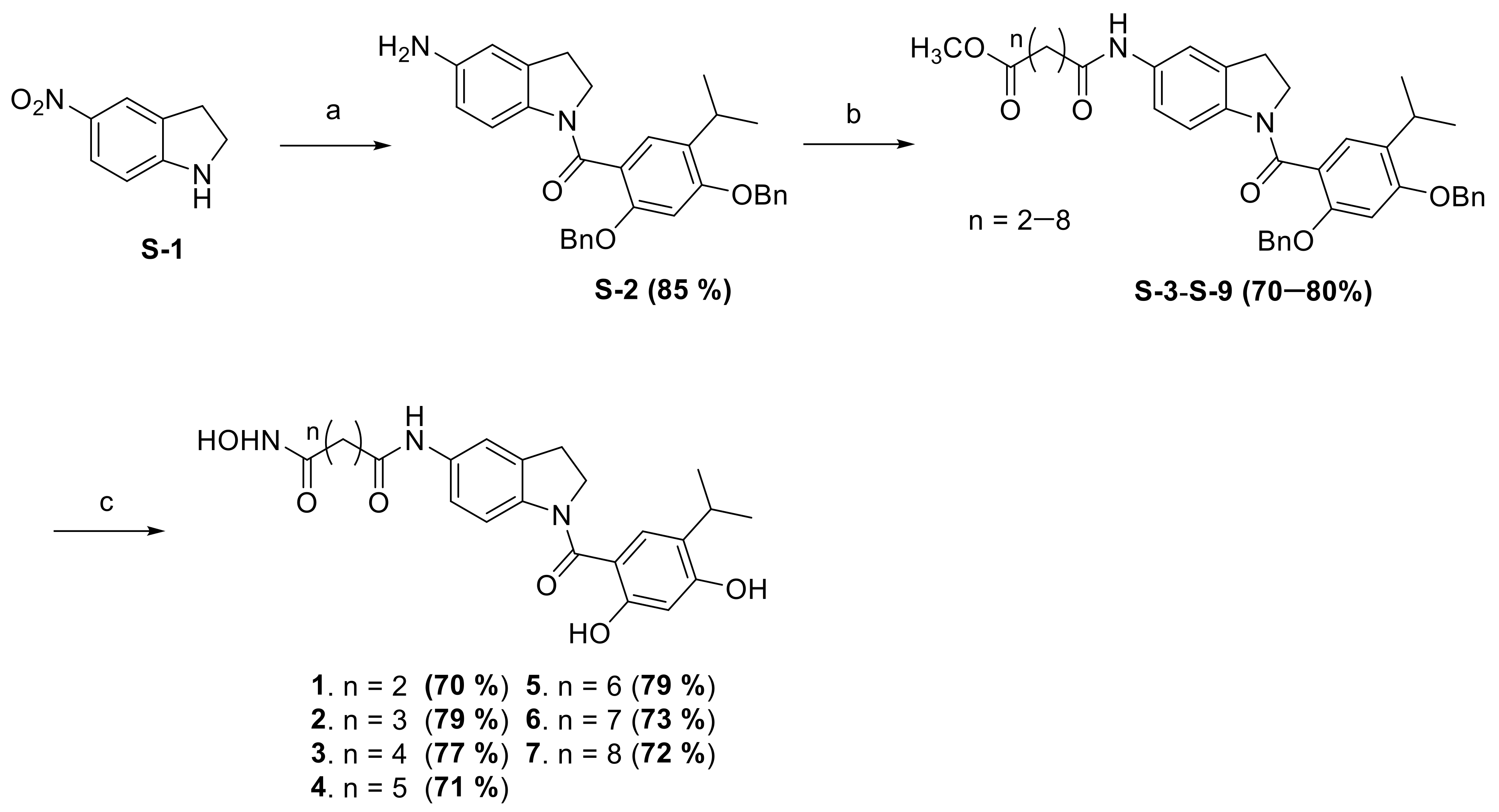
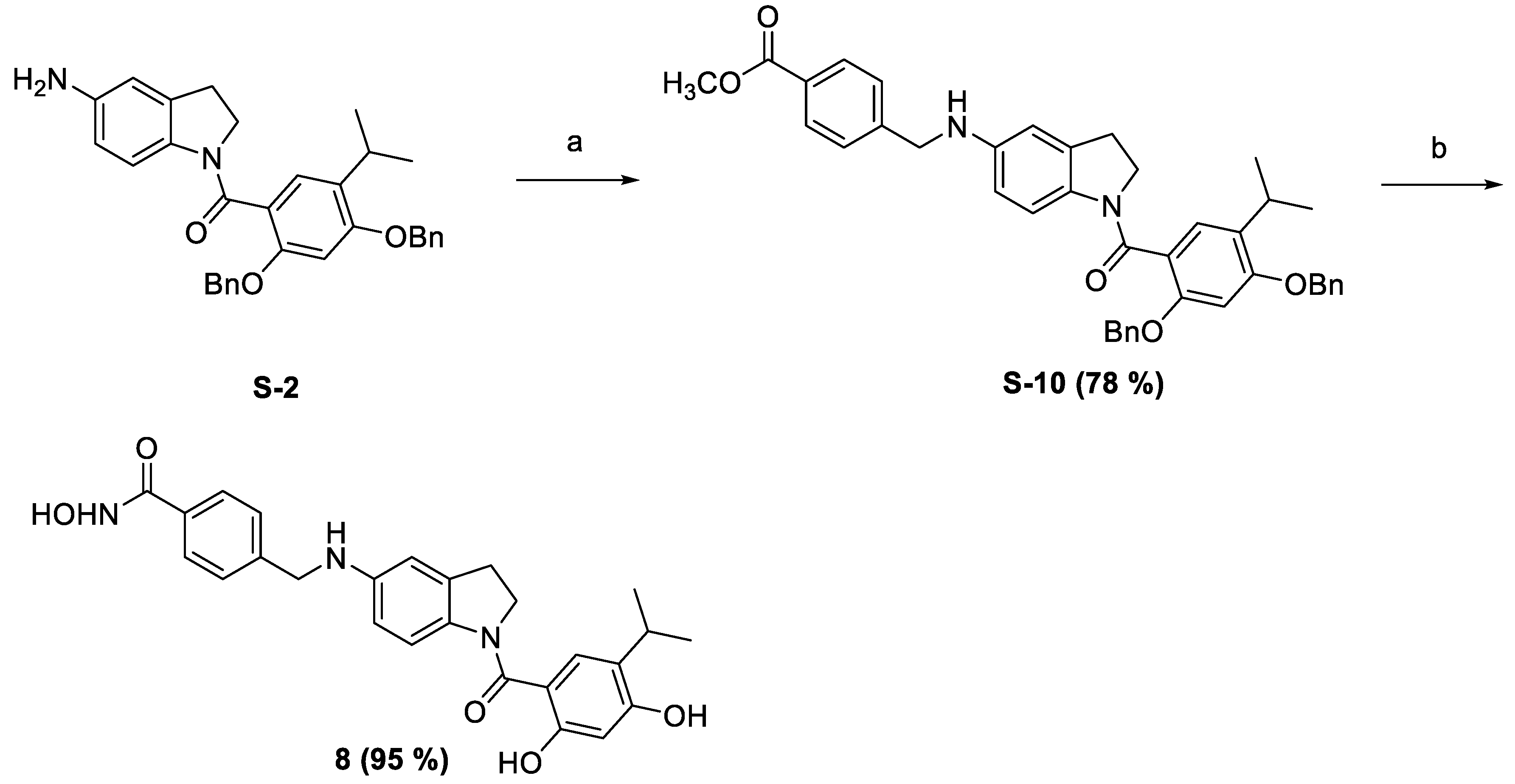
2.1. Synthesis of the Small Molecules
2.2. Inhibitory Activity of the Synthesized Small Molecules
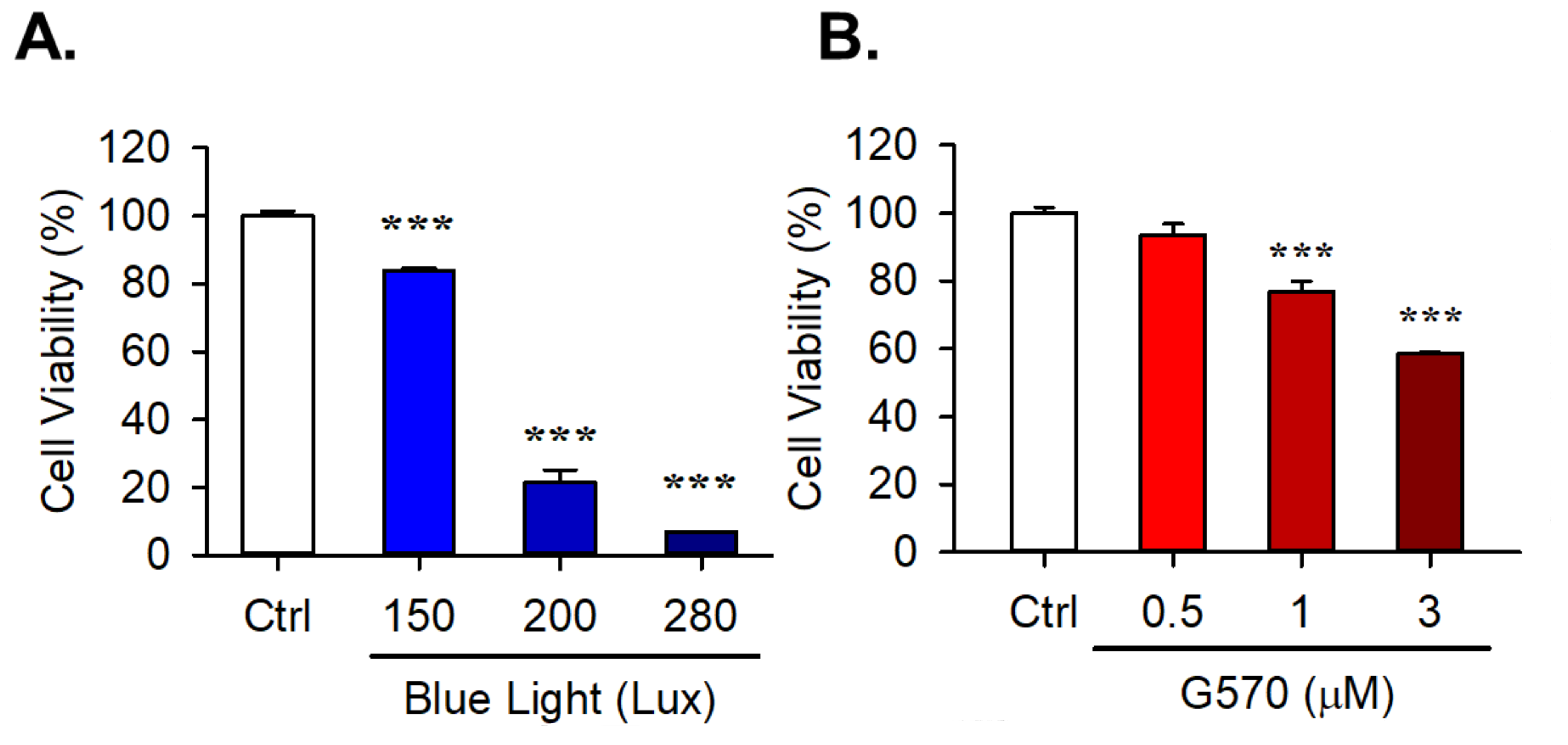
2.3. Effect of G570 and Blue Light (BL) Exposure on the Viability of ARPE-19 Cells
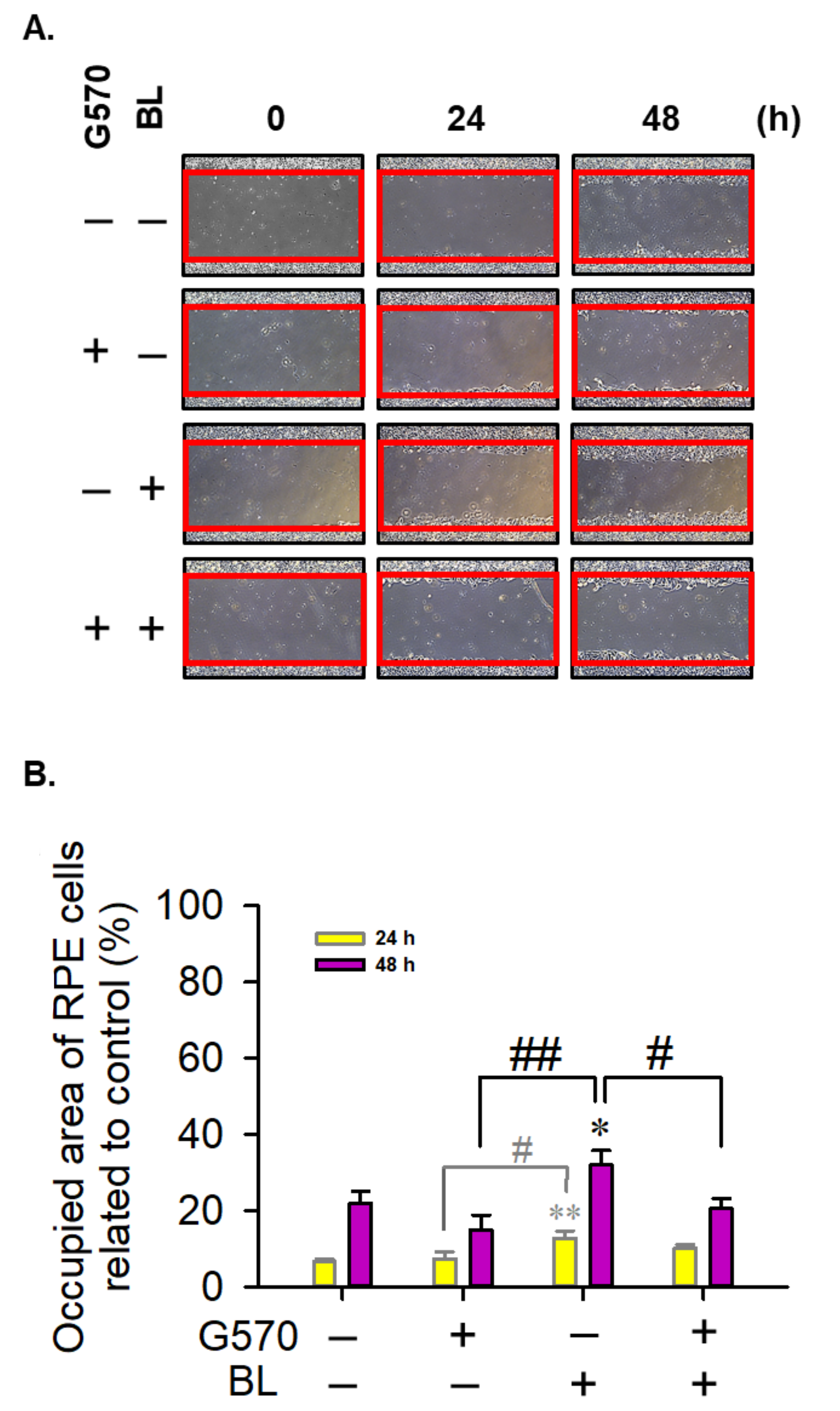
2.4. Effect of G570 on BL-Induced Migration in ARPE-19 Cells
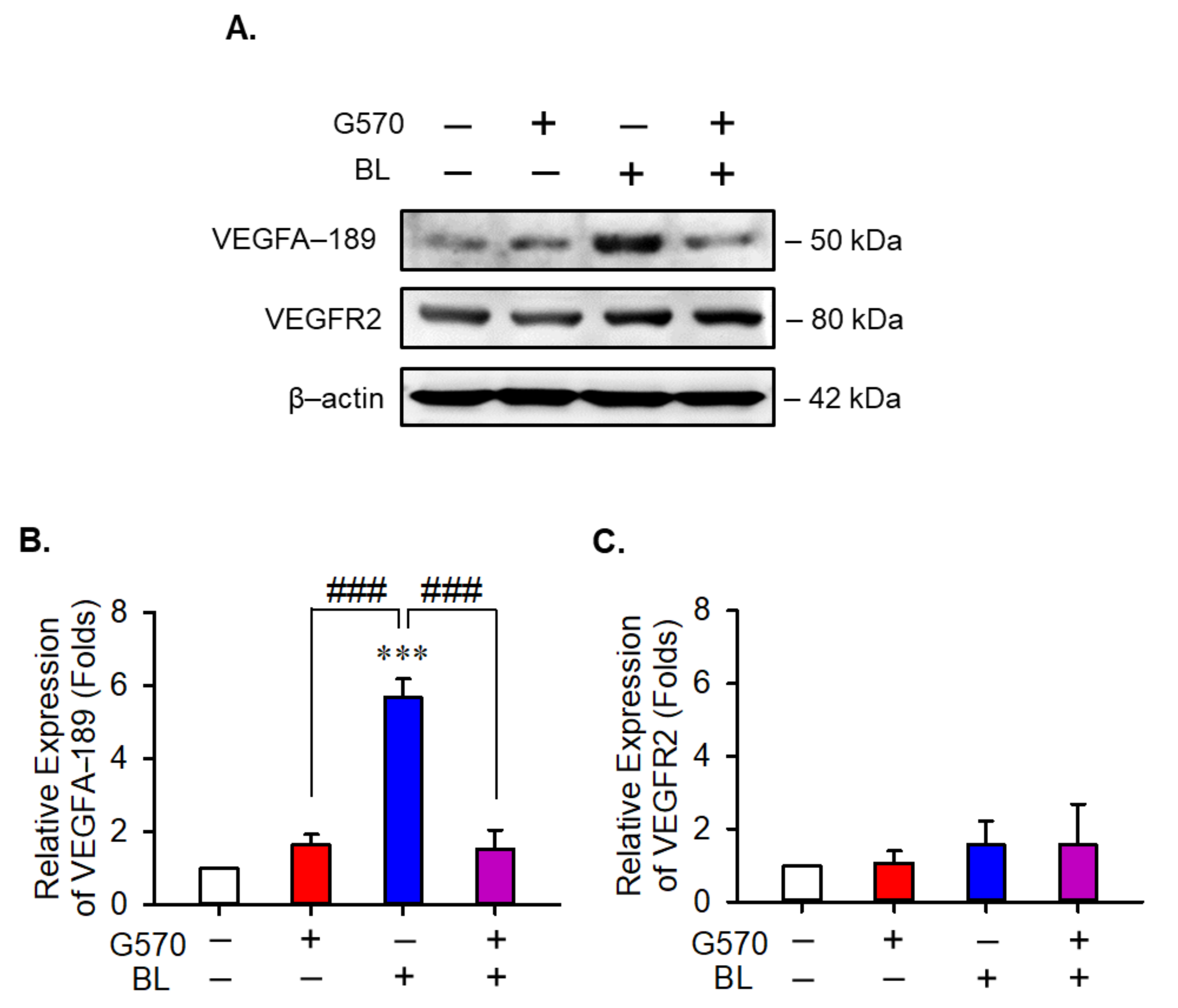
2.5. Effect of G570 on VEGF-189 and Its Receptor in BL-Exposed ARPE-19 Cells
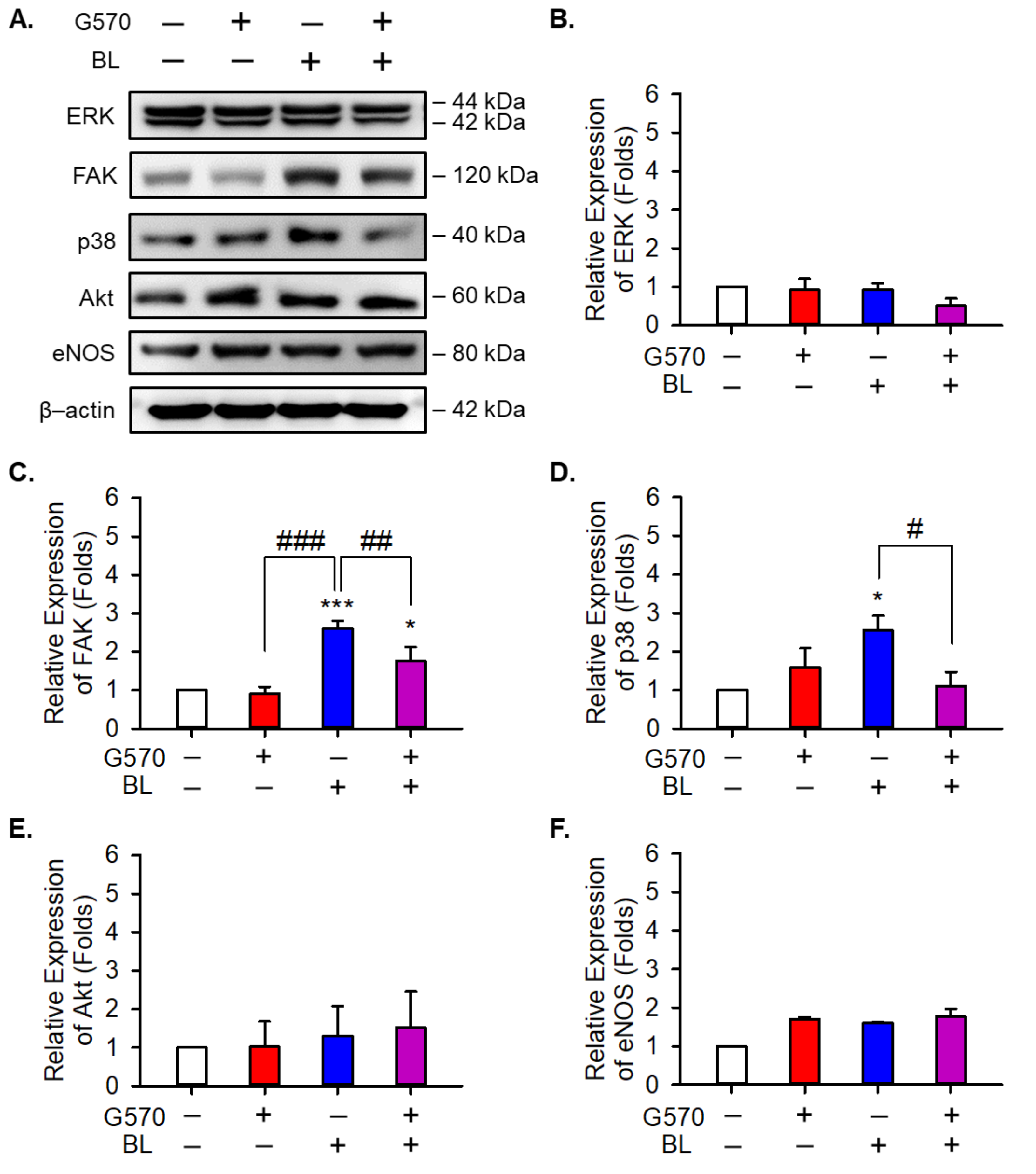
2.6. Effect of G570 on the Cell Migration and Proliferation Pathway of Angiogenesis in BL-Exposed ARPE-19 Cells
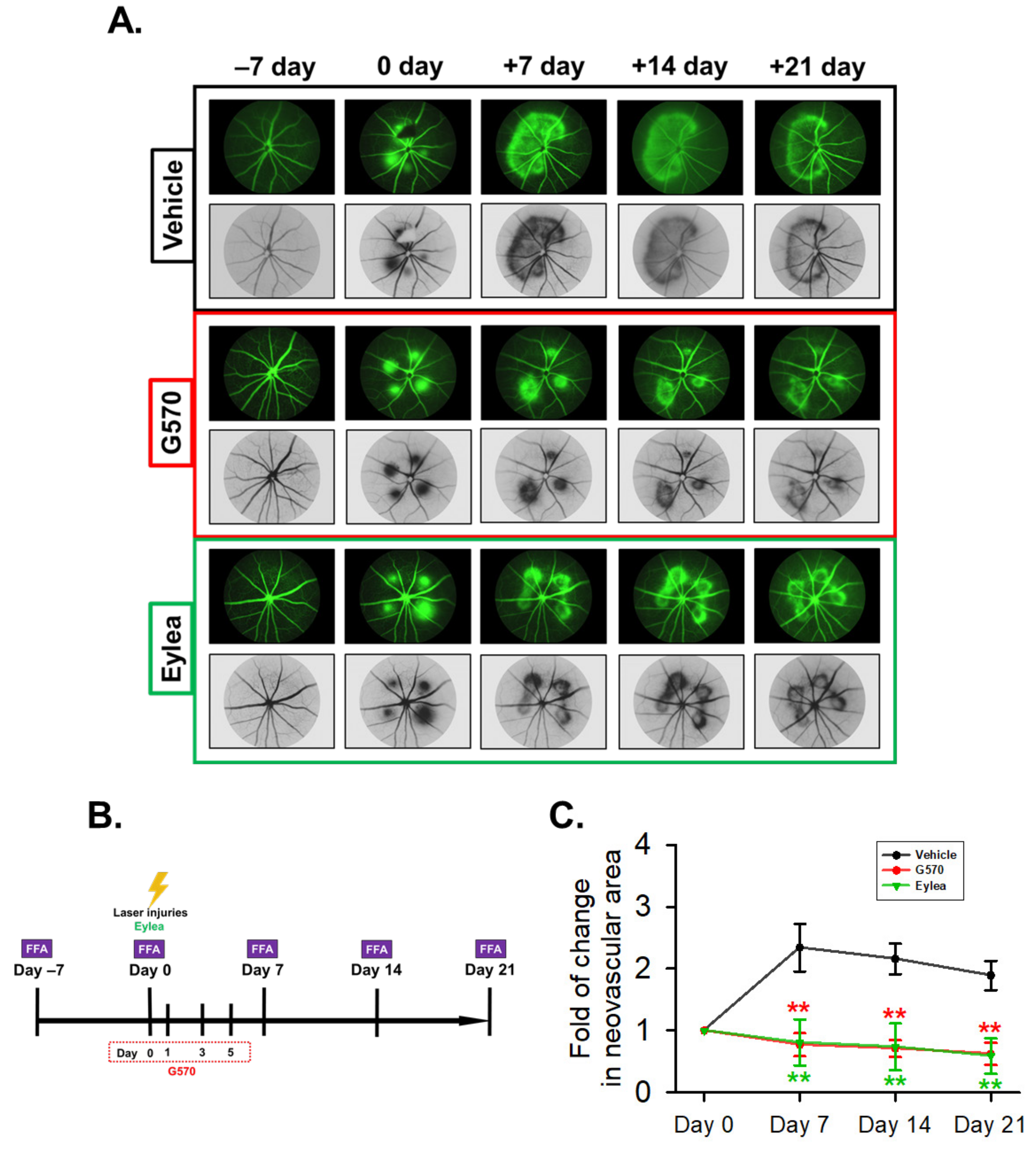
2.7. Effect of G570 on Laser-Induced Choroidal Neovascularization (CNV) in C57BL/6 Mice
3. Discussion
4. Materials and Methods
4.1. Synthesis of the Compounds
4.1.1. Synthesis of (5-Aminoindolin-1-yl)(2,4-bis(benzyloxy)-5-isopropylphenyl)methanone (S-2)
4.1.2. Synthesis of Methyl 4-((1-(2,4-Bis(benzyloxy)-5-isopropylbenzoyl)indolin-5-yl)amino)-4-oxobutanoate (S-3)
4.1.3. Synthesis of Methyl 5-((1-(2,4-Bis(benzyloxy)-5-isopropylbenzoyl)indolin-5-yl)amino)-5-oxopentanoate (S-4)
4.1.4. Synthesis of Methyl 6-((1-(2,4-Bis(benzyloxy)-5-isopropylbenzoyl)indolin-5-yl)amino)-6-oxohexanoate (S-5)
4.1.5. Synthesis of Methyl 7-((1-(2,4-Bis(benzyloxy)-5-isopropylbenzoyl)indolin-5-yl)amino)-7-oxohexanoate (S-6)
4.1.6. Synthesis of Methyl 8-((1-(2,4-Bis(benzyloxy)-5-isopropylbenzoyl)indolin-5-yl)amino)-8-oxohexanoate (S-7)
4.1.7. Synthesis of Methyl 9-((1-(2,4-Bis(benzyloxy)-5-isopropylbenzoyl)indolin-5-yl)amino)-9-oxohexanoate (S-8)
4.1.8. Synthesis of Methyl 10-((1-(2,4-Bis(benzyloxy)-5-isopropylbenzoyl)indolin-5-yl)amino)-10-oxohexanoate (S-9)
4.1.9. Synthesis of N1-(1-(2,4-dihydroxy-5-isopropylbenzoyl)indolin-5-yl)-N4-hydroxysuccinamide (1)
4.1.10. Synthesis of N1-(1-(2,4-dihydroxy-5-isopropylbenzoyl)indolin-5-yl)-N5-hydroxyglutaramide (2)
4.1.11. Synthesis of N1-(1-(2,4-dihydroxy-5-isopropylbenzoyl)indolin-5-yl)-N6-hydroxyadipamide (3)
4.1.12. Synthesis of N1-(1-(2,4-dihydroxy-5-isopropylbenzoyl)indolin-5-yl)-N7-hydroxyheptanediamide (4)
4.1.13. Synthesis of N1-(1-(2,4-dihydroxy-5-isopropylbenzoyl)indolin-5-yl)-N8-hydroxyoctanediamide (5)
4.1.14. Synthesis of N1-(1-(2,4-dihydroxy-5-isopropylbenzoyl)indolin-5-yl)-N9-hydroxynonanediamide (6)
4.1.15. Synthesis of N1-(1-(2,4-dihydroxy-5-isopropylbenzoyl)indolin-5-yl)-N10-hydroxydecanediamide (7)
4.1.16. Synthesis of Methyl 4-(((1-(2,4-Bis(benzyloxy)-5-isopropylbenzoyl)indolin-5-yAmino)methyl)benzoate (S-10)
4.1.17. Synthesis of 4-(((1-(2,4-Dihydroxy-5-isopropylbenzoyl)indolin-5-yl)amino)methyl)-N-hydroxybenzamide (8)
4.2. Cell Culture
4.3. MTT Assay
4.4. Cell Migration Assay
4.5. HeLa Nuclear Extract HDAC Activity Assay
4.6. Heat Shock Protein (Hsp) 90α Activity Assay
4.7. HDAC Enzyme Inhibition Assays
4.8. Western Blot Assay
4.9. Animals and Treatment
4.10. Laser-Induced Choroidal Neovascularization (CNV) Model and Injection
4.11. Fundus Angiography Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haddad, S.; Chen, C.A.; Santangelo, S.L.; Seddon, J.M. The genetics of age-related macular degeneration: A review of progress to date. Surv. Ophthalmol. 2006, 51, 316–363. [Google Scholar] [CrossRef]
- Rattner, A.; Nathans, J. Macular degeneration: Recent advances and therapeutic opportunities. Nat. Rev. Neurosci. 2006, 7, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.J.; Hsieh, C.M.; Nepali, K.; Liou, J.P. Ocular disease therapeutics: Design and delivery of drugs for diseases of the eye. J. Med. Chem. 2020, 63, 10533–10593. [Google Scholar] [CrossRef]
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, L.; Weinreb, R.N. Ophthalmic drug discovery: Novel targets and mechanisms for retinal diseases and glaucoma. Nat. Rev. Drug Discov. 2012, 11, 541–559. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okubo, A.; Rosa, R.H., Jr.; Bunce, C.V.; Alexander, R.A.; Fan, J.T.; Bird, A.C.; Luthert, P.J. The relationships of age changes in retinal pigment epithelium and Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 1999, 40, 443–449. [Google Scholar]
- Ferrara, N. Vascular endothelial growth factor and age-related macular degeneration: From basic science to therapy. Nat. Med. 2010, 16, 1107–1111. [Google Scholar] [CrossRef]
- Wang, H.; Han, X.; Wittchen, E.S.; Hartnett, M.E. TNF-α mediates choroidal neovascularization by upregulating VEGF expression in RPE through ROS-dependent β-catenin activation. Mol. Vis. 2016, 22, 116–128. [Google Scholar] [PubMed]
- Arjamaa, O.; Aaltonen, V.; Piippo, N.; Csont, T.; Petrovski, G.; Kaarniranta, K.; Kauppinen, A. Hypoxia and inflammation in the release of VEGF and interleukins from human retinal pigment epithelial cells. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1757–1762. [Google Scholar] [CrossRef]
- Mammadzada, P.; Corredoira, P.M.; André, H. The role of hypoxia-inducible factors in neovascular age-related macular degeneration: A gene therapy perspective. Cell Mol. Life Sci. 2020, 77, 819–833. [Google Scholar] [CrossRef] [Green Version]
- Ju, Y.; Tang, Z.; Dai, X.; Gao, H.; Zhang, J.; Liu, Y.; Yang, Y.; Ni, N.; Zhang, D.; Wang, Y. Protection against light-induced retinal degeneration via dual anti-inflammatory and anti-angiogenic functions of thrombospondin-1. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, T.; Negishi, K.; Miyake, S.; Hirasawa, M.; Osada, H.; Kurihara, T.; Tsubota, K.; Ozawa, Y. Blue light-induced inflammatory marker expression in the retinal pigment epithelium-choroid of mice and the protective effect of yellow intraocular lens material in vivo. Exp. Eye Res. 2015, 132, 48–51. [Google Scholar] [CrossRef] [Green Version]
- Falavarjani, K.G.; Nguyen, Q. Adverse events and complications associated with intravitreal injection of anti-VEGF agents: A review of the literature. Eye 2013, 27, 787–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraunfelder, F.T.; Fraunfelder, F.W. Oral anti-vascular endothelial growth factor drugs and ocular adverse events. J. Ocul. Pharmacol. Ther. 2018, 34, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Nickells, R.W.; Merbs, S.L. The potential role of epigenetics in ocular diseases. Arch. Ophthalmol. 2012, 130, 508–509. [Google Scholar] [PubMed]
- Alkozi, H.A.; Franco, R.; Pintor, J.J. Epigenetics in the eye: An overview of the most relevant ocular diseases. Front. Genet. 2017, 8, 144. [Google Scholar] [CrossRef] [Green Version]
- Crosson, C.E.; Mani, S.K.; Husain, S.; Alsarraf, O.; Menick, D.R. Inhibition of histone deacetylase protects the retina from ischemic injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3639–3645. [Google Scholar] [CrossRef] [PubMed]
- Samardzija, M.; Corna, A.; Gomez-Sintes, R.; Jarboui, M.A.; Armento, A.; Roger, J.E.; Petridou, E.; Haq, W.; Paquet-Durand, F.; Zrenner, E.; et al. HDAC inhibition ameliorates cone survival in retinitis pigmentosa mice. Cell Death Differ. 2020, 1–16. [Google Scholar] [CrossRef]
- Dahbash, M.; Sella, R.; Megiddo-Barnir, E.; Nisgav, Y.; Tarasenko, N.; Weinberger, D.; Rephaeli, A.; Livnat, T. The histone deacetylase inhibitor AN7, attenuates choroidal neovascularization in a mouse model. Int. J. Mol. Sci. 2019, 20, 714. [Google Scholar] [CrossRef] [Green Version]
- Leyk, J.; Daly, C.; Janssen-Bienhold, U.; Kennedy, B.N.; Richter-Landsberg, C. HDAC6 inhibition by tubastatin A is protective against oxidative stress in a photoreceptor cell line and restores visual function in a zebrafish model of inherited blindness. Cell Death Dis. 2017, 8, e3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaramurthi, H.; Roche, S.L.; Grice, G.L.; Moran, A.; Dillion, E.T.; Campiani, G.; Nathan, J.A.; Kennedy, B.N. Selective histone deacetylase 6 inhibitors restore cone photoreceptor vision or outer segment morphology in zebrafish and mouse models of retinal blindness. Front. Cell Dev. Biol. 2020, 8, 689. [Google Scholar] [CrossRef] [PubMed]
- Aguila, M.; Bevilacqua, D.; McCulley, C.; Schwarz, N.; Athanasiou, D.; Kanuga, N.; Novoselov, S.S.; Lange, C.A.; Ali, R.R.; Bainbridge, J.W.; et al. Hsp90 inhibition protects against inherited retinal degeneration. Hum. Mol. Genet. 2014, 23, 2164–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasiou, D.; Aguilà, M.; Bevilacqua, D.; Novoselov, S.S.; Parfitt, D.A.; Cheetham, M.E. The cell stress machinery and retinal degeneration. FEBS Lett. 2013, 587, 2008–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilà, M.; Cheetham, M.E. Hsp90 as a potential therapeutic target in retinal disease. In Retinal Degenerative Diseases; Advances in Experimental Medicine and Biology; Bowes Rickman, C., LaVail, M., Anderson, R., Grimm, C., Hollyfield, J., Ash, J., Eds.; Springer: Cham, Switzerland, 2016; Volume 854, pp. 161–167. [Google Scholar]
- Wu, W.C.; Kao, Y.H.; Hu, P.S.; Chen, J.H. Geldanamycin, a HSP90 inhibitor, attenuates the hypoxia-induced vascular endothelial growth factor expression in retinal pigment epithelium cells in vitro. Exp. Eye Res. 2007, 85, 721–731. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Abo-Ashour, M.F.; Nocentini, A.; El-Haggar, R.S.; Bua, S.; Bonardi, A.; Al-Rashood, S.T.; Hassan, G.S.; Gratteri, P.; Abdel-Aziz, H.A.; et al. Enhancement of the tail hydrophobic interactions within the carbonic anhydrase IX active site via structural extension: Design and synthesis of novel N-substituted isatins-SLC-0111 hybrids as carbonic anhydrase inhibitors and antitumor agents. Eur. J. Med. Chem. 2019, 162, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.Y.; Cho, T.Y.; Lu, C.K.; Liou, J.P.; Chen, M.C. Identification of 7-(4′-Cyanophenyl) indoline-1-benzenesulfonamide as a mitotic inhibitor to induce apoptotic cell death and inhibit autophagy in human colorectal cancer cells. Sci. Rep. 2017, 7, 12406. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Gettinger, S.; Senzer, N.N.; Martins, R.G.; Jänne, P.A.; Lilenbaum, R.; Gray, J.E.; Iafrate, A.J.; Katayama, R.; Hafeez, N. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non–small-cell lung cancer. J. Clin. Oncol. 2010, 28, 4953. [Google Scholar] [CrossRef]
- Felip, E.; Barlesi, F.; Besse, B.; Chu, Q.; Gandhi, L.; Kim, S.W.; Carcereny, E.; Sequist, L.V.; Brunsvig, P.; Chouaid, C.; et al. Phase 2 Study of the HSP-90 Inhibitor AUY922 in previously treated and molecularly defined patients with advanced non–small cell lung cancer. J. Thorac. Oncol. 2018, 13, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Goldman, J.W.; Raju, R.N.; Gordon, G.A.; El-Hariry, I.; Teofilivici, F.; Vukovic, V.M.; Bradley, R.; Karol, M.D.; Chen, Y.; Guo, W. A first in human, safety, pharmacokinetics, and clinical activity phase I study of once weekly administration of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid malignancies. BMC Cancer 2013, 13, 152. [Google Scholar] [CrossRef] [Green Version]
- Luan, Y.; Li, J.; Bernatchez, J.A.; Li, R. Kinase and histone deacetylase hybrid inhibitors for cancer therapy. J. Med. Chem. 2018, 62, 3171–3183. [Google Scholar] [CrossRef] [PubMed]
- Mamalis, A.; Garcha, M.; Jagdeo, J. Light emitting diode-generated blue light modulates fibrosis characteristics: Fibroblast proliferation, migration speed, and reactive oxygen species generation. Lasers Surg. Med. 2015, 47, 210–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Chen, C.Z.; Xing, Y.Q. Inhibition of cell proliferation, migration and apoptosis in blue-light illuminated human retinal pigment epithelium cells by down-regulation of HtrA1. Int. J. Ophthalmol. 2017, 10, 524–529. [Google Scholar]
- Hu, H.; Li, S.; Li, J.; Huang, C.; Zhou, F.; Zhao, L. Knockdown of fibromodulin inhibits proliferation and migration of RPE cell via the VEGFR2-AKT pathway. J. Ophthalmol. 2018, 2018, 5708537. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Bottcher, M.C.; Werner, J.A.; Mandic, R. Differential expression of VEGF-121, VEGF-165 and VEGF-189 in angiomas and squamous cell carcinoma cell lines of the head and neck. Anticancer Res. 2010, 30, 805–810. [Google Scholar]
- Kornberg, L.J.; Shaw, L.C.; Spoerri, P.E.; Caballero, S.; Grant, M.B. Focal adhesion kinase overexpression induces enhanced pathological retinal angiogenesis. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4463–4469. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Wu, H.J.; Guan, J.L. Nuclear FAK and its kinase activity regulate VEGFR2 transcription in angiogenesis of adult mice. Sci. Rep. 2018, 8, 2550. [Google Scholar] [CrossRef] [Green Version]
- Moiseev, R.V.; Morrison, P.W.J.; Steele, F.; Khutoryanskiy, V.V. Penetration enhancers in ocular drug delivery. Pharmaceutics 2019, 11, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; Chiang, B.; Wu, X.; Prausnitz, M.R. Ocular delivery of macromolecules. J. Control. Release 2014, 190, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Maciulaitiene, R.; Ragauskas, S.; Haapaniemi, A.M.; Kaja, S.; Januleviciene, I.; Kalesnykas, G. Aflibercept shows strong dose-dependency when administered intravitreally in the mouse CNV model. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2631. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | HSP90 IC50 (nM) | HeLa HDAC IC50 (nM) |
|---|---|---|
| 1 | 49.3 ± 1.6 | >1000 |
| 2 | 55.9 ± 3.2 | 862.9 ± 55.3 |
| 3 | N.D. | N.D. a |
| 4 | 40.2 ± 5.2 | 86.9 ± 14.4 |
| 5 | 46.3 ± 7.5 | 61.2 ± 2.0 |
| 6 | 52.1 ± 2.9 | 174.4 ± 5.9 |
| 7 | 58.6 ± 2.2 | 104.7 ± 14.8 |
| 8 | 85.7 ± 8.6 | >1000 |
| SAHA | >1000 | 99.4 ± 17.7 |
| BIIB021 | 65.7 ± 1.6 | >1000 |
| Compds | HDAC1 IC50 (µM) | HDAC3 IC50 (µM) | HDAC6 IC50 (nM) | HDAC8 IC50 (µM) | Selectivity Ratio | ||
|---|---|---|---|---|---|---|---|
| HDAC 1/6 | HDAC 3/6 | HDAC 8/6 | |||||
| 1 | - | - | >10,000 | - | - | - | - |
| 2 | 2.99 | 0.544 | 9.75 | 1.96 | 306 | 55 | 201 |
| 3 | 4.45 | 2.65 | 26.1 | 2.94 | 170 | 101 | 112 |
| 4 | 0.44 | 0.34 | 2.13 | 1.55 | 206 | 159 | 727 |
| 5 (MPT0G570) | 0.13 | 0.16 | 1.15 | 0.284 | 113 | 139 | 246 |
| 6 | 1.95 | 1.81 | 9.25 | 1.22 | 210 | 195 | 131 |
| 7 | 2.03 | 1.89 | 17.3 | 1.67 | 117 | 109 | 96 |
| 8 | - | - | 74.4 | 1.55 | - | - | 20 |
| Trichostatin A | 0.0069 | 0.0104 | 2.60 | 1.02 | 2.65 | 4 | 392 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, T.-J.; Nepali, K.; Tsai, C.-H.; Imtiyaz, Z.; Lin, F.-L.; Hsiao, G.; Lai, M.-J.; Cheng, Y.-W. The HDAC/HSP90 Inhibitor G570 Attenuated Blue Light-Induced Cell Migration in RPE Cells and Neovascularization in Mice through Decreased VEGF Production. Molecules 2021, 26, 4359. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144359
Hsu T-J, Nepali K, Tsai C-H, Imtiyaz Z, Lin F-L, Hsiao G, Lai M-J, Cheng Y-W. The HDAC/HSP90 Inhibitor G570 Attenuated Blue Light-Induced Cell Migration in RPE Cells and Neovascularization in Mice through Decreased VEGF Production. Molecules. 2021; 26(14):4359. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144359
Chicago/Turabian StyleHsu, Tai-Ju, Kunal Nepali, Chi-Hao Tsai, Zuha Imtiyaz, Fan-Li Lin, George Hsiao, Mei-Jung Lai, and Yu-Wen Cheng. 2021. "The HDAC/HSP90 Inhibitor G570 Attenuated Blue Light-Induced Cell Migration in RPE Cells and Neovascularization in Mice through Decreased VEGF Production" Molecules 26, no. 14: 4359. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144359