Aromatic Amines in Organic Synthesis. Part II. p-Aminocinnamaldehydes


Faculty of Chemical Technology and Engineering, UTP University of Sciences and Technology, Seminaryjna 3, 85-326 Bydgoszcz, Poland
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(14), 4360; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144360
Submission received: 24 June 2021
/
Revised: 9 July 2021
/
Accepted: 16 July 2021
/
Published: 19 July 2021
(This article belongs to the Section Organic Chemistry)
Abstract
:Ten derivatives of p-aminocinnamic aldehydes were prepared from the reaction of either aromatic amines with dimethylaminoacrolein or benzaldehydes with acetaldehyde. Their chemical structure and purity were verified by 1H NMR, 13C NMR and IR spectroscopic methods. We found that the synthesis applying dimethylaminoacrolein as the reagent gets better yields than the one based on the reaction with acetaldehyde. The yields of the cinnamic aldehydes varied according to the type of the amino group and the number and position of the substituents. The basic spectroscopic properties of the p-aminocinnamic aldehydes are also described since the compounds may be a precursor for the synthesis of dyes for diverse applications, e.g., in medicine and optoelectronics.
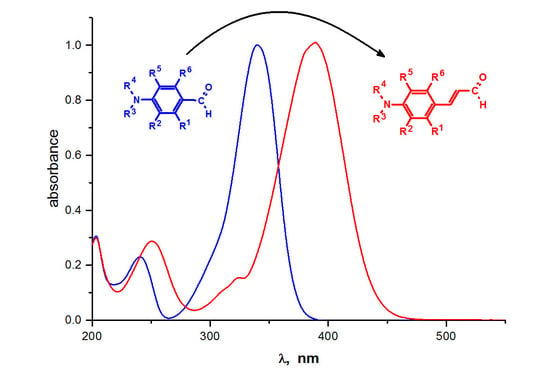
1. Introduction
Organic compounds containing different amino groups (or their derivatives) have attracted remarkable attention by virtue of their giant potential as dyes [1], solar cells [2,3], spectroscopic probes [4,5,6], one- [7,8] and multi-photon [9] polymerization initiators and drugs [10]. They have been studied for applications in various areas of photochemistry due to their structure versatility and facile modifications for electron-donating abilities [11,12,13]. In general, organic compounds bearing an alkylamino group may be synthesized through a related synthetic strategy using one of the following: Heck reaction [14], Knoevenagel condensation [13,15], Wittig reaction [16], Suzuki-Miyaura cross-coupling reaction [17] and several different reagents, such as aryl bromides with amino substituents [14], p-aminobenzaldehydes [13,15] and p-aminobenzoic acid derivatives [18,19].
Alternatively, organic compounds with an amino group, of particular interest for their nonlinear optical properties useful in modern technology, can be obtained using precursors with extended π-systems [20]. For the preparation of such compounds, standard p-aminobenzaldehydes should be replaced with p-aminocinnamaldehydes, their acids or esters [21,22].
There are different methods for the synthesis of cinnamaldehydes. One of them is based on the reaction between acetaldehyde with benzaldehydes having an electron-withdrawing substituent in the presence of KOH as a base [23]. The method is relatively simple and gives good yields. However, in the case of benzaldehydes with electron-donating substituents, in particular amino groups, this reaction has to be performed in concentrated sulfuric acid, and more drastic conditions are required. Moreover, products are isolated at a lower yield [23]. Therefore, alternative methods of synthesizing p-aminocinnamaldehydes with higher yields are sought. They can be obtained following the Heck reaction [24] or the reaction of aromatic amines with dimethylaminoacrolein [25].
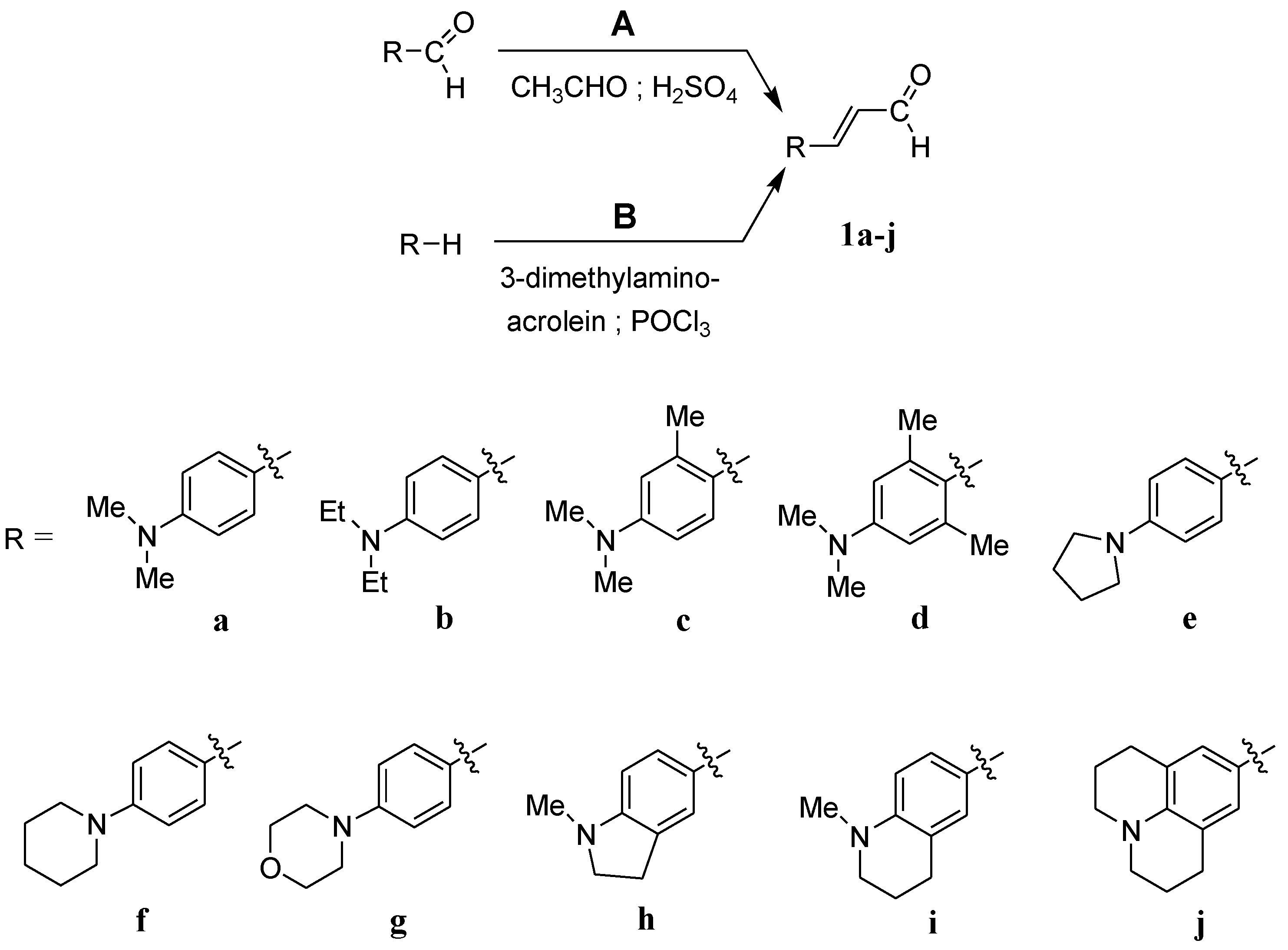
As part of our study on dyes with enlarged double bond systems that may be used as two-photon absorbers, [20] an optical amplifier, generator and modulator based on third order nonlinear optical phenomenon [26], photoinitiators of polymerization [8] or spectroscopic probes [6]. In the paper, we report synthesis, characterization and photophysical properties of the ten representatives of p-aminocinnamic aldehydes. They were obtained through two different procedures using corresponding benzaldehydes (Method A) or amines (Method B) as the starting materials (see Scheme 1). Method A is a single-step acid-catalyzed aldol condensation between acetaldehyde and appropriate benzaldehyde. This is a well-known method with moderate yields. Method B is analogous to the classical Vilsmeier–Haack formylation method; only 3-dimethylaminoacrolein is used instead of dimethylformamide. The mechanism of this reaction has been described by Ulrich and Brenmeier [25]. In this case, an electrophilic substitution takes place, and therefore, aromatic amines react readily.
2. Results
The synthetic routes for the preparation of ten p-aminocinnamic aldehydes are shown in Scheme 1. Six of these aldehydes (1c, 1d, 1e, 1f, 1h, 1i) have not been described in the literature yet. According to Method A, treatment of p-aminobenzaldehyde with acetaldehyde in concentrated sulfuric acid at a lower temperature (0 °C) followed by neutralization, afforded p-aminocinnamic aldehyde a good yield (32–54%). The yield of the synthesis was improved by ca. 6–16% for the reaction between aromatic amine and 3-dimethylaminoacrolein, in the presence of POCl3 conducted in chloroform at −20–0 °C (Method B). What is more, 4-(N,N-dimethylamino)-2,6-dimethylcinnamaldehyde (1d) can be obtained at a satisfactory yield only using Method B. This is probably due to the steric hindrance.
The structure and purity of all synthesized compounds were confirmed by spectroscopic methods, including NMR and IR spectra analysis (spectra are shown in the ESI file), as well as the elemental analysis. The NMR spectra confirmed the presence of signals from all protons and carbons in the synthesized molecules, as shown in the Experimental Section. For example, the proton signal of the aldehyde group is clearly visible at ca. 9.5 ppm, whereas the characteristic signals of the methine groups are given in the range from 7.8 to 6.2 ppm. The coupling constant for these protons (3JH,H = 16 Hz) indicates that the compounds are in the trans configuration. The signal of carbon from the aldehyde group occurs at ca. 193–195 ppm and is shifted by about 0.7 ppm compared to the analogous benzaldehydes. The presence of the carbonyl group is also confirmed by the characteristic, strong signal in the IR spectrum in the frequency range from 1667 (1e) to 1649 cm−1 (1b).
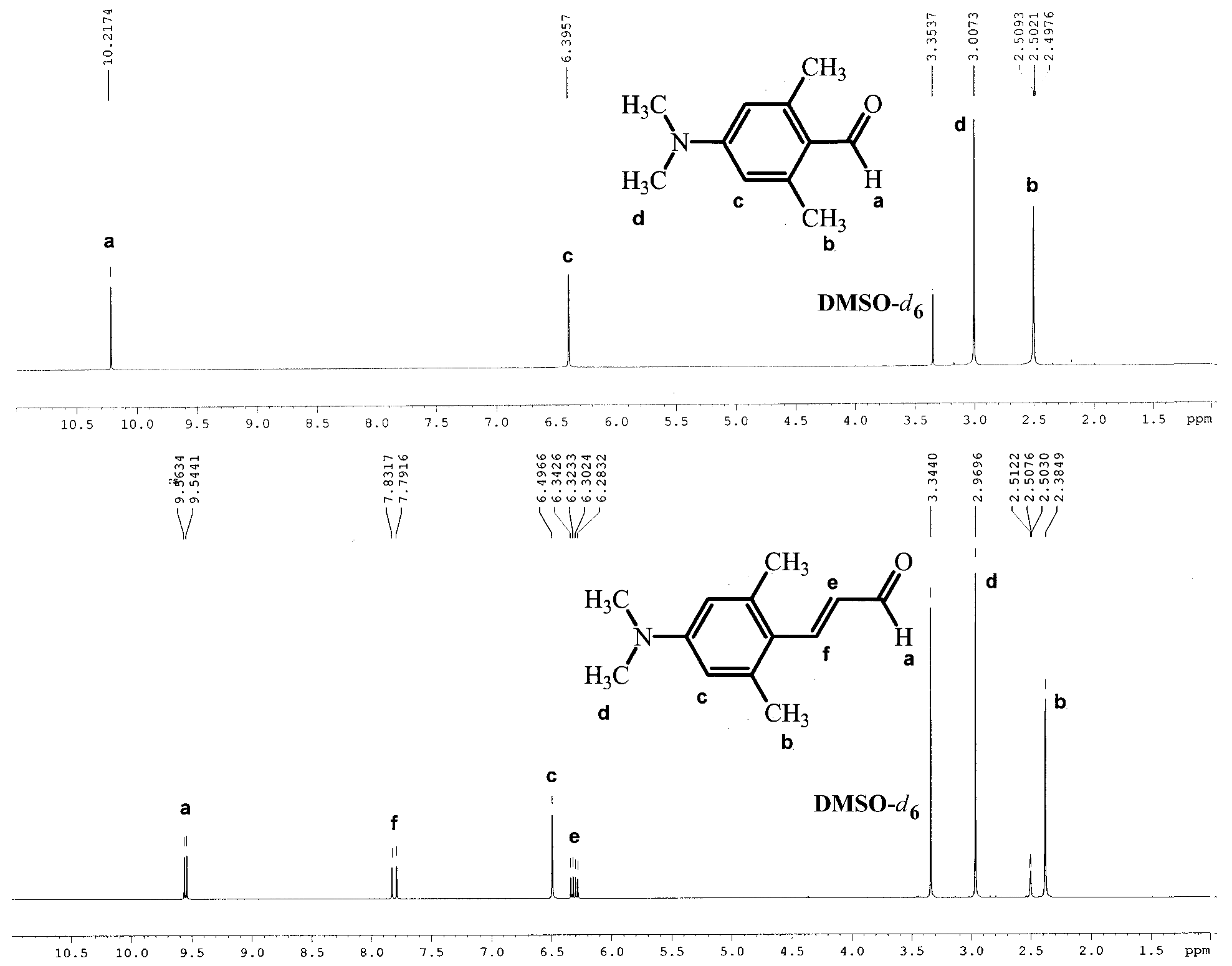
Figure 1 illustrates the 1H NMR spectra of 4-(dimethylamino)-2,6-dimethylcinnamaldehyde (1d) in DMSO-d6 and its corresponding benzaldehyde to show differences in signal positions associated with elongation of the π-system.
The comparative analysis of the 1H NMR spectra of the 4-(dimethylamino)-2,6-dimethylbenzaldehyde and 4-(dimethylamino)-2,6-dimethylcinnamaldehyde (Figure 1) shows that the signal of the CHO group is shifted from 10.2 to 9.5 ppm and split into doublet with a coupling constant of 8.0 Hz. Additionally, in cinnamic aldehyde, there are two characteristic signals from methine hydrogen at ca. 7.8 and 6.3 ppm, with the He signal visible as a doublet of doublets with unequal coupling constants.
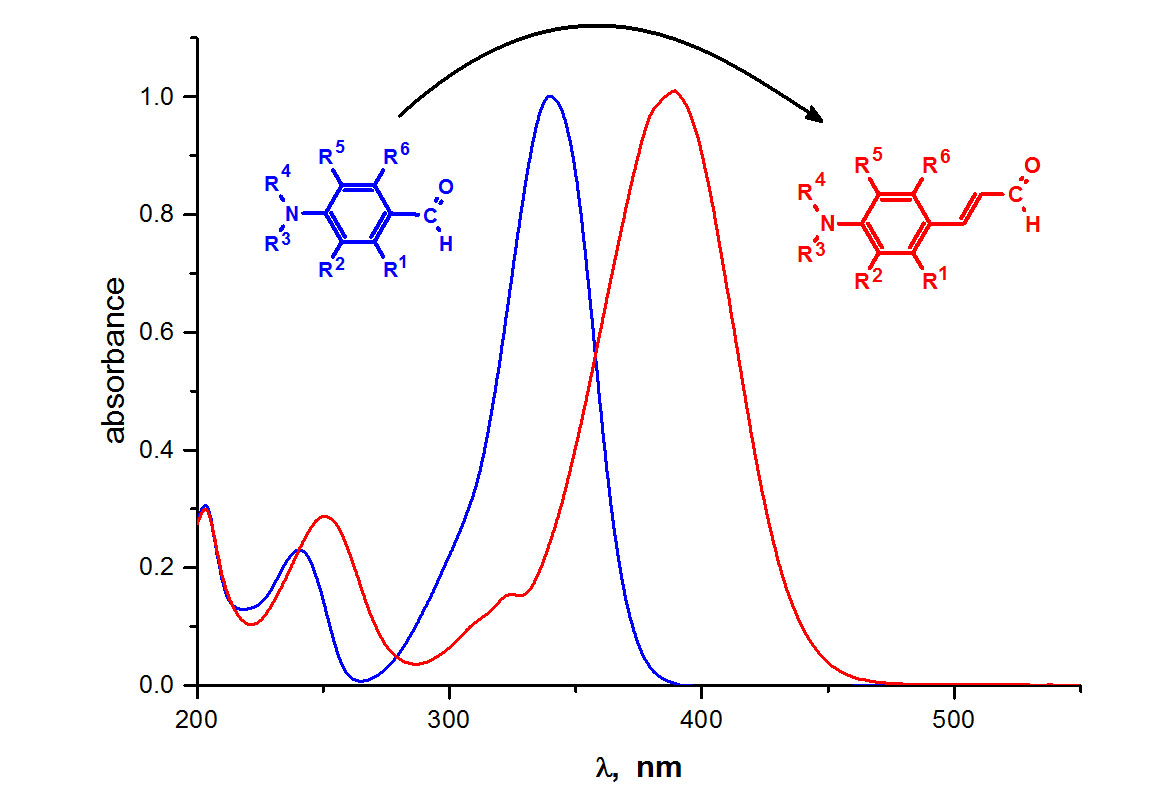
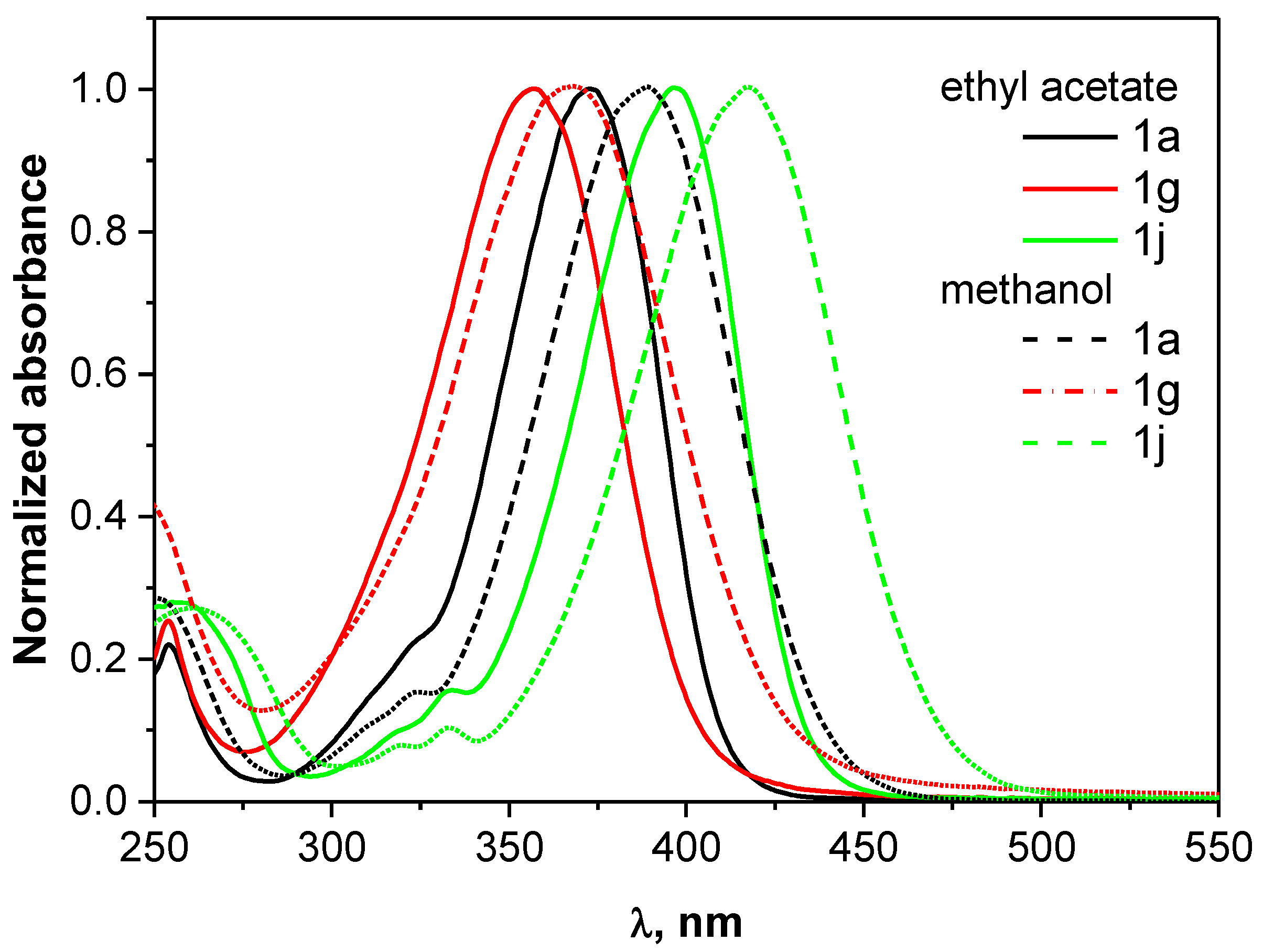
Since the compounds can be used for the preparation of different groups of dyes, their basic spectroscopic data are compiled in Table 1. They are determined in two solvents of varying properties, i.e., methanol and ethyl acetate. The normalized electronic absorption spectra of selected p-aminocinnamic aldehydes (1a, 1g and 1j) in ethyl acetate (EtOAc) and methanol (MeOH) are shown in Figure 2, whereas the spectra for all synthesized compounds are presented in the ESI file.
The UV–Vis absorption spectra display one absorption peak at ca. 390–400 nm, attributed to the π→π* transition. Compound 1j exhibits the most redshifted absorption band with a maximum at 417.5 and 460 nm in methanol and ethyl acetate, respectively, compared to the parent 1a cinnamic aldehyde (maximum at 389 and 372.5 nm). The significant redshift is attributed to the presence of the stiffening alkylamino group, which enhanced the extent of electron delocalization over the whole molecule. On the other hand, the strongest hypsochromic effect is observed in cinnamaldehyde with morpholine residue. The presence of the oxygen atom in the six-membered piperidine ring probably causes the coplanar alignment of the molecule, which results in a higher energy barrier for the S0-S1 transition and shifting the absorption maximum to the blue region. The molar extinction coefficients of the p-aminocinnamic aldehydes are in the range from 28,900 to 45,700 M−1cm−1 and 31,400 to 44,500 M−1cm−1 in MeOH and EtOAc, respectively. These values are rather high, indicating that the compounds have a good light-harvesting ability.
The fluorescence spectra of the compounds 1a–j (exited at 370 and 390 nm in EtOAc and MeOH, respectively) showed a sharp band in the range from 440 to 480 nm (see Figures S3 and S4 in ESI), corresponding to blue light emission. Similar to the UV-Vis absorption, the fluorescence maximum of 1j is redshifted by 14 and 23 nm in MeOH and EtOAc, respectively, relative to the parent p-N,N-dimethylaminocinnamic aldehyde, which arises from the different donating ability of the amino group. The bathochromic shift of the absorption and fluorescence spectra also occurs with increasing solvent polarity. However, the change of ethyl acetate to methanol causes a small (approx. 15 nm) redshift of the absorption maximum. A more pronounced shift with increasing solvent polarity (ca. 25–40 nm) is observed for the fluorescence band, which indicates better stabilization of the excited state of the compounds in a more polar environment and suggests an increase of the dipole moment upon excitation. The fluorescence quantum yield of the compounds is small and does not exceed 2% in ethyl acetate and 0.5% in methanol, which suggests that the deactivation of the S1 excited-state occurs mostly by non-radiative processes.
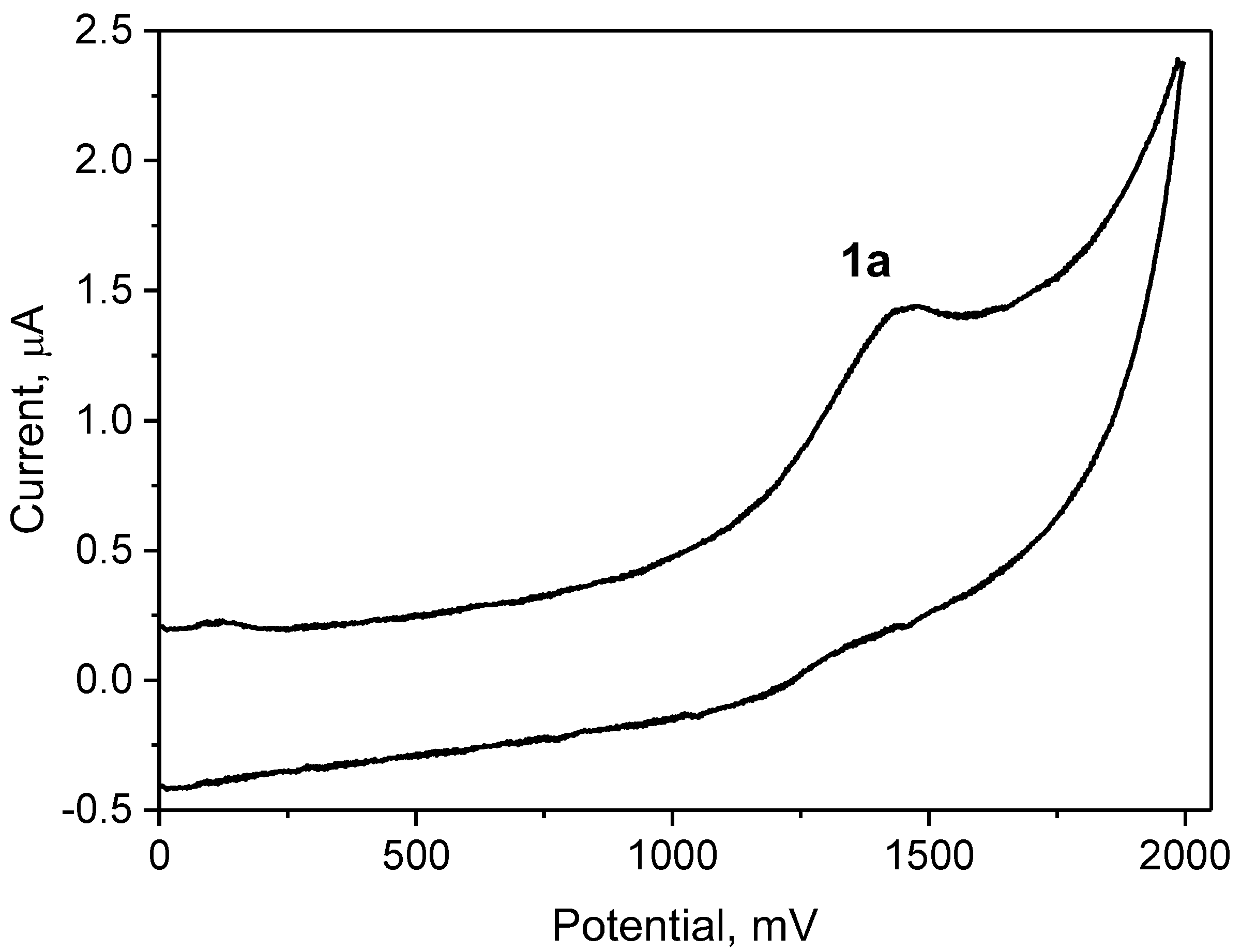
To reveal the substituent effect on an oxidation potential, the cyclic voltammograms of the aminocinnamic aldehydes were recorded in anhydrous acetonitrile as illustrated in Figure 3. The results of the electrochemical measurements for all compounds are collected in Table 2.
In all cases, the electrochemical oxidation of the aminocinnamic aldehydes is irreversible. The location of the oxidation peaks depends on the alkylamino substituent. Compared to the parent compound (1a), the aminocinnamic aldehydes are more easily oxidized. In the case of 1j containing the stiffened amine group, the oxidation proceeds at the lowest potential. Only compound 1g, which is a morpholine derivative, has a lower tendency to lose electrons.
3. Experimental Section
3.1. Materials and Methods
Starting reagents and solvents were purchased from Aldrich Chemical Co. Aldehydes were purified using Büchi Sepacore® Chromatography system (C-605, C-610) with a UV-Vis detector (C-640). Melting points were determined on the Büchi melting point apparatus MP-1. The 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded on a Bruker AscentTM 400 NMR spectrometer. Dimethylsulfoxide (DMSO-d6) was used as the solvent and tetramethylsilane as the internal standard. The IR spectra were recorded using a Bruker spectrophotometer Vector 22 in the range of 400–4500 cm−1 by the KBr pellet technique. Elemental analysis was carried out by an Elementar Vario MACRO apparatus. The UV-Vis absorption spectra were recorded on a Shimadzu UV-Vis Multispec-1501 spectrophotometer. The fluorescence spectra were obtained with a Hitachi F-7100 spectrofluorometer. The fluorescence quantum yield (FQY; ϕ) was calculated according to Equation (1) using coumarin I in ethanol as a reference [27].
where: I is the integrated intensity (area) (in units of photons); n is the refractive index. The absorbances (A) of both the sample (s) and reference (ref) solution at an excitation wavelength (370 nm) was ca. 0.1.
The oxidation potential (Eox) was measured by cyclic voltammetry using an Electrochemical Analyzer EA9C, MTM Cracow in anhydrous acetonitrile with 0.1 M of tetrabutylammonium perchlorate as the supporting electrolyte. The scan rate was 100 mV/s. The measurements were made in a typical three-electrode setup containing a platinum 1 mm electrode as the working electrode and platinum and Ag/AgCl as auxiliary and reference electrodes, respectively.
3.2. Synthesis
3.2.1. Method A
An appropriate p-aminobenzaldehyde (40 mmol) was dissolved in 30 mL of concentrated sulfuric acid under vigorous stirring. Then, the solution was cooled to 0 °C, and 120 mmol of acetaldehyde was added dropwise within 3 h. After another hour, the reaction mixture was poured onto ice and neutralized with 20% NaOH. The precipitate was filtered, dried, and recrystallized from ethanol. Only the aldehydes 1h, 1i and 1j needed to be purified by flash chromatography on silica gel (pore size 60 Å, 230–400 mesh particle size) using chloroform as the mobile phase [23].
3.2.2. Method B
Anhydrous chloroform (20 mL), an appropriate aromatic amine (65 mmol) and 3-dimethylaminoacrolein (55 mmol) were placed in a flask equipped with a thermometer and a dropping funnel. The mixture was cooled to −10 °C, and a solution of 55 mmol of POCl3 in 8 mL of chloroform was added, keeping the temperature below 0 °C. Then, the mixture was allowed to warm up to room temperature and stirred overnight. It was next heated to 60 °C and kept at this temperature for 2 h. Chloroform was distilled off under reduced pressure, and 25 mL of methanol was added with stirring and cooling. The reaction mixture was poured onto 50 g of crushed ice and neutralized with 20% NaOH. The crude product was filtered and recrystallized from ethanol. As before, the aldehydes 1h, 1i and 1j were purified by flash chromatography on silica gel (pore size 60 Å, 230–400 mesh particle size) using chloroform as the mobile phase [25].
- 3-[4-(Dimethylamino)phenyl]prop-2-enal (1a)
![Molecules 26 04360 i001]()

The compound was obtained as a brown solid; yield: 54% (method A), 61% (method B); mp 140 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.53 (d, J = 8.0 Hz, 1H, CHO), 7.59–7.55 (m, 3H), 6.75 (d, J = 9.0 Hz, 2H), 6.59 (dd, J = 8.0 Hz, J = 15.8 Hz, 1H), 3.01 (s, 6H, N(CH3)2); 13C NMR (100 MHz, DMSO-d6): δ 193.9 (CHO), 154.7 (CH), 152.7 (C), 131.1 (CH), 123.7 (CH), 121.8 (C), 112.2 (CH), 40.1 (N(CH3)2); IR (KBr): 1664, 1600, 1529, 1373, 1140, 973, 811.
Anal. calculated for C11H13NO: C, 75.40; H, 7.48; N, 7.99. Found: C, 75.51; H, 7.40; N, 7.94.
- 3-[4-(Diethylamino)phenyl]prop-2-enal (1b)
![Molecules 26 04360 i002]()

The compound was obtained as a brown solid; yield: 39% (method A), 55% (method B); mp 73 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.52 (d, J = 8.0 Hz, 1H, CHO), 7.56–7.52 (m, 3H), 6.71 (d, J = 9.0 Hz, 2H), 6.55 (dd, J = 8.0 Hz, J = 15.9 Hz, 1H), 3.42 (q, J = 6.9 Hz, 4H, NCH2), 1.12 (t, J = 7.0 Hz, 6H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 193.9 (CHO), 154.7 (CH), 150.3 (C), 131.5 (CH), 123.2 (CH), 121.0 (C), 111.6 (CH), 44.4 (NCH2), 12.9 (CH3); IR (KBr): 1649, 1596, 1525, 1415, 1187, 1135, 813.
Anal. calculated for C13H17NO: C, 76.81; H, 8.43; N, 6.89. Found: C, 76.90; H, 8.35; N, 6.86.
- 3-[4-(Dimethylamino)-2-methylphenyl]prop-2-enal (1c)
![Molecules 26 04360 i003]()

The compound was obtained as a dark red solid; yield: 49% (method A), 58% (method B); mp 110 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.57 (d, J = 8.0 Hz, 1H, CHO), 7.80 (d, J = 15.8 Hz, 1H, -CH=) 7.65 (d, J = 9.2 Hz, 1H), 6.61-6.48 (m, 3H), 2.99 (s, 6H, N(CH3)2), 2.40 (s, 3H, ArCH3); 13C NMR (100 MHz, DMSO-d6): δ 193.7 (CHO), 152.0 (C), 151.0 (CH), 139.9 (C), 128.6 (CH), 123.6 (CH), 119.8 (C), 113.0 (CH), 110.0 (CH), 39.6 (N(CH3)2), 19.9 (ArCH3); IR (KBr): 1650, 1597, 1580, 1374, 1296, 1212, 1154, 1099, 811.
Anal. calculated for C12H15NO: C, 76.16; H, 7.99; N, 7.40. Found: C, 76.23; H, 7.91; N, 7.38.
- 3-[4-(Dimethylamino)-2,6-dimethylphenyl]prop-2-enal (1d)
![Molecules 26 04360 i004]()

The compound was obtained as a dark yellow solid; yield: 0% (method A), 36% (method B); mp 111 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.55 (d, J = 7.7 Hz, 1H, CHO), 7.81 (d, J = 16 Hz, 1H, -CH=), 6.50 (s, 2H), 6.31 (dd, J = 7.7 Hz, J = 16.0 Hz, 1H), 2.97 (s, 6H, N(CH3)2), 2.38 (s, 6H, ArCH3); 13C NMR (100 MHz, DMSO-d6): δ 195.2 (CHO), 152.0 (CH), 151.4 (C), 140.6 (C), 128.8 (CH), 120.2 (C), 112.7 (CH), 40.0 (N(CH3)2), 22.8 (ArCH3); IR (KBr): 1656, 1583, 1510, 1366, 1146, 827.
Anal. calculated for C13H17NO: C, 76.81; H, 8.43; N, 6.89. Found: C, 76.93; H, 8.41; N, 6.88.
- 3-[4-(Pyrrolidin-1-yl)phenyl]prop-2-enal (1e)
![Molecules 26 04360 i005]()

The compound was obtained as a brown solid; yield: 45% (method A), 54% (method B); mp 131 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.52 (d, J = 8.0 Hz, 1H, CHO), 7.57–7.54 (m, 3H), 6.60-6.54 (m, 3H), 3.32 (t, J = 4.5 Hz, 4H, NCH2), 1.97 (m, 4H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 193.8 (CHO), 155.0 (CH), 150.2 (C), 131.3 (CH), 123.2 (CH), 121.3 (C), 112.2 (CH), 47.7 (NCH2), 25.4 (CH2); IR (KBr): 1667, 1594, 1526, 1398, 1187, 1136, 971, 810.
Anal. calculated for C13H15NO: C, 77.58; H, 7.51; N, 6.96. Found: C, 77.61; H, 7.43; N, 6.98.
- 3-[4-(Piperidin-1-yl)phenyl]prop-2-enal (1f)
![Molecules 26 04360 i006]()

The compound was obtained as a dark brown solid; yield: 52% (method A), 60% (method B); mp 87 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.55 (d, J = 7.9 Hz, 1H, CHO), 7.59–7.55 (m, 3H), 6.96 (d, J = 9.0 Hz, 2H), 6.61 (dd, J = 8.0 Hz, J = 15.8 Hz, 1H), 3.33 (t, J = 8.7 Hz, 4H, NCH2), 1.58 (m, 6H); 13C NMR (100 MHz, DMSO-d6): δ 194.1 (CHO), 154.3 (CH), 153.3 (C), 131.1 (CH), 124.5 (CH), 123.2 (C), 114.5 (CH), 48.4 (NCH2), 25.3 (CH2), 24.4 (CH2); IR (KBr): 1653, 1600, 1516, 1243, 1137, 1124, 811.
Anal. calculated for C14H17NO: C, 78.11; H, 7.96; N, 6.51. Found: C, 78.19; H, 7.88; N, 6.45.
- 3-[4-(Morpholin-4-yl)phenyl]prop-2-enal (1g)
![Molecules 26 04360 i007]()

The compound was obtained as a dark brown solid; yield: 52% (method A), 58% (method B); mp 109 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.57 (d, J = 7.9 Hz, 1H, CHO), 7.62–7.58 (m, 3H), 6.99 (d, J = 9.0 Hz, 2H), 6.66 (dd, J = 7.9 Hz, J = 15.7 Hz, 1H), 3.73 (t, J = 4.9 Hz, 4H, OCH2), 3.26 (t, J = 4.9 Hz, 4H, NCH2); 13C NMR (100 MHz, DMSO-d6): δ 194.3 (CHO), 154.1 (CH), 153.4 (C), 130.9 (CH), 125.2 (CH), 124.5 (C), 114.5 (CH), 66.2 (OCH2), 47.4 (NCH2); IR (KBr): 1659, 1601, 1518, 1234, 1137, 1123, 927, 811, 629.
Anal. calculated for C13H15NO2: C, 71.87; H, 6.96; N, 6.45. Found: C, 71.93; H, 6.92; N, 6.41.
- 3-(1-Methyl-2,3-dihydro-1H-indol-5-yl)prop-2-enal (1h)
![Molecules 26 04360 i008]()

Purified by flash chromatography (silica gel, chloroform as eluent). The compound was obtained as a yellow solid; yield: 37% (method A), 52% (method B); mp 65 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.50 (d, J = 7.9 Hz, 1H, CHO), 7.53 (d, J = 15.6 Hz, 1H, -CH=), 7.43 (s, 1H), 7.38 (d, J = 8.2 Hz, 1H), 6.54 (dd, J = 8.0 Hz, J=15.8 Hz, 1H), 6.51 (d, J = 7.8 Hz, 1H), 3.45 (t, J = 8.4 Hz, 2H, NCH2), 2.95 (t, J = 8.3 Hz, 2H), 2.81 (s, 3H, NCH3); 13C NMR (100 MHz, DMSO-d6): δ 193.9 (CHO), 156.4 (C), 155.1 (CH), 132.0 (CH), 131.2 (C), 124.4 (CH), 123.2 (CH), 123.2 (C), 106.0 (CH), 55.0 (NCH2), 34.6 (NCH3), 27.7 (ArCH2); IR (KBr): 1657, 1595, 1505, 1329, 1301, 1130, 1081, 813.
Anal. calculated for C12H13NO: C, 76.98; H, 7.00; N, 7.48. Found: C, 77.04; H, 7.08; N, 7.41.
- 3-(1-Methyl-1,2,3,4-tetrahydroquinolin-6-yl)prop-2-enal (1i)
![Molecules 26 04360 i009]()

Purified by flash chromatography (silica gel, chloroform as eluent). The compound was obtained as a brown solid; yield: 43% (method A), 55% (method B); mp 65 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.50 (d, J = 8.0 Hz, 1H, CHO), 7.49 (d, J = 15.6 Hz, 1H, -CH=), 7.37 (d, J = 8.5 Hz, 1H), 7.29, (s, 1H), 6.59 (d, J = 8.6 Hz, 1H), 6.53 (dd, J = 8.0 Hz, J = 15.5 Hz, 1H), 3.32 (t, J = 5.7 Hz, 2H, NCH2), 2.94 (s, 3H, NCH3), 2.71 (t, J = 6.2 Hz, 2H, ArCH2), 1.88 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 193.8 (CHO), 154.9 (CH), 149.4 (C), 130.1 (CH), 129.5 (CH), 123.1 (CH), 122.6 (C), 121.5 (C), 110.6 (CH), 50.9 (NCH2), 38.8 (NCH3), 27.6 (ArCH2), 21.7 (CH2); IR (KBr): 1664, 1595, 1525, 1322, 1205, 1137, 807.
Anal. calculated for C13H15NO: C, 77.58; H, 7.51; N, 6.96. Found: C, 77.52; H, 7.43; N, 6.91.
- 3-(9-Julolidyl)prop-2-enal (1j)
![Molecules 26 04360 i010]()

Purified by flash chromatography (silica gel, chloroform as eluent). The compound was obtained as a brown solid; yield: 32% (method A), 47% (method B); mp 125 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.47 (d, J = 8.0 Hz, 1H, CHO), 7.42 (d, J = 15.5 Hz, 1H, -CH=), 7.11 (s, 1H), 6.47 (dd, J = 8.0 Hz, J = 15.5 Hz, 1H), 3.24 (t, J = 5.8 Hz, 4H, NCH2), 2.68 (t, J = 6.3 Hz, 4H, ArCH2), 1.86 (m, 4H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 193.6 (CHO), 155.1 (CH), 145.9 (C), 128.6 (CH), 122.6 (CH), 121.0 (C), 120.6 (C), 49.7 (NCH2), 27.5 (CH2), 21.4 (CH2); IR (KBr): 1654, 1588, 1523, 1319, 1128, 969.
Anal. calculated for C15H17NO: C, 79.26; H, 7.54; N, 6.16. Found: C, 79.21; H, 7.50; N, 6.19.
4. Conclusions
p-Aminocinnamic aldehydes were prepared from the reaction of either acetaldehyde with aromatic aldehydes bearing appropriate substituents (Method A) or aromatic amine and 3-dimethylaminoacrolein in the presence of POCl3 (Method B). The desired products were obtained at a low to moderate yield with slightly higher values of ca. 6–16% achieved using Method B. The yields of the reactions carried out according to method A are 32–54%, while for Method B, they are in the range of 36–61%. In addition to the better yield, an important advantage of Method B is the use of relatively readily available aromatic amines instead of the often difficult to obtain benzaldehydes. Moreover, the 4-(N,N-dimethylamino)-2,6-dimethylcinnamaldehyde (1d), probably due to spatial hindrance, could only be synthesized by Method B. It should also be noted that six of the synthesized aldehydes (1c, 1d, 1e, 1f, 1h, 1i) have not been described in the literature so far.
Spectroscopic analysis showed that the absorption and fluorescence maxima of the cinnamic aldehydes, due to the additional double bond, are redshifted by approximately 45 and 75 nm, respectively, relative to the corresponding benzaldehydes. Moreover, their spectroscopic properties depend on the amine substituent present in the phenyl ring and solvent used. The structure of the cinnamaldehydes also influences the location of the oxidation peaks.
Supplementary Materials
Supplementary data related to NMR, IR, UV-Vis and fluorescence spectroscopy data for all tested compounds can be found online.
Author Contributions
M.P. conceived of the presented idea, carried out the synthesis and experiments, drafted the manuscript in Polish and designed the tables and figures. B.J. contributed to the analysis of the results and wrote the English version of the manuscript. All authors discussed the results and contributed to the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was founded by the National Science Center of Poland (Decision No. DEC-2012/07/B/ST4/01417).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data is provided by Marek Pietrzak (UTP University of Science and Technology), please contact [email protected]. The data is not publicly available apart from the data contained in the article or Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Sample Availability
Samples of the compounds are available from the author (M. Pietrzak).
References
- Li, X.; Gao, X.; Shi, W.; Ma, H. Design strategies for water-soluble small molecular chromogenic and fluorogenic probes. Chem. Rev. 2014, 114, 590–659. [Google Scholar] [CrossRef]
- Jiang, X.; Marinado, T.; Gabrielsson, E.; Hagberg, D.P.; Sun, L.; Hagfeldt, A. Structural modification of organic dyes for efficient coadsorbent-free dye-sensitized solar cells. J. Phys. Chem. C. 2010, 114, 2799–2805. [Google Scholar] [CrossRef]
- Chen, S.-L.; Yang, L.-N.; Li, Z.-S. How to design more efficient organic dyes for dye-sensitized solar cells? Adding more sp2-hybridized nitrogen in the triphenylamine donor. J. Power Sources. 2013, 223, 86–93. [Google Scholar] [CrossRef]
- Lavis, D.L.; Raines, R.T. Bright Ideas for Chemical Biology. ACS Chem. Biol. 2008, 3, 142–155. [Google Scholar] [CrossRef] [Green Version]
- Costero, A.M.; Banuls, M.J.; Aurell, M.J.; Ochando, L.E.; Domenech, A. Cation and anion fluorescent and electrochemical sensors derived from 4,4′-substituted biphenyl. Tetrahedron 2005, 61, 10309–10320. [Google Scholar] [CrossRef]
- Krawczyk, P.; Pietrzak, M.; Janek, T.; Jędrzejewska, B.; Cysewski, P. Spectroscopic and nonlinear optical properties of new chalcone fluorescent probes for bioimaging applications: A theoretical and experimental study. J. Mol. Model. 2016, 22, 125–135. [Google Scholar] [CrossRef]
- Cook, W.D.; Chen, F. Enhanced photopolymerization of dimethacrylates with ketones, amines, and iodonium salts: The CQ system. J. Poym. Sci. Pol. Chem. 2011, 49, 5030–5041. [Google Scholar] [CrossRef]
- Jędrzejewska, B.; Pietrzak, M.; Rafiński, Z. Phenyltrialkylborates as co-initiators with cyanine dyes in visible light polymerization of acrylates. Polymer 2011, 52, 2110–2119. [Google Scholar] [CrossRef]
- Hrobarik, P.; Hrobarikova, V.; Sigmundova, I.; Zahradnik, P.; Fakis, M.; Polyzos, I.; Persephonis, P. Benzothiazoles with tunable electron-withdrawing strength and reverse polarity: A route to triphenylamine-based chromophores with enhanced two-photon absorption. J. Org. Chem. 2011, 76, 8726–8736. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science. 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, Z.R.; Rotkiewicz, K.; Rettig, W. Structural changes accompanying intramolecular electron transfer: Focus on twisted intramolecular charge-transfer states and structures. Chem. Rev. 2003, 103, 3899–4031. [Google Scholar] [CrossRef]
- Zakrzewska, A.; Gawinecki, R.; Kolehmainen, E.; Ośmiałowski, B. 13C-NMR based evaluation of the electronic and steric interactions in aromatic amines. Int. J. Mol. Sci. 2005, 6, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Pietrzak, M.; Bajorek, A. 5-Phenyl-1,2,3,4-tetrahydronaphthalene derivatives: Synthesis, spectroscopic and electrochemical investigation. Dyes Pigments 2013, 96, 63–70. [Google Scholar] [CrossRef]
- Heck, R.F.; Nolley, J.P. Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar] [CrossRef]
- Pietrzak, M.; Jędrzejewska, B.; Mądrzejewska, D.; Bajorek, A. Convenient synthesis of p-aminobenzoic acids and their methyl esters. Org. Prep. Proced. Int. 2017, 49, 45–52. [Google Scholar] [CrossRef]
- Gadigennavar, S.; Ranganathan, M.; Sankararaman, S. Which isomer is it, 1,2,5,6- or 1,4,5,8-tetrasubstituted cycloocta-1,3,5,7-tetraene? Synthesis of symmetrically tetrasubstituted cycloocta-1,3,5,7-tetraene derivatives. Org. Biomol. Chem. 2020, 45, 9284–9291. [Google Scholar] [CrossRef]
- Wan, J.; Fan, B.; Thang, S.H. Sonochemical preparation of polymer-metal nanocomposites with catalytic and plasmonic properties. Nanoscale Adv. 2021, 11, 3306–3315. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X.; Yan, H.; Wu, Y.; Li, Z.; Gong, S.; Liu, P.; Liu, Z. Benzothiazole-enamide-based BF2 complexes: Luminophores exhibiting aggregation-induced emission, tunable emission and highly efficient solid-state emission. J. Mater. Chem. C 2015, 3, 2953–2959. [Google Scholar] [CrossRef]
- Ośmiałowski, B.; Petrusevich, E.F.; Antoniak, M.A.; Grela, I.; Bin Jassar, M.A.; Nyk, M.; Luis, J.M.; Jędrzejewska, B.; Zaleśny, R.; Jacquemin, D. Controlling two-photon action cross section by changing a single heteroatom position in fluorescent dyes. J. Phys. Chem. Lett. 2020, 11, 5920–5925. [Google Scholar] [CrossRef] [PubMed]
- Jędrzejewska, B.; Krawczyk, P.; Pietrzak, M.; Gordel, M.; Matczyszyn, K.; Samoć, M.; Cysewski, P. Styryl dye possessing donor-π-acceptor structure-synthesis, spectroscopic and computational studies. Dyes Pigments 2013, 99, 673–685. [Google Scholar] [CrossRef]
- Matsumura, K.; Ono, M.; Yoshimura, M.; Kimura, H.; Watanabe, H.; Okamoto, Y.; Ihara, M.; Takahashi, R.; Saji, H. Synthesis and biological evaluation of novel styryl benzimidazole derivatives as probes for imaging of neurofibrillary tangles in Alzheimer’s disease. Bioorg. Med. Chem. 2013, 21, 3356–3362. [Google Scholar] [CrossRef]
- Shi, P.C.; Jiang, X.D.; Gao, R.N.; Dou, Y.Y.; Zhao, W.L. Synthesis and application of Vis/NIR dialkylaminophenylbuta-1,3-dienyl borondipyrromethene dyes. Chin. Chem. Lett. 2015, 26, 834–838. [Google Scholar] [CrossRef]
- Wang, N.; Wang, C. The Effect of Para Substitution on the Rate of Alkaline Hydrolysis of Ethyl 5-Ethyl-2,4-pentadienoates. J. Org. Chem. 1971, 36, 3178–3179. [Google Scholar] [CrossRef]
- Togninelli, A.; Gevariya, H.; Alongi, M.; Botta, M. An improved general method for palladium catalyzed alkenylations and alkynylations of aryl halides under microwave conditions. Tetrahedron Lett. 2007, 48, 4801–4803. [Google Scholar] [CrossRef]
- Ullrich, F.W.; Breitmaier, E. Vinylogous vilsmeier formylation with 3-(N,N-dimethylamino)-acroleins. Synthesis 1983, 641–645. [Google Scholar] [CrossRef]
- Szukalski, A.; Jędrzejewska, B.; Krawczyk, P.; Bajorek, A. An optical modulator on the pyrazolone-BASED bi-component system. Dyes Pigments 2020, 172, 107805. [Google Scholar] [CrossRef]
- Olmsted, J. Calorimetric determinations of absolute fluorescence quantum yields. J. Phys. Chem. 1979, 83, 2581–2584. [Google Scholar] [CrossRef]
Scheme 1.
General routes for the preparation of derivatives of p-aminocinnamic aldehyde.

Figure 1.
1H NMR spectra of 4-(dimethylamino)-2,6-dimethylbenzaldehyde and 4-(dimethylamino)-2,6-dimethylcinnamaldehyde (1d) in DMSO-d6.
Figure 1.
1H NMR spectra of 4-(dimethylamino)-2,6-dimethylbenzaldehyde and 4-(dimethylamino)-2,6-dimethylcinnamaldehyde (1d) in DMSO-d6.

Figure 2.
Normalized electronic absorption spectra of selected p-aminocinnamaldehydes in ethyl acetate and methanol.
Figure 2.
Normalized electronic absorption spectra of selected p-aminocinnamaldehydes in ethyl acetate and methanol.

Figure 3.
Cyclic voltammperogram of compound 1a in anhydrous acetonitrile. Solution contained tetra-n-butylammonium perchlorate as the supporting electrolyte and was deoxygenated prior to analysis by purging with helium for 15 min. Scan rate was 100 mVs−1.
Figure 3.
Cyclic voltammperogram of compound 1a in anhydrous acetonitrile. Solution contained tetra-n-butylammonium perchlorate as the supporting electrolyte and was deoxygenated prior to analysis by purging with helium for 15 min. Scan rate was 100 mVs−1.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Absorption maxima wavelength (λmax abs; nm), molar absorption coefficient (εmax; M−1 cm−1), fluorescence maxima wavelength (λmax fl; nm), fluorescence quantum yield (ϕfl; %) and Stokes shift (∆νSt; cm−1) in ethyl acetate and methanol.
Table 1.
Absorption maxima wavelength (λmax abs; nm), molar absorption coefficient (εmax; M−1 cm−1), fluorescence maxima wavelength (λmax fl; nm), fluorescence quantum yield (ϕfl; %) and Stokes shift (∆νSt; cm−1) in ethyl acetate and methanol.
| Abbr. | Ethyl Acetate | Methanol | ||||
|---|---|---|---|---|---|---|
| λmax abs (εmax) | λmax fl (ϕfl) | ∆νSt | λmax abs (εmax) | λmax fl (ϕfl) | ∆νSt | |
| 1a | 372.5 (43,000) | 437 (0.42) | 3962 | 389 (37,700) | 474 (0.33) | 4610 |
| 1b | 379.5 (44,500) | 438 (0.69) | 3519 | 395.5 (45,700) | 477 (0.20) | 4320 |
| 1c | 378 (35,500) | 444 (0.33) | 3933 | 395.5 (36,900) | 474 (0.14) | 4187 |
| 1d | 374 (35,200) | 474 (0.71) | 5641 | 392 (34,900) | not fluorescent | - |
| 1e | 379.5 (43,900) | 440 (0.61) | 3623 | 396.5 (40,200) | 476 (0.30) | 4212 |
| 1f | 368.5 (33,800) | 440 (0.99) | 4410 | 385 (31,500) | 481 (0.28) | 5184 |
| 1g | 357 (31,400) | 438 (0.51) | 5180 | 368 (28,900) | 486 (0.38) | 6598 |
| 1h | 378 (33,800) | 460 (1.19) | 4716 | 395.5 (33,900) | 485 (0.18) | 4666 |
| 1i | 385 (40,500) | 453 (1.14) | 3899 | 402.5 (35,600) | 486 (0.26) | 4269 |
| 1j | 396.5 (41,800) | 460 (1.88) | 3482 | 417.5 (38,300) | 488 (0.24) | 3460 |
Table 2.
The measured oxidation potential of the tested aminocinnamaldehydes.
| Abbr. | Eox (mV) | Abbr. | Eox (mV) |
|---|---|---|---|
| 1a | 1285 | 1f | 1228 |
| 1b | 1219 | 1g | 1300 |
| 1c | 1194 | 1h | 1093 |
| 1d | 1081 | 1i | 1132 |
| 1e | 1186 | 1j | 870 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pietrzak, M.; Jędrzejewska, B. Aromatic Amines in Organic Synthesis. Part II. p-Aminocinnamaldehydes. Molecules 2021, 26, 4360. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144360
AMA Style
Pietrzak M, Jędrzejewska B. Aromatic Amines in Organic Synthesis. Part II. p-Aminocinnamaldehydes. Molecules. 2021; 26(14):4360. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144360
Chicago/Turabian StylePietrzak, Marek, and Beata Jędrzejewska. 2021. "Aromatic Amines in Organic Synthesis. Part II. p-Aminocinnamaldehydes" Molecules 26, no. 14: 4360. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144360