
Application of Experimental Design Methodologies in the Enantioseparation of Pharmaceuticals by Capillary Electrophoresis: A Review

 , , , and
, , , and
Abstract
:1. Introduction
2. Experimental Designs Applied in CE Chiral Separations
2.1. Selection of Experimental Design
2.2. Screening Designs
2.2.1. Two-Level Full Factorial Design
2.2.2. Two-Level Fractional Factorial Design
Plackett-Burman Design
2.2.3. Asymmetric and Symmetric Screening Designs
2.3. Optimization Designs
- Three-level full factorial design
- Orthogonal array design
2.3.1. Central Composite Design
2.3.2. Box-Behnken Design
2.3.3. Doehlert Design
2.3.4. D-Optimal Designs
2.4. Model Evaluation and Validation
2.5. Application of DoE in Analytical QbD
2.6. Analytical Design Space and Method Operable Design Region
3. Application of DoE in the Development of Chiral CE Methods
3.1. Capillary Zone Electrophoresis (CZE)
3.2. Capillary Electrophoresis—Mass Spectrometry (CE-MS)
3.3. Micellar Electrokinetic Chromatography (MEKC)
3.4. Non-Aqueous Capillary Electrophoresis (NACE)
3.5. Capillary Electrochromatography (CEC)
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| AQbD | Analytical Quality by Design |
| ASD | Asymmetric screening design |
| BBD | Box-Behnken design |
| BGE | background electrolyte |
| CCC | Central composite circumscribed design |
| CCD | Central composite design |
| CD | cyclodextrin |
| CE | capillary electrophoresis |
| CEC | capillary electrochromatography |
| CE-MS | capillary electrophoresis—mass spectrometry |
| CE-ESI-MS | capillary electrophoresis coupled with electrospray ionization mass spectrometry |
| CMA | Critical Method Attribute |
| CMP | Critical Method Parameter |
| CM-β-CD | carboxymethyl-β-cyclodextrin |
| CPP | critical process parameter |
| CQA | critical quality attributes |
| CS | chiral selector |
| CSP | chiral stationary phase |
| CZE | capillary zone electrophoresis |
| D-oD | D-optimal design |
| D-Phe | D-Phenylalanine |
| DoE | Design of Experiments |
| DD | Doehlert design |
| DM-β-CD | dimethyl-β-cyclodextrin |
| DS | Design Space |
| EMA | European Medicines Agency |
| EOF | electroosmotic flow |
| FDA | Food and Drug Administration |
| FCCD | Face centered central composite design |
| FFD | Full factorial design |
| FLEC | l-(9-fluorenyl)ethyl chloroformate |
| FMOC | 9-fluorenylmethyl chloroformate |
| FrFD | Fractional factorial design |
| HDAS-β-CD | heptakis(2,3-di-O-acetyl-6-O-sulfo)-β-cyclodextrin |
| HDM-β-CD | heptakis-2,6-di-O-methyl-β-cyclodextrin |
| HDMS-β-CD | heptakis-(2,3-di-O-methyl-6-O-sulfo)-β-cyclodextrin |
| HES-β-CD | heptakis-6-sulfato-β-CD |
| HMAS-β-CD | heptakis(2-O-methyl-3-O-acetyl-6-O-sulfo)-β-cyclodextrin |
| HPLC | high performance liquid chromatography |
| HP-β-CD | hydroxypropyl-β-cyclodextrin |
| HP-γ-CD | hydroxypropyl-γ-cyclodextrin |
| HS-β-CD | highly sulphated-β-cyclodextrin |
| HTM-β-CD | heptakis (2,3,6-tri-O-methyl)-β-cyclodextrin |
| HSA | human serum albumin |
| ICH | International Council for Harmonisation |
| LIF | laser induced fluorescence |
| LOD | limit of detection |
| LOQ | limit of quantification |
| M-α-CD | methyl-α-cyclodextrin |
| M-β-CD | methyl-β-cyclodextrin |
| M-γ-CD | randomly methylated γ-cyclodextrin |
| MDA | 3,4-methylenedioxyamphetamine |
| MDEA | 3,4-methylenedioxyethylamphetamine |
| MDMA | 3,4-methylenedioxymethamphetamine |
| MEKC | micellar electrokinetic chromatography |
| MODR | Method operable design region |
| NACE | nonaqueous capillary electrophoresis |
| OAD | Orthogonal array design |
| ODMS-γ-CD | octakis(2,3-di-O-methyl-6-O-sulfo)-γ-cyclodextrin |
| OFAT | one factor at a time |
| PBD | Plackett-Burman design |
| PEG | polyethylene glycol |
| PO | polar organic |
| QbD | Quality by Design |
| RP | reverse phase |
| RSM | Response Surface Methodology |
| S-β-CD | sulfated-β-CD |
| S-γ-CD | sulfated-γ-CD |
| SBE-β-CD | sulfobutylether-β-cyclodextrin |
| SDS | sodium dodecyl sulfate |
| S/N | signal-to-noise |
| SSD | Symmetric screening design |
| SNRI | serotonin and norepinephrine reuptake inhibitor |
| SSRI | selective serotonin reuptake inhibitor |
References
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef]
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef]
- Ward, T.J.; Ward, K.D. Chiral separations: A review of current topics and trends. Anal. Chem. 2012, 84, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Chankvetadze, B. Contemporary theory of enantioseparations in capillary electrophoresis. J. Chromatogr. A 2018, 1567, 2–25. [Google Scholar] [CrossRef]
- Krait, S.; Konjaria, M.L.; Scriba, G.K.E. Advances of capillary electrophoresis enantioseparations in pharmaceutical analysis (2017–2020). Electrophoresis 2021. [Google Scholar] [CrossRef]
- Fanali, S.; Chankvetadze, B. Some thoughts about enantioseparations in capillary electrophoresis. Electrophoresis 2019, 40, 2420–2437. [Google Scholar] [CrossRef]
- Yu, R.B.; Quirino, J.P. Chiral Selectors in Capillary Electrophoresis: Trends during 2017–2018. Molecules 2019, 24, 1135. [Google Scholar] [CrossRef] [Green Version]
- Pharmacopoeial Discussion Group. European Pharmacopoeia, 10th ed.; Council of Europe: Strasbourg, France, 2020. [Google Scholar]
- Fukuda, I.M.; Pinto, C.F.F.; Moreira, C.D.S.; Saviano, A.M.; Lourenço, F.R. Design of experiments (DoE) applied to pharmaceutical and analytical quality by design (QbD). Braz. J. Pharm. Sci. 2018, 54, 1–16. [Google Scholar] [CrossRef]
- Sangshetti, J.N.; Deshpande, M.; Zaherr, Z.; Shinde, D.B.; Arote, R. Quality by design approach: Regulatory need. Arab. J. Chem. 2017, 10, 3412–3425. [Google Scholar] [CrossRef] [Green Version]
- Politis, S.N.; Colombo, P.; Colombo, G.; Rekkas, D.M. Design of experiments (DoE) in pharmaceutical development. Drug. Dev. Ind. Pharm. 2017, 43, 889–901. [Google Scholar] [CrossRef]
- Siouffi, A.M.; Phan-Tan-Luu, R. Optimization methods in chromatography and capillary electrophoresis. J. Chromatogr. A 2000, 892, 75–106. [Google Scholar] [CrossRef]
- Sentellas, S.; Saurina, J. Chemometrics in capillary electrophoresis. Part A: Methods for optimization. J. Sep. Sci. 2003, 26, 875–885. [Google Scholar] [CrossRef]
- Sentellas, S.; Saurina, J. Chemometrics in capillary electrophoresis. Part B: Methods for data analysis. J. Sep. Sci. 2003, 26, 1395–1402. [Google Scholar] [CrossRef]
- Hanrahan, G.; Montes, R.; Gomez, F.A. Chemometric experimental design-based optimization techniques in capillary electrophoresis: A critical review of modern applications. Anal. Bioanal. Chem. 2008, 390, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Orlandini, S.; Gotti, R.; Furlanetto, S. Multivariate optimization of capillary electrophoresis methods: A critical review. J. Pharm. Biomed. Anal. 2014, 87, 290–307. [Google Scholar] [CrossRef]
- Bezerra, M.A.; Santelli, R.E.; Oliveira, E.P.; Villar, L.S.; Escaleira, L.A. Response surface methodology (RSM) as a tool for optimization in analytical chemistry. Talanta 2008, 76, 965–977. [Google Scholar] [CrossRef]
- Vera Candioti, L.; De Zan, M.M.; Cámara, M.S.; Goicoechea, H.C. Experimental design and multiple response optimization. Using the desirability function in analytical methods development. Talanta 2014, 124, 123–138. [Google Scholar] [CrossRef]
- Eriksson, L.; Johansson, E.; Kettaneh-Wold, N.; Wikström, C.; Wold, S. Design of Experiments-Principles and Applications, 3rd ed.; Umetrics AB: Umeå, Sweden, 2008. [Google Scholar]
- ICH. ICH Harmonised Tripartite Guideline; Validation of Analytical Procedures: Text and Methodology Q2(R1) International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; ICH: Geneva, Switzerland, 2005. [Google Scholar]
- Brynn Hibbert, D. Experimental design in chromatography: A tutorial review. J. Chromatogr. B 2012, 910, 2–13. [Google Scholar] [CrossRef]
- Montgomery, D.C. Design and Analysis of Experiments, 4th ed.; John Wiley & Sons: New York, NY, USA, 1997. [Google Scholar]
- Dejaegher, B.; Mangelings, D.; Heyden, Y.V. Experimental design methodologies in the optimization of chiral CE or CEC separations: An overview. Methods Mol. Biol. 2013, 970, 409–427. [Google Scholar] [CrossRef]
- Massart, D.L.; Vandeginste, B.G.M.; Buydens, L.M.C.; De Jong, S.; Lewi, P.J.; Smeyers-Verbeke, J. Handbook of Chemometrics and Qualimetrics: Part. A, 1st ed.; Elsevier Science B.V.: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Dejaegher, B.; Vander Heyden, Y. Experimental designs and their recent advances in set-up, data interpretation, and analytical applications. J. Pharm. Biomed. Anal. 2011, 56, 141–158. [Google Scholar] [CrossRef]
- Goupy, J.L. Methods for Experimental Design, 1st ed.; Elsevier Science Publishers B.V.: Amsterdam, The Netherlands, 1993. [Google Scholar]
- Lee, R. Statistical design of experiments for screening and optimization. Chem. Ing. Tech. 2019, 91, 191–200. [Google Scholar] [CrossRef]
- Lundstedt, T.; Seifert, E.; Abramo, L.; Thelin, B.; Nyström, A.; Pettersen, J.; Bergman, R. Experimental design and optimization. Chemom. Intell. Lab. Syst. 1998, 42, 3–40. [Google Scholar] [CrossRef]
- Deming, S.N.; Morgan, S.L. Experimental Design: A Chemometric Approach, 2nd ed.; Elsevier Science Publishers B.V.: Amsterdam, The Netherlands, 1993. [Google Scholar]
- Ranade, S.S.; Thiagarajan, P. Selection of a design for response surface. IOP Conf. Ser. Mater. Sci. Eng. 2017, 263, 022043. [Google Scholar] [CrossRef] [Green Version]
- Brereton, R.G. Chemometrics-Data Analysis for the Laboratory and Chemical Plant, 1st ed.; John Wiley & Sons Ltd.: Chichester, UK, 2003. [Google Scholar]
- Atkinson, A.C.; Donev, A.N. Optimum Experimental Designs; Oxford Science Publications: Oxford, UK, 1992. [Google Scholar]
- De Aguiar, P.F.; Bourguignon, B.; Khots, M.S.; Massart, D.L.; Phan-Than-Luu, R. D-optimal designs. Chemom. Intell. Lab. Syst. 1995, 30, 199–210. [Google Scholar] [CrossRef]
- Siebertz, K.; van Bebber, D.; Hochkirchen, T. Grundlagen. In Statistische Versuchsplanung; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- ICH. ICH Harmonised Tripartite Guideline. Pharmaceutical Development Q8(R2) International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; ICH: Geneva, Switzerland, 2009. [Google Scholar]
- ICH. ICH Harmonised Tripartite Guideline. Quality Risk Management Q9 International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; ICH: Geneva, Switzerland, 2005. [Google Scholar]
- ICH. ICH Harmonised Tripartite Guideline. Pharmaceutical Quality System Q10 International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; ICH: Geneva, Switzerland, 2008. [Google Scholar]
- ICH. ICH Harmonised Tripartite Guideline. Development and Manufacture of Drug Substances Q11 International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; ICH: Geneva, Switzerland, 2012. [Google Scholar]
- Borman, P.; Nethercote, P.; Chatfield, M.; Thompson, D.; Truman, K. The application of Quality by Design to Analytical Methods. Pharm. Tech. 2007, 31, 142–152. [Google Scholar]
- Vogt, F.G.; Kord, A.S. Development of Quality-By-Design Analytical Methods. J. Pharm. Sci. 2011, 100, 797–812. [Google Scholar] [CrossRef] [PubMed]
- Rozet, E.; Lebrun, P.; Debrus, B.; Boulanger, B.; Hubert, P. Design Spaces for analytical methods. Trends Anal. Chem. 2013, 42, 157–167. [Google Scholar] [CrossRef]
- Orlandini, S.; Pinzauti, S.; Furlanetto, S. Application of quality by design to the development of analytical separation methods. Anal. Bioanal. Chem. 2013, 405, 443–450. [Google Scholar] [CrossRef]
- Deidda, R.; Orlandini, S.; Hubert, P.; Hubert, C. Risk-based approach for method development in pharmaceutical quality control context: A critical review. J. Pharm. Biomed. Anal. 2018, 161, 110–121. [Google Scholar] [CrossRef]
- Dispas, A.; Avohou, H.T.; Lebrun, P.; Hubert, P.; Hubert, C. ‘Quality by Design’ approach for the analysis of impurities in pharmaceutical drug products and drug substances. Trends Anal. Chem. 2018, 101, 24–33. [Google Scholar] [CrossRef]
- Derringer, G.; Suich, R. Simultaneous optimization of several response variables. J. Qual. Technol. 1980, 12, 214–219. [Google Scholar] [CrossRef]
- Altria, K.D.; Goodall, D.M.; Rogan, M.M. Chiral separation of β-amino alcohols by capillary electrophoresis using cyclodextrins as buffer additives. I. Effect of varying operating parameters. Chromatographia 1992, 34, 19–24. [Google Scholar] [CrossRef]
- Rogan, M.M.; Altria, K.D.; Goodall, D.M. Plackett-Burman experimental design in chiral analysis using capillary electrophoresis. Chromatographia 1994, 38, 723–729. [Google Scholar] [CrossRef]
- Small, T.S.; Fell, A.F.; Coleman, M.W.; Berridge, J.C. Central composite design for the rapid optimisation of ruggedness and chiral separation of amlodipine in capillary electrophoresis. Chirality 1995, 7, 226–234. [Google Scholar] [CrossRef]
- Bechet, I.; Paques, P.; Fillet, M.; Hubert, P.; Crommen, J. Chiral separation of basic drugs by capillary zone electrophoresis with cyclodextrin additives. Electrophoresis 1994, 15, 818–823. [Google Scholar] [CrossRef]
- Boonkerd, S.; Detaevernier, M.R.; Vander Heyden, Y.; Vindevogel, J.; Michotte, Y. Determination of the enantiomeric purity of dexfenfluramine by capillary electrophoresis: Use of a Plackett-Burman design for the optimization of the separation. J. Chromatogr. A 1996, 736, 281–289. [Google Scholar] [CrossRef]
- Varesio, E.; Gauvrit, J.-Y.; Longeray, R.; Lantéri, P.; Veuthey, J.-L. Central composite design in the chiral analysis of amphetamines by capillary electrophoresis. Electrophoresis 1997, 18, 931–937. [Google Scholar] [CrossRef]
- Fanali, S.; Furlanetto, S.; Aturki, Z.; Pinzauti, S. Experimental design methodologies to optimize the CE separation of epinephrine enantiomers. Chromatographia 1998, 48, 395–401. [Google Scholar] [CrossRef]
- Guillaume, Y.C.; Peyrin, E. Chemometric method to optimize chiral separation of imidazole derivatives by capillary electrophoresis. Talanta 1999, 50, 533–540. [Google Scholar] [CrossRef]
- Vargas, M.G.; Vander Heyden, Y.; Maftouh, M.; Massart, D.L. Rapid development of the enantiomeric separation of β-blockers by capillary electrophoresis using an experimental design approach. J. Chromatogr. A 1999, 855, 681–693. [Google Scholar] [CrossRef]
- Daali, Y.; Cherkaoui, S.; Christen, P.; Veuthey, J.-L. Experimental design for enantioselective separation of celiprolol by capillary electrophoresis using sulfated β-cyclodextrin. Electrophoresis 1999, 20, 3424–3431. [Google Scholar] [CrossRef]
- De Boer, T.; Bijma, R.; Ensing, K. Modelling of conditions for the enantiomeric separation of β2-adrenergic sympathicomimetics by capillary electrophoresis using cyclodextrins as chiral selectors in a polyethylene glycol gel. J. Pharm. Biomed. Anal. 1999, 19, 529–537. [Google Scholar] [CrossRef]
- Zhu, X.; Lin, B.; Jakob, A.; Epperlein, U.; Koppenhoefer, B. Optimization and parameter study for chiral separation by capillary electrophoresis. HRC J. High. Resolut. Chromatogr. 1999, 22, 449–453. [Google Scholar] [CrossRef]
- Perrin, C.; Vargas, M.G.; Vander Heyden, Y.; Maftouh, M.; Massart, D.L. Fast development of separation methods for the chiral analysis of amino acid derivatives using capillary electrophoresis and experimental designs. J. Chromatogr. A 2000, 883, 249–265. [Google Scholar] [CrossRef]
- Gotti, R.; Furlanetto, S.; Andrisano, V.; Cavrini, V.; Pinzauti, S. Design of experiments for capillary electrophoretic enantioresolution of salbutamol using dermatan sulfate. J. Chromatogr. A 2000, 875, 411–422. [Google Scholar] [CrossRef]
- Mateus, L.; Cherkaoui, S.; Christen, P.; Veuthey, J.-L. Enantioseparation of atropine by capillary electrophoresis using sulfated β-cyclodextrin: Application to a plant extract. J. Chromatogr. A 2000, 868, 285–294. [Google Scholar] [CrossRef]
- Loukas, Y.L.; Sabbah, S.; Scriba, G.K.E. Method development and validation for the chiral separation of peptides in the presence of cyclodextrins using capillary electrophoresis and experimental design. J. Chromatogr. A 2001, 931, 141–152. [Google Scholar] [CrossRef]
- Brunnkvist, H.; Karlberg, B.; Granelli, I. Enantiomeric separation of TAPP, H-Tyr-(D)Ala-Phe-Phe-NH2, by capillary electrophoresis using 18-crown-6-tetracarboxylic acid as a chiral selector. J. Chromatogr. B 2003, 793, 343–350. [Google Scholar] [CrossRef]
- Saavedra, L.; Barbas, C. Optimization of the separation lactic acid enantiomers in body fluids by capillary electrophoresis. J. Chromatogr. B 2002, 766, 235–242. [Google Scholar] [CrossRef]
- Harang, V.; Tysk, M.; Westerlund, D.; Isaksson, R.; Johansson, G. A statistical experimental design to study factors affecting enantioseparation of propranolol by capillary electrophoresis with cellobiohydrolase (Cel7A) as chiral selector. Electrophoresis 2002, 23, 2306–2319. [Google Scholar] [CrossRef]
- Ficarra, R.; Cutroneo, P.; Aturki, Z.; Tommasini, S.; Calabrò, M.L.; Phan-Tan-Luu, R.; Fanali, S.; Ficarra, P. An experimental design methodology applied to the enantioseparation of a non-steroidal anti-inflammatory drug candidate. J. Pharm. Biomed. Anal. 2002, 29, 989–997. [Google Scholar] [CrossRef]
- Awadallah, B.; Schmidt, P.C.; Wahl, M.A. Quantitation of the enantiomers of ofloxacin by capillary electrophoresis in the parts per billion concentration range for in vitro drug absorption studies. J. Chromatogr. A 2003, 988, 135–143. [Google Scholar] [CrossRef]
- Marchesini, A.F.; Williner, M.R.; Mantovani, V.E.; Goicoechea, H.C.; Robles, J.C. Optimization of the enantioseparation of ketamine by using capillary electrophoresis and β-carboxy-cyclodextrins. Acta Farm. Bonaer. 2004, 23, 201–205. [Google Scholar]
- Perrin, C.; Fabre, H.; Massart, D.L.; Vander Heyden, Y. Influence of peak measurement parameters on the quality of chiral electrophoretic separations. Electrophoresis 2003, 24, 2469–2480. [Google Scholar] [CrossRef]
- Perrin, C.; Fabre, H.; Maftouh, M.; Massart, D.L.; Vander Heyden, Y. Robustness testing of chiral separations by capillary electrophoresis using highly sulfated cyclodextrins. J. Chromatogr. A 2003, 1007, 165–177. [Google Scholar] [CrossRef]
- Jimidar, M.I.; Van Ael, W.; Van Nyen, P.; Peeters, M.; Redlich, D.; De Smet, M. A screening strategy for the development of enantiomeric separation methods in capillary electrophoresis. Electrophoresis 2004, 25, 2772–2785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-P.; Lee, K.-P.; Kim, S.-H.; Choi, S.-H.; Gopalan, A.I.; Yuan, Z.-B. Comparative study on the chiral separation of phenyl alcohols by capillary electrophoresis and liquid chromatography. Electrophoresis 2004, 25, 2711–2719. [Google Scholar] [CrossRef]
- Jimidar, M.I.; Vennekens, T.; Van Ael, W.; Redlich, D.; De Smet, M. Optimization and validation of an enantioselective method for a chiral drug with eight stereoisomers in capillary electrophoresis. Electrophoresis 2004, 25, 2876–2884. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gomez, M.A.; Villanueva-Camañas, R.M.; Sagrado, S.; Medina-Hernández, M.J. Multivariate optimization approach for chiral resolution of drugs using human serum albumin in affinity electrokinetic chromatography-partial filling technique. Electrophoresis 2005, 26, 4116–4126. [Google Scholar] [CrossRef]
- Elek, J.; Mangelings, D.; Iványi, T.; Lázár, I.; Vander Heyden, Y. Enantioselective capillary electrophoretic separation of tryptophane- and tyrosine-methylesters in a dual system with a tetra-oxadiaza-crown-ether derivative ad a cyclodextrin. J. Pharm. Biomed. Anal. 2005, 38, 601–608. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Jun Zhang, Y.; Jun Gong, W.; Ming Wang, S.; Yong Xue, H.; Pill Lee, K. Design of experiments for capillary electrophoretic enantioresolution of tamsulosin using sulfated-β-cyclodextrin as chiral selector. J. Liq. Chromatogr. Rel. Technol. 2007, 30, 215–234. [Google Scholar] [CrossRef]
- Danel, C.; Chaminade, P.; Odou, P.; Bartélémy, C.; Azarzar, D.; Bonte, J.-P.; Vaccher, C. Enantioselective analysis of the antipsychotic 9-hydroxyrisperidone, main metabolite of risperidone, by chiral capillary EKC using dual CDs. Electrophoresis 2007, 28, 2683–2692. [Google Scholar] [CrossRef]
- Sungthong, B.; Jáč, P.; Scriba, G.K.E. Development and validation of a capillary electrophoresis method for the simultaneous determination of impurities of escitalopram including the R-enantiomer. J. Pharm. Biomed. Anal. 2008, 46, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Borges, K.B.; Pupo, M.T.; De Freitas, L.A.P.; Bonato, P.S. Box-Behnken design for the optimization of an enantioselective method for the simultaneous analysis of propranolol and 4-hydroxypropranolol by CE. Electrophoresis 2009, 30, 2874–2881. [Google Scholar] [CrossRef]
- Gomis, D.B.; Velasco, C.B.; Sanchez, I.H.; Alvarez, M.D.G. Optimization by factorial design of a capillary electrophoresis method for the chiral separation and determination of zopiclone and its synthesis precursor. J. Liq. Chrom. Rel. Technol. 2009, 32, 2654–2668. [Google Scholar] [CrossRef]
- Fayed, A.S.; Weshahy, S.A.; Shehata, M.A.; Hassan, N.Y.; Pauwels, J.; Hoogmartens, J.; Van Schepdael, A. Separation and determination of clopidogrel and its impurities by capillary electrophoresis. J. Pharm. Biomed. Anal. 2009, 49, 193–200. [Google Scholar] [CrossRef]
- Liu, M.; Zheng, Y.; Ji, Y.; Zhang, C. Development and validation of a capillary electrophoresis method for the enantiomeric purity determination of RS86017 using experimental design. J. Pharm. Biomed. Anal. 2011, 55, 93–100. [Google Scholar] [CrossRef]
- Neumajer, G.; Sohajda, T.; Darcsi, A.; Tóth, G.; Szente, L.; Noszál, B.; Béni, S. Chiral recognition of dapoxetine enantiomers with methylated-gamma-cyclodextrin: A validated capillary electrophoresis method. J. Pharm. Biomed. Anal. 2012, 62, 42–47. [Google Scholar] [CrossRef]
- Harnisch, H.; Scriba, G.K.E. Capillary electrophoresis method for the determination of (R)-dapoxetine, (3S)-3-(dimethylamino)-3-phenyl-1-propanol, (S)-3-amino-3-phenyl-1-propanol and 1-naphthol as impurities of dapoxetine hydrochloride. J. Pharm. Biomed. Anal. 2019, 162, 257–263. [Google Scholar] [CrossRef]
- Zhang, Q.; Du, Y. Evaluation of the enantioselectivity of glycogen-based synergistic system with amino acid chiral ionic liquids as additives in capillary electrophoresis. J. Chromatogr. A 2013, 1306, 97–103. [Google Scholar] [CrossRef]
- Asensi-Bernardi, L.; Escuder-Gilabert, L.; Martín-Biosca, Y.; Medina-Hernández, M.J.; Sagrado, S. Modeling the chiral resolution ability of highly sulfated β-cyclodextrin for basic compounds in electrokinetic chromatography. J. Chromatogr. A 2013, 1308, 152–160. [Google Scholar] [CrossRef]
- Rogez-Florent, T.; Foulon, C.; Six, P.; Goossens, L.; Danel, C.; Goossens, J.F. Optimization of the enantioseparation of a diaryl-pyrazole sulphonamide derivative by capillary electrophoresis in a dual CD mode using experimental design. Electrophoresis 2014, 35, 2765–2771. [Google Scholar] [CrossRef] [PubMed]
- Wahl, O.; Holzgrabe, U. Evaluation of enantiomeric purity of magnesium-L-aspartate dihydrate. J. Pharm. Biomed. Anal. 2015, 102, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Orlandini, S.; Pasquini, B.; Del Bubba, M.; Pinzauti, S.; Furlanetto, S. Quality by design in the chiral separation strategy for the determination of enantiomeric impurities: Development of a capillary electrophoresis method based on dual cyclodextrin systems for the analysis of levosulpiride. J. Chromatogr. A 2015, 1380, 177–185. [Google Scholar] [CrossRef]
- Szabó, Z.-I.; Tóth, G.; Völgyi, G.; Komjáti, B.; Hancu, G.; Szente, L.; Sohajda, T.; Béni, S.; Muntean, D.L.; Noszál, B. Chiral separation of asenapine enantiomers by capillary electrophoresis and characterization of cyclodextrin complexes by NMR spectroscopy, mass spectrometry and molecular modelling. J. Pharm. Biomed. Anal. 2016, 117, 398–404. [Google Scholar] [CrossRef]
- Szabó, Z.-I.; Szöcs, L.; Muntean, D.L.; Noszál, B.; Tõth, G. Chiral separation of uncharged pomalidomide enantiomers using carboxymethyl-β-cyclodextrin: A validated capillary electrophoretic method. Chirality 2016, 28, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Szabó, Z.-I.; Foroughbakhshfasaei, M.; Gál, R.; Horváth, P.; Komjáti, B.; Noszál, B.; Tóth, G. Chiral separation of lenalidomide by liquid chromatography on polysaccharide-type stationary phases and by capillary electrophoresis using cyclodexrin selectors. J. Sep. Sci. 2018, 41, 1414–1423. [Google Scholar] [CrossRef]
- Kazsoki, A.; Fejos, I.; Sohajda, T.; Zhou, W.; Hu, W.; Szente, L.; Béni, S. Development and validation of a cyclodextrin-modified capillary electrophoresis method for the enantiomeric separation of vildagliptin enantiomers. Electrophoresis 2016, 37, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Krait, S.; Douša, M.; Scriba, G.K.E. Quality by Design-guided development of a capillary electrophoresis method for the chiral purity determination of ambrisentan. Chromatographia 2016, 79, 1343–1350. [Google Scholar] [CrossRef]
- Meng, R.; Kang, J. Determination of the stereoisomeric impurities of sitafloxacin by capillary electrophoresis with dual chiral additives. J. Chromatogr. A 2017, 1506, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Cârcu-Dobrin, M.; Budău, M.; Hancu, G.; Gagyi, L.; Rusu, A.; Kelemen, H. Enantioselective analysis of fluoxetine in pharmaceutical formulations by capillary zone electrophoresis. Saudi Pharm. J. 2017, 25, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Harnisch, H.; Chien, Y.; Scriba, G.K.E. Capillary electrophoresis method for the chiral purity determination of pregabalin derivatized with dansyl chloride. Chromatographia 2018, 81, 719–725. [Google Scholar] [CrossRef]
- Krait, S.; Scriba, G.K.E. Quality by design-assisted development of a capillary electrophoresis method for the chiral purity determination of dexmedetomidine. Electrophoresis 2018, 39, 2575–2580. [Google Scholar] [CrossRef]
- Krait, S.; Heuermann, M.; Scriba, G.K.E. Development of a capillary electrophoresis method for the determination of the chiral purity of dextromethorphan by a dual selector system using quality by design methodology. J. Sep. Sci. 2018, 41, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, B.; Orlandini, S.; Villar-Navarro, M.; Caprini, C.; Del Bubba, M.; Douša, M.; Giuffrida, A.; Gotti, R.; Furlanetto, S. Chiral capillary zone electrophoresis in enantioseparation and analysis of cinacalcet impurities: Use of Quality by Design principles in method development. J. Chromatogr. A 2018, 1568, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Martínez, M.G.; Ramírez-Galicia, G. Molecular modeling and chiral separation of benzodiazepines by capillary electrophoresis using highly sulfated cyclodextrins. J. Mex. Chem. Soc. 2018, 62, 358–370. [Google Scholar] [CrossRef]
- Wahl, J.; Holzgrabe, U. Capillary electrophoresis separation of phenethylamine enantiomers using amino acid based ionic liquids. J. Pharm. Biomed. Anal. 2018, 148, 245–250. [Google Scholar] [CrossRef]
- De Cock, B.; Mangelings, D.; Vander Heyden, Y. Interinstrumental transfer of a chiral capillary electrophoretic method: The use of robustness test information to overcome differences in detector and data-handling specifications. Chromatographia 2018, 81, 335–348. [Google Scholar] [CrossRef]
- Abdel-Megied, A.M.; Hanafi, R.S.; Aboul-Enein, H.Y. A chiral enantioseparation generic strategy for anti-Alzheimer and antifungal drugs by short end injection capillary electrophoresis using an experimental design approach. Chirality 2018, 30, 165–176. [Google Scholar] [CrossRef]
- Papp, L.A.; Hancu, G.; Gyéresi, Á.; Kelemen, H.; Szabó, Z.-I.; Noszál, B.; Dubský, P.; Tóth, G. Chiral separation of lansoprazole and rabeprazole by capillary electrophoresis using dual cyclodextrin systems. Electrophoresis 2019, 40, 2799–2805. [Google Scholar] [CrossRef]
- Sarkany, A.; Hancu, G.; Cârje, A.; Drăguț, C.; Papp, L.A. Chiral separation of tramadol enantiomers by capillary electrophoresis using cyclodextrins as chiral selectors and experimental design method optimization. Chem. Pap. 2019, 73, 2363–2370. [Google Scholar] [CrossRef]
- Niedermeier, S.; Scriba, G.K.E. Quality by Design-based development of a chiral capillary electrophoresis method for the determination of dextrodropropizine and 1-phenylpiperazine as impurities of levodropropizine. Chromatographia 2020, 83, 123–129. [Google Scholar] [CrossRef]
- Cârcu-Dobrin, M.; Sabău, A.G.; Hancu, G.; Árpád, G.; Rusu, A.; Kelemen, H.; Papp, L.A.; Cârie, A. Chiral discrimination of amlodipine from pharmaceutical products using capillary electrophoresis. Braz. J. Pharm. Sci. 2020, 56, e18259. [Google Scholar] [CrossRef] [Green Version]
- Budău, M.; Hancu, G.; Muntean, D.L.; Papp, L.A.; Cârje, A.G.; Garaj, V. Enantioseparation of citalopram enantiomers by capillary electrophoresis: Method development through experimental design and computational modelling. Chirality 2020, 32, 1119–1128. [Google Scholar] [CrossRef]
- Milan, A.; Hancu, G.; Lupu, D.; Budău, M.; Garaj, V.; Kelemen, H. Venlafaxine chiral separation by capillary electrophoresis using cyclodextrin derivatives as chiral selector and experimental design method optimization. Symmetry 2020, 12, 849. [Google Scholar] [CrossRef]
- Rudaz, S.; Cherkaoui, S.; Gauvrit, J.-Y.; Lantéri, P.; Veuthey, J.-L. Experimental designs to investigate capillary electrophoresis-electrospray ionization-mass spectrometry enantioseparation with the partial-filling technique. Electrophoresis 2001, 22, 3316–3326. [Google Scholar] [CrossRef]
- Wan, H.; Andersson, P.E.; Engström, A.; Blomberg, L.G. Direct and indirect chiral separation of amino acids by capillary electrophoresis. J. Chromatogr. A 1995, 704, 179–193. [Google Scholar] [CrossRef]
- Orlandini, S.; Pasquini, B.; Caprini, C.; Del Bubba, M.; Douša, M.; Pinzauti, S.; Furlanetto, S. Enantioseparation and impurity determination of ambrisentan using cyclodextrin-modified micellar electrokinetic chromatography: Visualizing the design space within quality by design framework. J. Chromatogr. A 2016, 1467, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Flor, S.; Huala Juan, M.; Tripodi, V.; Lucangioli, S. Development of an enantioselective capillary electrophoresis method for the simultaneous determination of montelukast enantiomeric and diastereoisomeric forms and its main degradation product. Electrophoresis 2016, 37, 2420–2428. [Google Scholar] [CrossRef]
- Niedermeier, S.; Scriba, G.K.E. A quality by design-based approach to a capillary electrokinetic assay for the determination of dextromepromazine and levomepromazine sulfoxide as impurities of levomepromazine. J. Pharm. Biomed. Anal. 2017, 146, 402–409. [Google Scholar] [CrossRef]
- Servais, A.C.; Fillet, M.; Chiap, P.; Dewè, W.; Hubert, P.; Crommen, J. Influence of the nature of the electrolyte on the chiral separation of basic compounds in nonaqueous capillary electrophoresis using heptakis(2,3-di-O-methyl-6-O-sulfo)-β-cyclodextrin. J. Chromatogr. A 2005, 1068, 143–150. [Google Scholar] [CrossRef]
- Marini, R.D.; Servais, A.C.; Rozet, E.; Chiap, P.; Boulanger, B.; Rudaz, S.; Crommen, J.; Hubert, P.; Fillet, M. Nonaqueous capillary electrophoresis method for the enantiomeric purity determination of S-timolol using heptakis(2,3-di-O-methyl-6-O-sulfo)-beta-cyclodextrin: Validation using the accuracy profile strategy and estimation of uncertainty. J. Chromatogr. A. 2006, 1120, 102–111. [Google Scholar] [CrossRef]
- Marini, R.D.; Rozet, E.; Vander Heyden, Y.; Ziemons, E.; Boulanger, B.; Bouklouze, A.; Servais, A.-C.; Fillet, M.; Crommen, J.; Hubert, P.H. Robustness testing of a chiral NACE method for R-timolol determination in S-timolol maleate and uncertainty assessment from quantitative data. J. Pharm. Biomed. Anal. 2007, 44, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Chiap, P.; Oprean, R.; Crommen, J.; Fillet, M.; Servais, A.-C. Effect of the nature of the single-isomer anionic CD and the BGE composition on the enantiomeric separation of β-blockers in NACE. Electrophoresis 2009, 30, 2862–2868. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Gillotin, F.; Chiap, P.; Bodoki, E.; Crommen, J.; Fillet, M.; Servais, A.-C. Generic systems for the enantioseparation of basic drugs in NACE using single-isomer anionic CDs. J. Pharm. Biomed. Anal. 2011, 54, 154–159. [Google Scholar] [CrossRef]
- Niedermeier, S.; Scriba, G.K.E. Chiral separation of four phenothiazines by nonaqueous capillary electrophoresis and quality by design-based method development for quantification of dextromepromazine as chiral impurity of levomepromazine. J. Chromatogr. A 2020, 1624, 461232. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.; Wikström, H.; Armstrong, D.W.; Owens, P.K. Enantioselective reversed-phase and non-aqueous capillary electrochromatography using a teicoplanin stationary phase. J. Chromatogr. A 2000, 897, 349–363. [Google Scholar] [CrossRef]
- Wikström, H.; Svensson, L.A.; Torstensson, A.; Owens, P.K. Immobilisation and evaluation of a vancomycin chiral stationary phase for capillary electrochromatography. J. Chromatogr. A 2000, 869, 395–409. [Google Scholar] [CrossRef]
- Mangelings, D.; Tanret, I.; Matthijs, N.; Maftouh, M.; Massart, D.L.; Vander Heyden, Y. Separation strategy for acidic chiral pharmaceuticals with capillary electrochromatography on polysaccharide stationary phases. Electrophoresis 2005, 26, 818–832. [Google Scholar] [CrossRef]
- Vander Heyden, Y.; Khots, M.S.; Massart, D.L. Three-level screening designs for the optimisation or the ruggedness testing of analytical procedures. Anal. Chim. Acta 1993, 276, 189–195. [Google Scholar] [CrossRef]



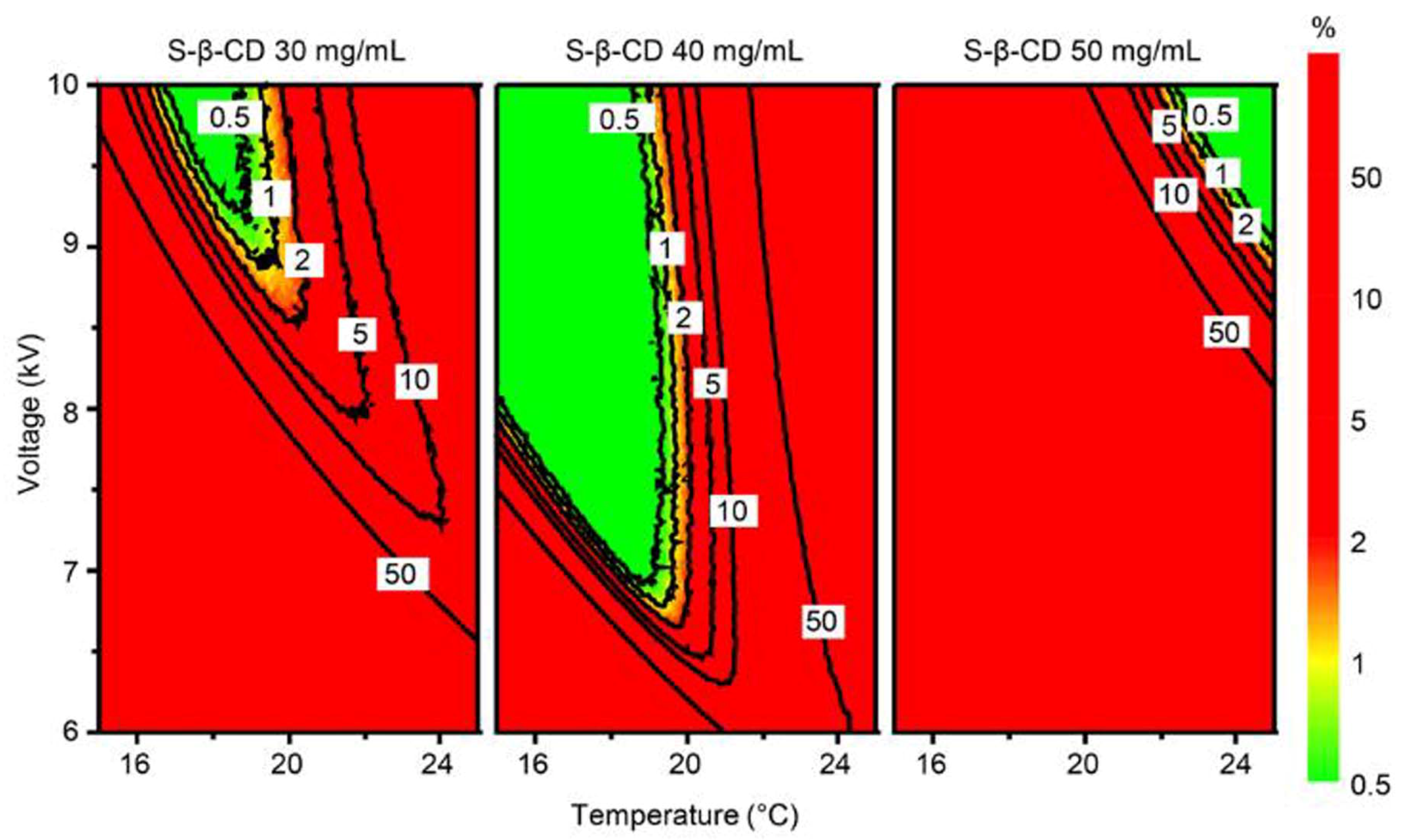{kind=link}
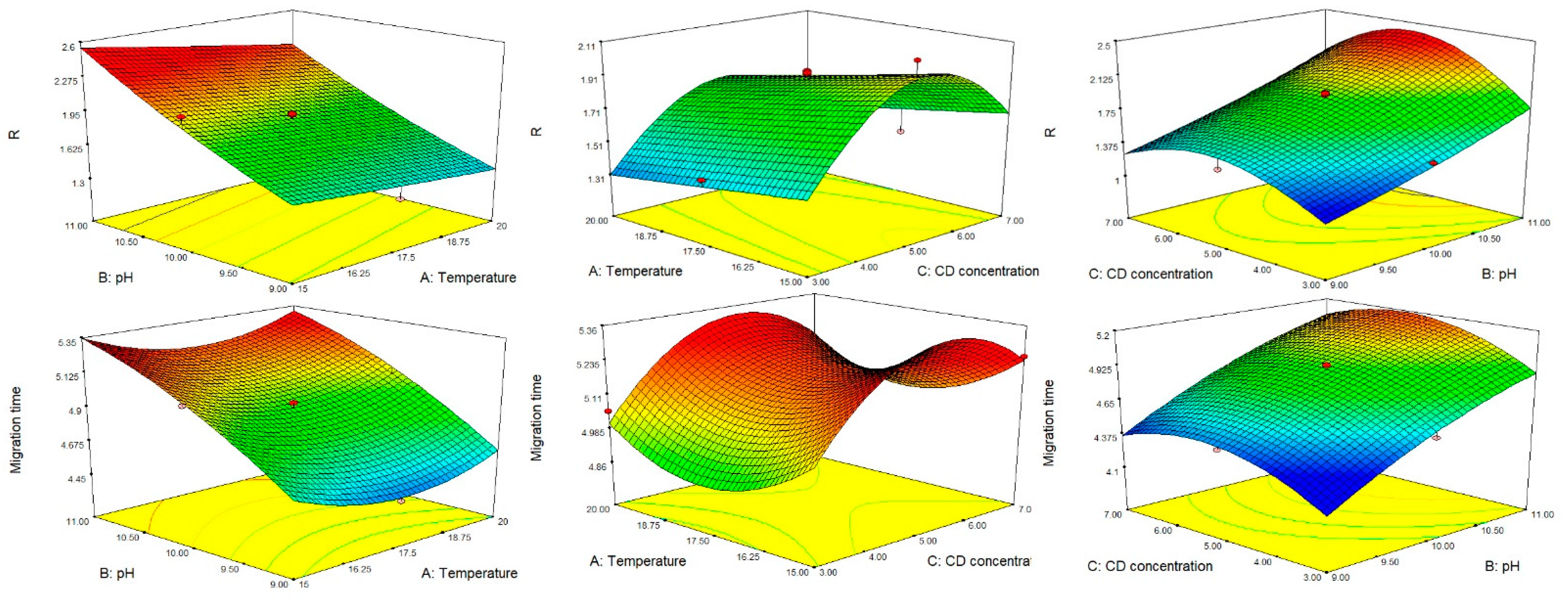{kind=link}
| Analytes | Sample | Experimental Design Type | Factors | Responses | Optimized Conditions | References |
|---|---|---|---|---|---|---|
| Clenbuterol | bulk substance | PBD | BGE pH, BGE concentration, CD concentration, methanol concentration, injection time | resolution, migration time, number of theoretical plates | 0.1 M citric acid/0.2 M phosphate BGE, pH 4.0, 16 mM β-CD, 19 °C, 13 kV, 214 nm | [47] |
| Amlodipine | bulk substance | CCD | BGE pH, CD concentration, temperature | resolution, Kaiser peak separation function | 50 mM phosphate BGE, pH 3.16, 18.2 mM α-CD, 17.2 °C, 25 kV, 214 nm | [48] |
| Dexfenfluramine-chiral purity (levofenfluramine) | bulk substance | PBD | BGE pH, CD concentration, methanol concentration, temperature, voltage | resolution, migration time, peak symmetry | 100 mM phosphate BGE, pH 3.0, 10 mM DM-β-CD, 40% methanol, 20 °C, 25 kV, 214 nm | [50] |
| Amphetamines (amphetamine, methamphetamine, MDA, MDMA, MDEA) | bulk substance | CCD | BGE concentration, BGE pH, CD concentration, temperature, voltage | amphetamine resolution, total resolution, migration time, generated power | 118 mM phosphate BGE, pH 3.5, 16 mM HP-β-CD, 28 °C, 25 kV, 200 nm | [51] |
| Epinephrine | bulk substance | FrFD CCD | BGE concentration, BGE pH, CD concentration, temperature, voltage BGE concentration, CD concentration, voltage | resolution, migration time | 111 mM phosphate BGE, pH 7.0, 9 mM CM-β-CD, 20 °C, 18 kV, 206 nm | [52] |
| Imidazole derivatives (bifonazole, econazole, miconazole, sulconazole) | bulk substance | FrFD | BGE pH, acetonitrile concentration, CD concentration, temperature | resolution, migration time, number of theoretical plates | 100 mM phosphate BGE, pH 4.70, 5.2% acetonitrile, 5.80 mM β-CD, 35 °C, 30 kV, 230 nm | [53] |
| Celiprolol | bulk substance, tablet, urine | FCCD | BGE concentration, BGE pH, CD concentration, temperature | resolution, migration time, current | 52 mM acetate BGE, pH 4.0, 3 mM S-β-CD, 19.5 °C, 30 kV, 220 nm | [55] |
| Salbutamol | spiked urine | PBD DD | BGE concentration, BGE pH, CS concentration, methanol concentration, voltage BGE pH, CS concentration, voltage | resolution, migration time | 30 mM Tris BGE, pH 5.3, 1.75% dermatan sulphate, 5% methanol, 15 °C, 24 kV, 220 nm | [59] |
| Atropine | ophthalmic solution, plant extract | CCD | BGE concentration, BGE pH, CD concentration | resolution, migration time, current | 55 mM phosphate BGE, pH 7.0, 2.9 mM S-β-CD, 20 °C, 20 kV, 195 nm | [60] |
| Methadone | bulk substance | FFD FCCD | CD concentration, percentage of capillary filled with CD, gas nebulization pressure | resolution | CE-ESI-MS: 20 mM ammonium acetate BGE, pH 4.0, 18 mg/mL HP-β-CD/ 0.25 mg/mL CM-β-CD/ 0.4 mg/mL SBE-β-CD, 90/70/90% capillary-filled, nebulization pressure 2 psi | [110] |
| Lactic acid | body fluids (plasma, urine, amniotic and cerebrospinal fluid) | FFD FFD | BGE concentration, BGE pH, CS concentration BGE concentration, CS concentration | resolution | 200 mM phosphate BGE, pH 6.0, 413 mM HP-β-CD, 20 °C,−20 kV, 200 nm | [63] |
| Propranolol | bulk substance | FCCD | BGE concentration, BGE pH, acetonitrile concentration | effective mobility, efficiency, symmetry, selectivity, relative mobility difference, resolution, difference in effective mobility | 15 mM bistris-acetate, pH 6.5, 31.2 µM Cel7A, 17% (v/v) acetonitrile, 22 °C, 20 µA, 200 nm | [64] |
| 2-[(4′-benzoyloxy-2′-hydroxy)phenyl-propionic acid] | bulk substance | CCD | CS concentration, BGE pH, temperature | resolution, migration time | 50 mM Britton-Robinson BGE, pH 6.4, 7 mM vancomycin, 22 °C, 20 kV, 195 nm | [65] |
| Ofloxacin | bulk substance | FCCD | BGE concentration, BGE pH, CD concentration, temperature | resolution, migration time, peak area, current | 50 mM phosphate BGE, pH 2.8, 4% (w/v) M-β-CD, 25 °C, 20 kV, 280 nm | [66] |
| Ketamine | injection solution | PBD CCD | BGE concentration, BGE pH, CD concentration, methanol concentration, voltage BGE pH, methanol concentration | resolution | 50 mM phosphate BGE, pH 5.20, 2% (w/v) CM-β-CD, 30% methanol, 20 °C, 15 kV, 206 nm | [67] |
| Tamsulosin | bulk substance | BBD FCCD CCC | CD concentration, voltage, temperature | resolution, migration time | 100 mM Tris BGE, pH 2.5, 0.15% (w/v) S-β-CD, 25 °C, 25 kV, 210 nm | [75] |
| Risperidone, 9-hydroxyrisperidone | bulk substance | CCD | CD1 concentration, CD2 concentration, BGE concentration | resolution, migration time, selectivity, number of theoretical plates | 80 mM phosphate BGE, pH 2.5, 37 mM HP- β-CD, 3.7% S-α-CD, 25 °C, 20 kV, 270 nm | [76] |
| S-Citalopram--chiral purity (R-Citalopram) | bulk substance, tablets | FCCD | BGE concentration, CD2 concentration, voltage, temperature | resolution, migration time, current | 20 mM phosphate BGE, pH 2.5, 0.5 mg/mL β-CD, 22 mg/mL S-α-CD, 28 °C, −20 kV, 205 nm | [77] |
| Propranolol, 4-hydroxypropranolol | bulk substance | BBD | BGE concentration, BGE pH, CD concentration, voltage | resolution, migration time | 25 mM triethylamine/phosphate BGE, pH 9.0, 4% (w/v) CM-β-CD, 25 °C, 17 kV, 208 nm | [78] |
| Zopiclone | bulk substance, tablets | FFD | BGE concentration, BGE pH, CD concentration, voltage | resolution, migration time | 60.2 mM phosphate BGE, pH 2.0, 1 M urea, 20 mM β-CD, 25 °C, 30 kV, 215 nm | [79] |
| Clopidogrel (chiral purity R-clopidogrel) | bulk substance | reduced CCD | BGE concentration, BGE pH, CD concentration, voltage | resolution, migration time | 10 mM triethylamine–phosphoric acid BGE, pH 2.3, 5% S-β-CD, 20 °C, −12 kV, 195 nm | [80] |
| Dapoxetine | bulk substance | OAD | BGE concentration, BGE pH, CD concentration, methanol concentration, temperature, voltage | resolution | 70 mM acetate BGE, pH 4.5, 3 mM M-γ-CD, 20% (v/v) methanol, 15 °C, 15 kV, 215 nm | [82] |
| Magnesium-L-aspartate (chiral purity D-aspartate) | bulk substance, tablets | FFD | BGE concentration, BGE pH, CD concentration | resolution, migration time | 50 mM phosphate, pH 7, 18 mM HP-β-CD, 18% (v/v) DMSO, 20 °C, 30 kV, LIF detection | [87] |
| Asenapine | bulk substance | OAD | BGE concentration, BGE pH, CD concentration, methanol concentration, temperature, voltage | resolution | 160 mM Tris-acetate BGE, pH 3.5, 7 mM β-CD, 20 °C, 15 kV, 214 nm | [89] |
| Pomalidomide | bulk substance | OAD | BGE concentration, BGE pH, temperature, voltage | resolution | 50 mM Tris-acetate BGE, pH 6.5, 15 mM CM-β-CD, 20 °C, 15 kV, 215 nm | [90] |
| Lenalidomide (chiral purity R-lenalidomide) | bulk substance | FCCD | BGE concentration, temperature, voltage | resolution | 30 mM phosphate BGE, pH 6.5, 30 mM SBE-β-CD, 10 °C, 12 kV, 210 nm | [91] |
| Vildagliptin (chiral purity R-vildagliptin) | bulk substance, tablets | OAD | BGE concentration, BGE pH, CD concentration, temperature, voltage, injection parameters | resolution | 75 mM Tris-acetate BGE, pH 4.75, 20 mM SBE-α-CD, 15 °C, 25 kV, 40 mbar × 4 s hydrodynamic injection, 200 nm | [92] |
| Levosulpiride, (chiral purity R-sulpiride) | bulk substance, tablets | Asymmetric screening matrix DD | BGE concentration, BGE pH, type of neutral cyclodextrin, charged CD concentration, neutral CD concentration, voltage BGE pH, charged CD concentration, neutral CD concentration, voltage | resolution, migration time | 5 mM Britton-Robinson BGE, pH 3.45, 10 mM S-β-CD, 34 mM M-β-CD, 16 °C, −14 kV, 214 nm | [88] |
| Ambrisentan (chiral purity R-ambrisentan) | bulk substance, coated tablets | Asymmetric screening matrix FCCD | BGE concentration, BGE pH, CD concentration, SDS concentration, temperature, voltage, capillary length BGE pH, CD concentration, voltage | resolution, migration time | capillary total length 64.5 cm, 100 mM borate BGE, pH 9.20, 100 mM SDS, 50 mM γ-CD, 22 °C, 30 kV, 200 nm | [112] |
| S-Ambrisentan (chiral purity R-ambrisentan) | bulk substance | FrFD FCCD | concentration, BGE pH, CD concentration, temperature, voltage BGE concentration, temperature, voltage | resolution, migration time | 50 mM sodium acetate BGE, pH 4.0, 30 mM γ-CD, 25 °C, 25 kV, 200 nm | [93] |
| Montelukast (chiral purity R-trans-montelukast, R,S-cis-montelukast, S,R-cis-montelukast) | bulk substance, chewable tablets, oral granules | FFD | BGE concentration, voltage | resolution | 20 mM borate BGE, 10 mM SDS, pH 9.0, 10 mM HTM-β-CD, 12 mM SBE-β-CD, 15 °C, 18 kV, 254 nm | [113] |
| Levomepromazine (chiral purity R-mepromazine) | bulk substance, injection solution | FrFD FCCD | BGE concentration, BGE pH, CD concentration, temperature, voltage BGE concentration, BGE pH, CD concentration | resolution, migration time, selectivity | 100 mM citric acid BGE, pH 2.85, 3.6 mg/mL HP-γ-CD, 15 °C, 25 kV, 253 nm | [114] |
| S-Dapoxetine (chiral purity R-Dapoxetine) | bulk substance, tablets | FrFD FCCD | BGE concentra-tion, BGE pH, CD1-CD2 1:1 mix concentration, temperature, voltage CD1 concentra-tion, CD2 concentration, voltage | resolution, migration time, peak symmetry, current | 50 mM phosphate BGE, pH 6.3, 45 mg/mL S-γ-CD, 40.2 mg/mL DM-β-CD, 15 °C, 9 kV, 215 nm | [83] |
| Sitafloxacin, chiral impurities | bulk substance | FrFD FCCD | BGE concentration, BGE pH, CD concentration, Cu2+ concentration, D-Phe concentration, temperature, voltage BGE pH, CD concentration, Cu2+ concentration, D-Phe concentration | resolution, migration time | 15 mM phosphate BGE, pH 4.5, 15 mM D-Phe, 20 mM CuSO4, 20 mM γ-CD, 25 °C, 15 kV, 297 nm | [94] |
| Fluoxetine | bulk substance, tablets | OAD | BGE concentration, BGE pH, CD concentration, temperature, voltage, injection parameters | resolution | 50 mM phosphate BGE, pH 5.0, 10 mM HTM-β-CD, 15 °C, 20 kV, 50 mbar × 1 s hydrodynamic injection, 230 nm | [95] |
| Pregabalin | bulk substance, capsules | D-oD FCCD | BGE concentration, BGE pH, CD concentration, temperature, voltage CD concentration, temperature, voltage | resolution, migration time | 100 mM phosphate BGE concentration, pH 2.5, 40 mg/mL HTM-β-CD, 25 °C, 15 kV, 220 nm | [96] |
| Dexmedetomidine (chiral purity R-medetomidine) | bulk substance, tablets | FrFD FCCD | BGE concentration, BGE pH, CD concentration, temperature, voltage CD concentration, temperature, voltage | selectivity, migration time, current | 50 mM phosphate BGE, pH 6.5, 40 mg/mL S-β-CD, 17 °C, 10 kV, 200 nm | [97] |
| Dextrometorphan (chiral purity R-metorphan) | bulk substance, tablets | FrFD FCCD | BGE concentration, BGE pH, CD concentration, temperature, voltage CD concentration, voltage | resolution, migration time | 30 mM phosphate BGE, pH 6.5, 16 mg/mL S-β-CD, 14 mg/mL M-α-CD, 20 °C, 20 kV, 200 nm | [98] |
| Cinacalcet (chiral purity S-cinacalcet) | bulk substance, tablets | BBD | BGE pH, CD concentration, methanol concentration, voltage | resolution, migration time | 150 mM phosphate BGE, pH 2.7, 3.1 mM HP-γ-CD, 2% (v/v) methanol, 18 °C, 26 kV, 220 nm | [99] |
| Benzodiazepines (lorazepam, lormetazepam, oxazepam, temazepam) | bulk substance | FrFD | BGE pH, CD type, CD concentration, methanol concentration | resolution | 20 mM borate BGE, pH 9.0, 5% HES-β-CD, 15% methanol, 15 °C, 20 kV, 230 nm | [100] |
| Lansoprazole, Rabeprazole | bulk substance, capsules | FrFD CCD | BGE concentration, BGE pH, CD1 concentration, temperature, voltage CD1 concentration, voltage, temperature | resolution, migration time | Lansoprazole: 25 mM phosphate BGE, pH 7.0, 10 mM SBE-β-CD, 20 mM γ-CD, 17 °C, 20 kV, 210 nm Rabeprazole: 25 mM phosphate BGE, pH 7.0, 15 mM SBE-β-CD, 30 mM γ-CD, 18 °C, 20 kV, 210 nm | [104] |
| Levodropropizine (chiral purity R-dropropizine) | bulk substance, pharmaceutical drops | FFD FCCD | CD concentration, temperature, voltage, 2-propanol concentration CD concentration, temperature | separation factors, migration time, current | 25 mM phosphate BGE, pH 7.0, 23.5 mg/mL S-β-CD, 10% (v/v) 2-propanol, 16.3 °C, 16.5 kV, 200 nm | [106] |
| Levomepromazine (chiral purity R-mepromazine) | bulk substance, tablets | FrFD FCCD | BGE concentration, CD concentration, temperature, voltage BGE concentration, CD concentration, temperature | resolution, migration time, current | 750 mM acetic acid, 55 mM ammonium acetate in methanol BGE, 27.5 mg/mL HDMS-β-CD, 15 °C, 22 kV, 250 nm | [120] |
| Tramadol | bulk substance, capsules | FCCD | BGE pH, CD concentration, temperature | resolution, migration time | 25 mM tetraborate BGE, pH 11.0, 5 mM CM-β-CD, 15 °C, 17.5 kV, 210 nm | [105] |
| Amlodipine | bulk substance, tablets | OAD | BGE concentration, BGE pH, CD concentration, temperature, voltage, injection parameters | resolution | 25 mM phosphate BGE, pH 9.0, 15 mM CM-β-CD, 15 °C, 25 kV, 30 mbar × 1 s hydrodynamic injection, 230 nm | [107] |
| Citalopram | bulk substance, tablets | FrFD FCCD | BGE concentration, BGE pH, CD concentration, temperature, voltage, injection pressure CD concentration, temperature, voltage | resolution, migration time | 25 mM phosphate BGE, pH 7.0, 3 mM CM-β-CD, 17.5 °C, 15 kV, 50 mbar injection pressure, 230 nm | [108] |
| Venlafaxine | bulk substance, capsules | FCCD | CD concentration, temperature, voltage | resolution, migration time | 25 mM phosphate BGE, pH 2.5, 10 mM CM-β-CD, 15 °C, 25 kV, 230 nm | [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hancu, G.; Orlandini, S.; Papp, L.A.; Modroiu, A.; Gotti, R.; Furlanetto, S. Application of Experimental Design Methodologies in the Enantioseparation of Pharmaceuticals by Capillary Electrophoresis: A Review. Molecules 2021, 26, 4681. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154681
Hancu G, Orlandini S, Papp LA, Modroiu A, Gotti R, Furlanetto S. Application of Experimental Design Methodologies in the Enantioseparation of Pharmaceuticals by Capillary Electrophoresis: A Review. Molecules. 2021; 26(15):4681. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154681
Chicago/Turabian StyleHancu, Gabriel, Serena Orlandini, Lajos Attila Papp, Adriana Modroiu, Roberto Gotti, and Sandra Furlanetto. 2021. "Application of Experimental Design Methodologies in the Enantioseparation of Pharmaceuticals by Capillary Electrophoresis: A Review" Molecules 26, no. 15: 4681. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154681