
Doubly Decarboxylative Synthesis of 4-(Pyridylmethyl)chroman-2-ones and 2-(Pyridylmethyl)chroman-4-ones under Mild Reaction Conditions †


Abstract
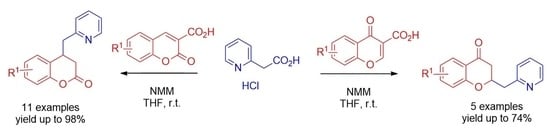
:
1. Introduction
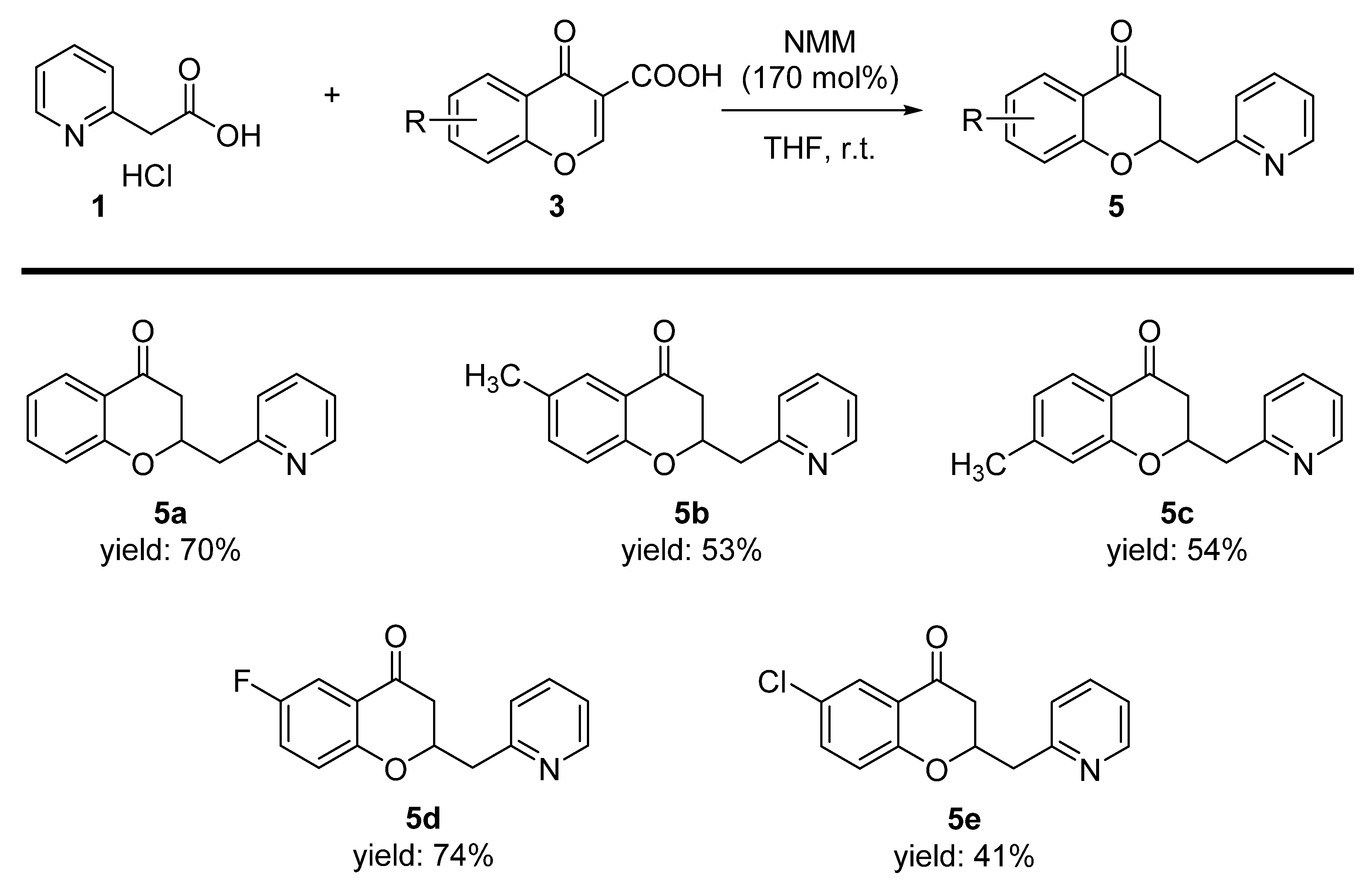
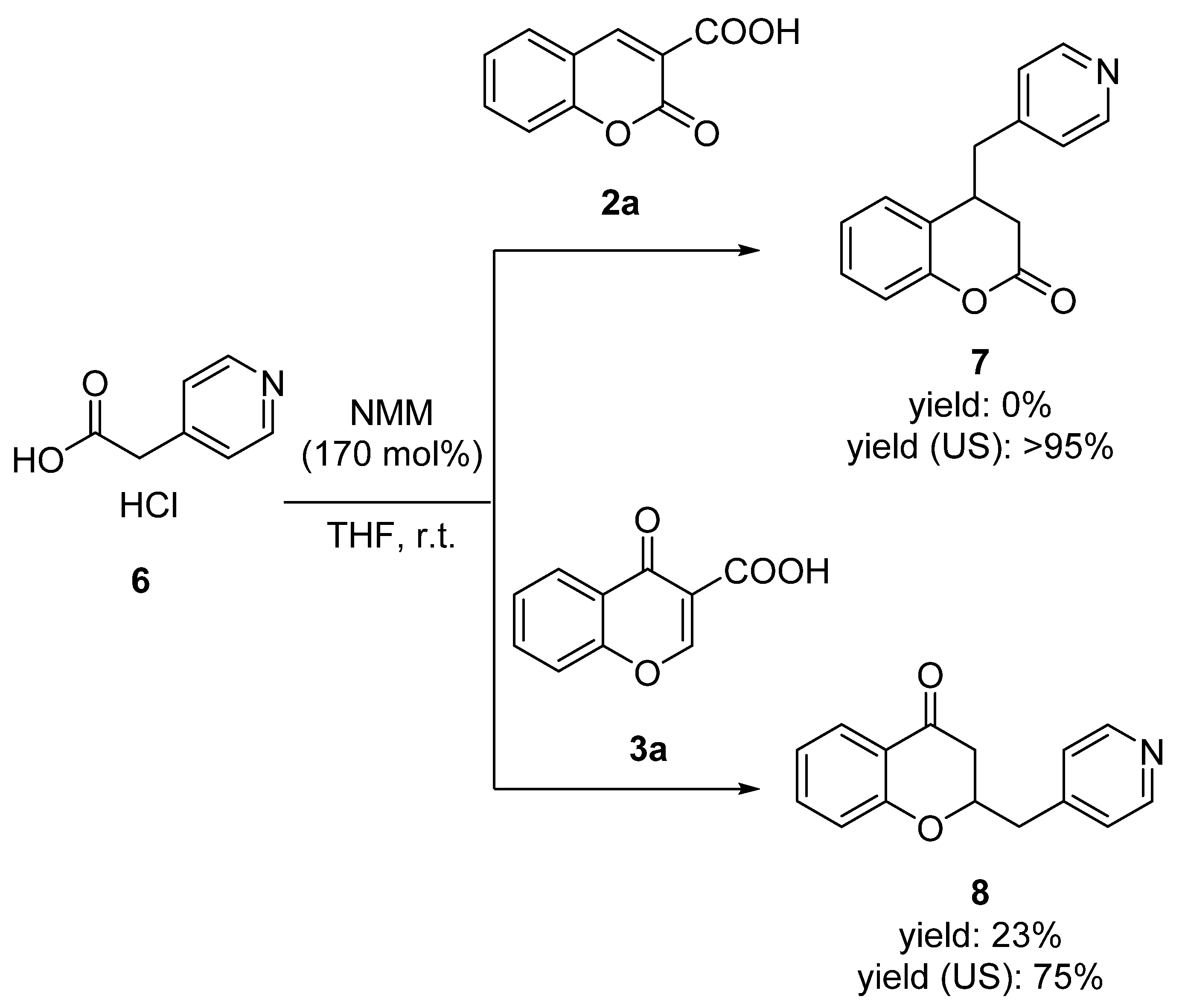
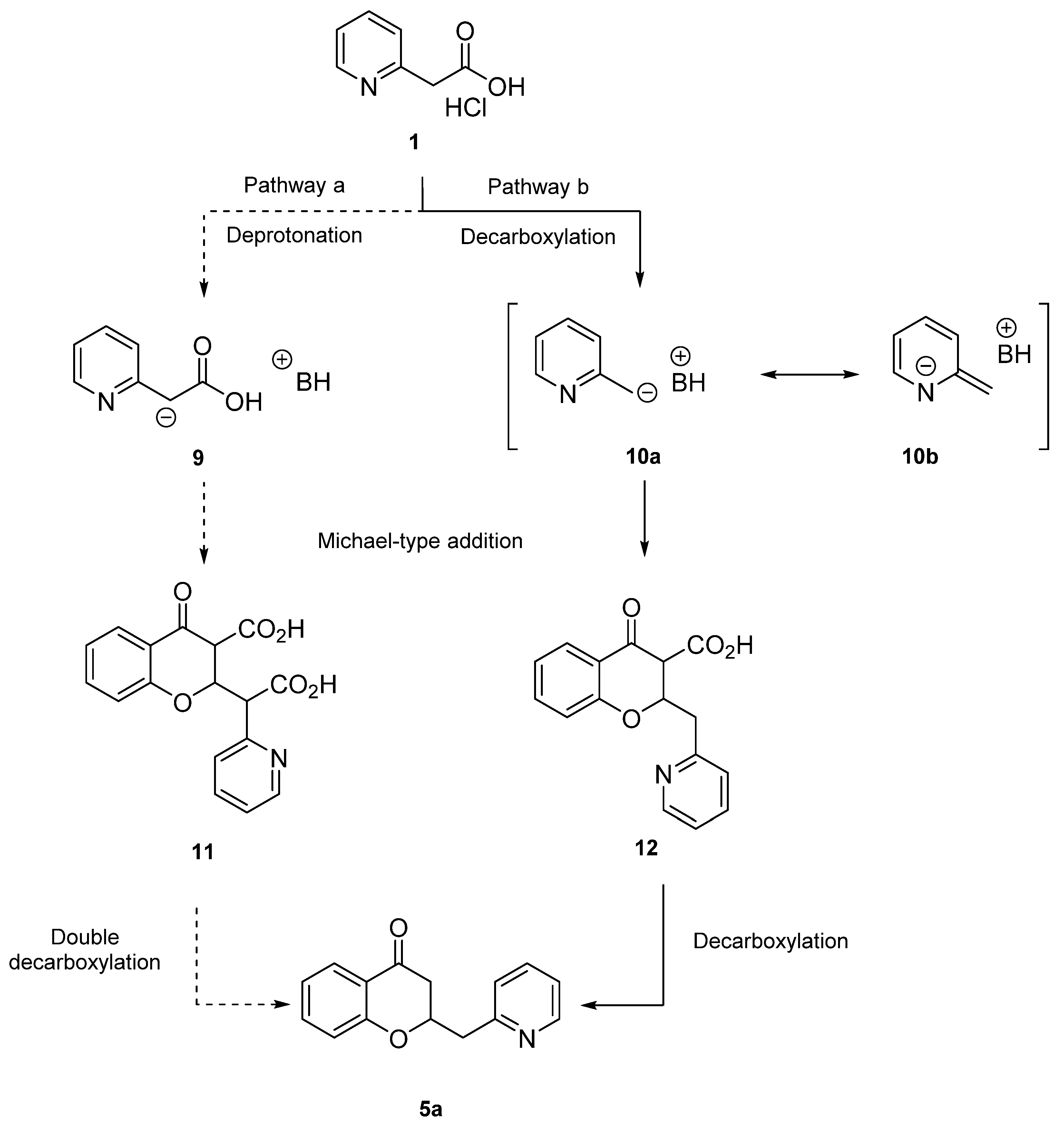
2. Results
3. Conclusions
4. Materials and Methods
4.1. General Methods
4.2. General Procedure
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Tokoroyama, T. Discovery of the Michael Reaction. Eur. J. Org. Chem. 2010, 2010, 2009–2016. [Google Scholar] [CrossRef]
- Bhanja, C.; Jena, S.; Nayak, S.; Mohapatra, S. Organocatalytic tandem Michael addition reactions: A powerful access to the enantioselective synthesis of functionalized chromenes, thiochromenes and 1,2-dihydroquinolines. Beilstein J. Org. Chem. 2012, 8, 1668–1694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, W. Recent advances in organocatalytic asymmetric Michael reactions. Catal. Sci. Technol. 2011, 2, 42–53. [Google Scholar] [CrossRef]
- Kürti, L.; Czakó, B. Strategic Applications of Named Reactions in Organic Synthesis; Elsevier: Amsterdam, The Netherlands, 2005; pp. 286–287. [Google Scholar]
- Perlmutter, P. Conjugate Addition Reactions in Organic Synthesis; Pergamon: Oxford, UK, 1992. [Google Scholar]
- Sibi, M.P.; Manyem, S. Enantioselective Conjugate Additions. Tetrahedron 2000, 56, 8033–8061. [Google Scholar] [CrossRef]
- Krause, N.; Hoffmann-Röder, A. Recent Advances in Catalytic Enantioselective Michael Additions. Synthesis 2001, 2001, 0171–0196. [Google Scholar] [CrossRef]
- Berner, O.M.; Tedeschi, L.; Enders, D. Asymmetric Michael Additions to Nitroalkenes. Eur. J. Org. Chem. 2002, 2002, 1877–1894. [Google Scholar] [CrossRef]
- Vicario, J.; Badía, D.; Carrillo, L. Organocatalytic Enantioselective Michael and Hetero-Michael Reactions. Synthesis 2007, 2007, 2065–2092. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem. 2007, 2007, 1701–1716. [Google Scholar] [CrossRef]
- Almaşi, D.; Alonso, D.A.; Nájera, C. Organocatalytic asymmetric conjugate additions. Tetrahedron Asymmetry 2007, 18, 299–365. [Google Scholar] [CrossRef]
- Tan, C.-H.; Pan, Y. Catalytic Decarboxylative Reactions: Biomimetic Approaches Inspired by Polyketide Biosynthesis. Synthesis 2011, 2011, 2044–2053. [Google Scholar] [CrossRef]
- Wang, Z.-L. Recent Advances in Catalytic Asymmetric Decarboxylative Addition Reactions. Adv. Synth. Catal. 2013, 355, 2745–2755. [Google Scholar] [CrossRef]
- Nakamura, S. Catalytic enantioselective decarboxylative reactions using organocatalysts. Org. Biomol. Chem. 2014, 12, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Bojanowski, J.; Albrecht, A. Carboxylic-Acid-Activated Olefins in Decarboxylative Reactions. Asian J. Org. Chem. 2019, 8, 746–754. [Google Scholar] [CrossRef]
- Nakamura, S.; Toda, A.; Sano, M.; Hatanaka, T.; Funahashi, Y. Organocatalytic Enantioselective Conjugate Addition of Malonic Acid Half Thioesters to Coumarin-3-carboxylic Acids Using N-HeteroarenesulfonylCinchonaAlkaloid Amides. Adv. Synth. Catal. 2016, 358, 1029–1034. [Google Scholar] [CrossRef]
- Ren, Q.; Gao, T.; Li, W.; Wan, L.; Hu, Y.; Peng, Y.; Sun, S.; Hu, L.; Wu, M.; Guo, H.; et al. A highly enantioselective Michael reaction between α,β-unsaturated ketones and malonic acid half-thioesters. New J. Chem. 2015, 39, 5100–5103. [Google Scholar] [CrossRef]
- Ren, Q.; Sun, S.; Huang, J.; Li, W.; Wu, M.; Guo, H.; Wang, J. An enantioselective cascade reaction between α,β-unsaturated aldehydes and malonic half-thioesters: A rapid access to chiral δ-lactones. Chem. Commun. 2014, 50, 6137–6140. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Liu, Q.; Liu, H.; Yan, L.; Jiang, Z. Asymmetric Decarboxylative 1,4-Addition of Malonic Acid Half Thioesters to Vinyl Sulfones: Highly Enantioselective Synthesis of 3-Monofluoromethyl-3-Arylpropanoic Esters. Chem. Asian J. 2014, 9, 1252–1256. [Google Scholar] [CrossRef]
- Kang, Y.K.; Lee, H.J.; Moon, H.W.; Kim, D.Y. Organocatalytic enantioselective decarboxylative Michael addition of β-ketoacids to α,β-unsaturated ketones. RSC Adv. 2013, 3, 1332–1335. [Google Scholar] [CrossRef]
- Bae, H.Y.; Some, S.; Lee, J.H.; Kim, J.-Y.; Song, M.J.; Lee, S.; Zhang, Y.J.; Song, C.E. Organocatalytic Enantioselective Michael-Addition of Malonic Acid Half-Thioesters to β-Nitroolefins: From Mimicry of Polyketide Synthases to Scalable Synthesis of γ-Amino Acids. Adv. Synth. Catal. 2011, 353, 3196–3202. [Google Scholar] [CrossRef]
- Furutachi, M.; Mouri, S.; Matsunaga, S.; Shibasaki, M. A Heterobimetallic Ni/La-salan Complex for Catalytic Asymmetric Decarboxylative 1,4-Addition of Malonic Acid Half-Thioester. Chem. Asian J. 2010, 5, 2351–2354. [Google Scholar] [CrossRef]
- Lubkoll, J.; Wennemers, H. Mimicry of Polyketide Synthases—Enantioselective 1,4-Addition Reactions of Malonic Acid Half-Thioesters to Nitroolefins. Angew. Chem. Int. Ed. 2007, 46, 6841–6844. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.W. Conjugate addition to chromones: Synthesis of substituted 4-chromanones. Tetrahedron Lett. 1984, 25, 4299–4302. [Google Scholar] [CrossRef]
- Neo, A.G.; Díaz, J.; Marcaccini, S.; Marcos, C.F. Conjugate addition of isocyanides to chromone 3-carboxylic acid: An efficient one-pot synthesis of chroman-4-one 2-carboxamides. Org. Biomol. Chem. 2012, 10, 3406–3416. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Wang, L.; Xu, L.; Zhao, H.; Xiao, J. Facile synthesis of azaarene-2-substituted chromanone derivatives via tandem sp3 C–H functionalization/decarboxylation of azaarenes with 4-oxo-4H-chromene-3-carboxylic acid. RSC Adv. 2014, 4, 53188–53191. [Google Scholar] [CrossRef]
- Xu, L.; Shao, Z.; Wang, L.; Xiao, J. Tandem sp3 C–H Functionlization/Decarboxylation of 2-Alkylazaarenes with Coumarin-3-carboxylic Acids. Org. Lett. 2014, 16, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wang, L.; Guo, H.; Sun, S.; Wang, J. Facile synthesis of 4-substituted 3,4-dihydrocoumarins via an organocatalytic double decarboxylation process. Org. Biomol. Chem. 2012, 10, 2537–2541. [Google Scholar] [CrossRef]
- Frankowski, S.; Kowalska, J.; Albrecht, A. Pyridylacetic acids and related systems as alkylheteroarene surrogates in asymmetric decarboxylative Michael addition. Chem. Commun. 2021, 57, 3387–3390. [Google Scholar] [CrossRef]
- Andersen, Ø.M.; Markham, K.R. Flavonoids: Chemistry, Biochemistry and Applications; CRC Press, Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Saengchantara, S.T.; Wallace, T.W. Chromanols, chromanones, and chromones. Nat. Prod. Rep. 1986, 3, 465–475. [Google Scholar] [CrossRef]
- Kamat, D.P.; Tilve, S.G.; Kamat, V.P.; Kirtany, J.K. Syntheses and Biological Activities of Chroman-2-ones. A Review. Org. Prep. Proced. Int. 2015, 47, 1–79. [Google Scholar] [CrossRef]
- Masters, K.-S.; Braese, S. Xanthones from Fungi, Lichens, and Bacteria: The Natural Products and Their Synthesis. Chem. Rev. 2012, 112, 3717–3776. [Google Scholar] [CrossRef]
- Ee, G.C.L.; Mah, S.H.; Teh, S.S.; Rahmani, M.; Go, R.; Taufiq-Yap, Y.H. Soulamarin, a New Coumarin from Stem Bark of Calophyllum soulattri. Molecules 2011, 16, 9721. [Google Scholar] [CrossRef]
- Picker, K.; Ritchie, E.; Taylor, W. The chemical constituents of Australian Flindersia species. XXI. An examination of the bark and the leaves of F. laevicarpa. Aust. J. Chem. 1976, 29, 2023–2036. [Google Scholar] [CrossRef]
- Zhao, D.-L.; Shao, C.-L.; Gan, L.-S.; Wang, M.; Wang, C.-Y. Chromone Derivatives from a Sponge-Derived Strain of the Fungus Corynespora cassiicola. J. Nat. Prod. 2015, 78, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Nibbs, A.E.; Scheidt, K.A. Asymmetric Methods for the Synthesis of Flavanones, Chromanones, and Azaflavanones. Eur. J. Org. Chem. 2011, 2012, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, B.R.; Scheidt, K.A. Pyranone Natural Products as Inspirations for Catalytic Reaction Discovery and Development. Acc. Chem. Res. 2015, 48, 1172–1183. [Google Scholar] [CrossRef] [Green Version]
- Moffett, R.B. Central Nervous System Depressants. VII.1 Pyridyl Coumarins. J. Med. Chem. 1964, 7, 446–449. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Wang, S.; Hong, K.; Li, X.; Liu, P.; Wang, Y.; Zhu, W. Cytotoxic Bipyridines from the Marine-Derived Actinomycete Actinoalloteichus cyanogriseus WH1-2216-6. J. Nat. Prod. 2011, 74, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Olbe, L.; Carlsson, E.; Lindberg, P. A proton-pump inhibitor expedition: The case histories of omeprazole and esomeprazole. Nat. Rev. Drug Discov. 2003, 2, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Shtyrlin, N.V.; Pavelyev, R.S.; Pugachev, M.V.; Sysoeva, L.P.; Musin, R.Z.; Shtyrlin, Y.G. Synthesis of novel 6-substituted sulfur-containing derivatives of pyridoxine. Tetrahedron Lett. 2012, 53, 3967–3970. [Google Scholar] [CrossRef]
- Reynolds, R.D. Bioavailability of vitamin B-6 from plant foods. Am. J. Clin. Nutr. 1988, 48, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, P.T.; Athmaram, T.N.; Arunkumar, G.R. Novel nicotine analogues with potential anti-mycobacterial activity. Bioorganic Med. Chem. 2016, 24, 1637–1647. [Google Scholar] [CrossRef]
- Minisci, F.; Vismara, E.; Fontana, F. Recent Developments of Free-Radical Substitutions of Heteroaromatic Bases. Heterocycles 1989, 28, 489. [Google Scholar] [CrossRef] [Green Version]
- Schlosser, M.; Mongin, F. Pyridine elaboration through organometallic intermediates: Regiochemical control and completeness. Chem. Soc. Rev. 2007, 36, 1161–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, J.A.; Mousseau, J.J.; Pelletier, G.; Charette, A.B. Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines. Chem. Rev. 2012, 112, 2642–2713. [Google Scholar] [CrossRef] [PubMed]
- Chelucci, G. Metal-complexes of optically active amino- and imino-based pyridine ligands in asymmetric catalysis. Coord. Chem. Rev. 2013, 257, 1887–1932. [Google Scholar] [CrossRef]
- Albrecht, A. Utilization of Chromone-3-Carboxylic Acids as Acceptors in the Michael-Type Decarboxylative Addition. Eur. J. Org. Chem. 2018, 2018, 6482–6485. [Google Scholar] [CrossRef]
- Ishizuka, N.; Matsumura, K.-I.; Sakai, K.; Fujimoto, M.; Mihara, S.-I.; Yamamori, T. Structure−Activity Relationships of a Novel Class of Endothelin-A Receptor Antagonists and Discovery of Potent and Selective Receptor Antagonist, 2-(Benzo[1,3]dioxol-5-yl)-6-isopropyloxy-4-(4-methoxyphenyl)-2H-chromene-3- carboxylic Acid (S-1255). 1. Study on Structure−Activity Relationships and Basic Structure Crucial for ETA Antagonism. J. Med. Chem. 2002, 45, 2041–2055. [Google Scholar] [CrossRef] [PubMed]
- Song, A.; Wang, X.; Lam, K.S. A convenient synthesis of coumarin-3-carboxylic acids via Knoevenagel condensation of Meldrum’s acid with ortho-hydroxyaryl aldehydes or ketones. Tetrahedron Lett. 2003, 44, 1755–1758. [Google Scholar] [CrossRef]








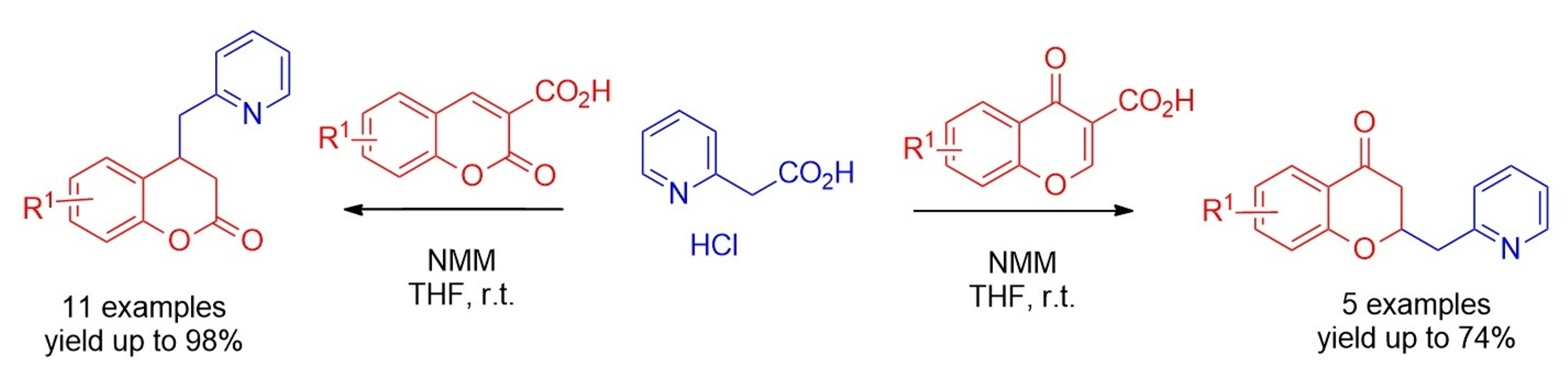{kind=link}
{kind=link}
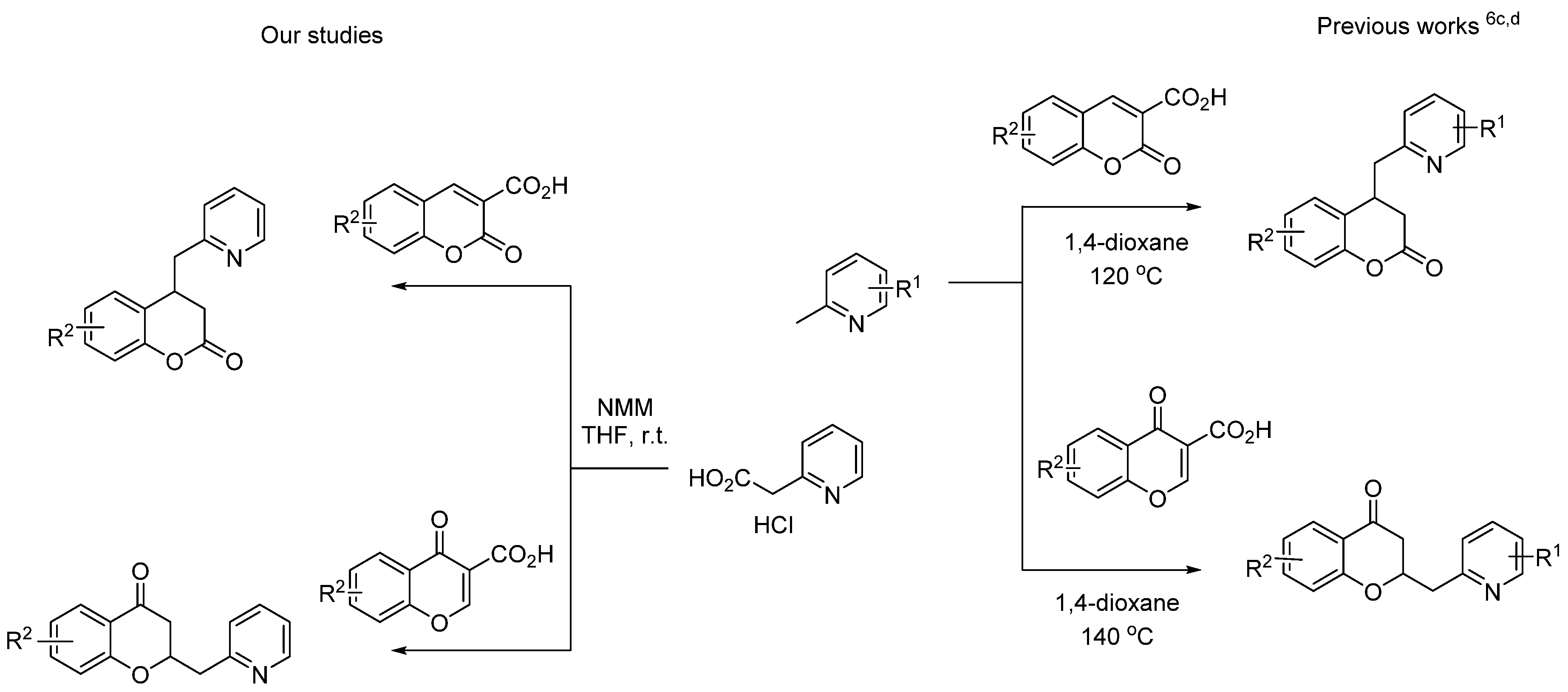{kind=link}
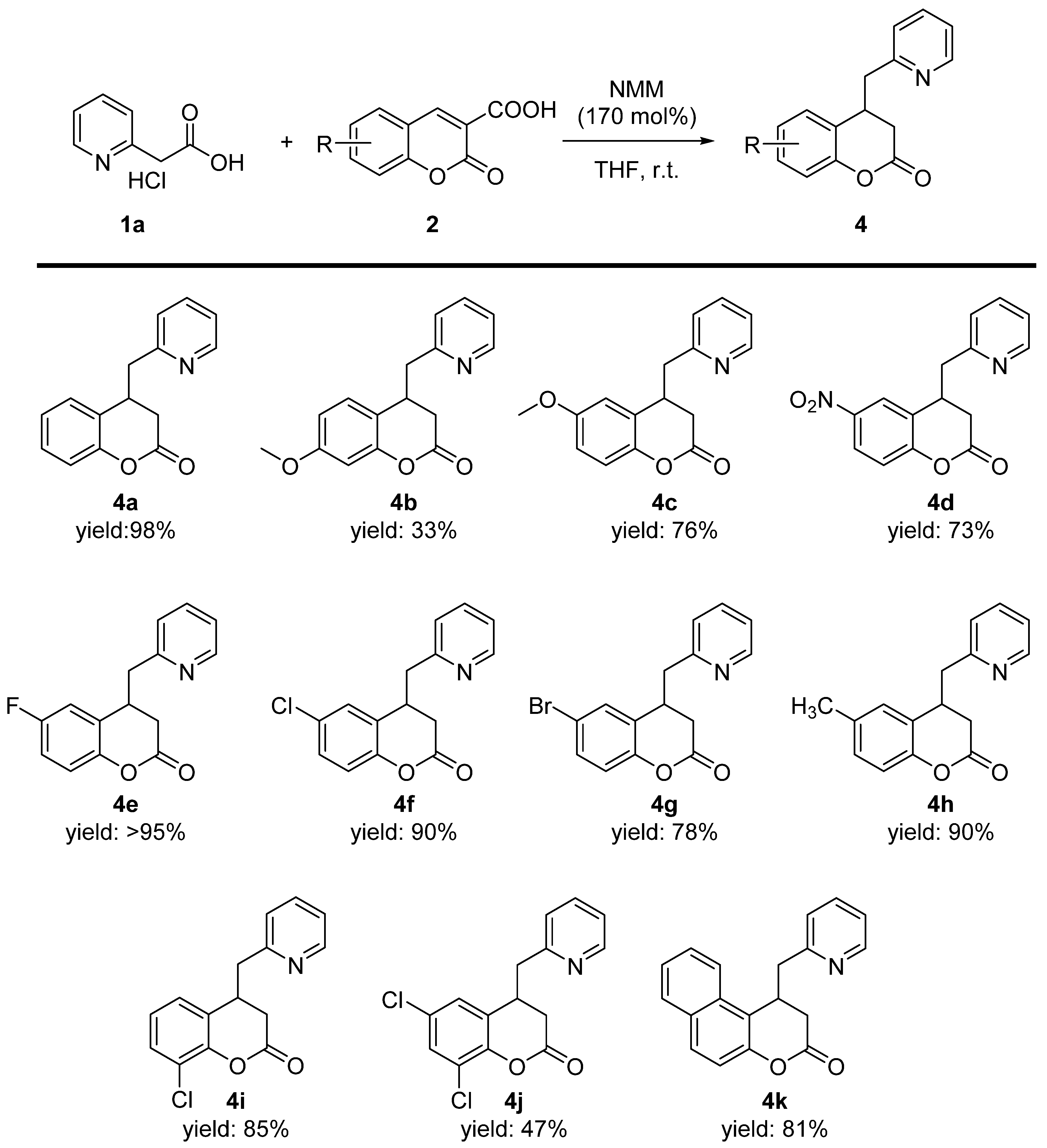{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| No. | Solvent | Base (equiv.) | X | Y | 2 or 3 (equiv) | 1a (equiv.) | Yield of 4a | Yield of 5a |
| 1 | THF | DABCO (1.2) | COOH | COOH | 1.0 | 1.0 | 69 | 23 |
| 2 | THF | DMAP (1.2) | COOH | COOH | 1.0 | 1.0 | 62 | 50 |
| 3 | THF | Pyrrolidine (1.2) | COOH | COOH | 1.0 | 1.0 | 65 | 50 |
| 4 | THF | 2,6-lutidine (1.2) | COOH | COOH | 1.0 | 1.0 | 54 | 38 |
| 5 | THF | TEA (1.2) | COOH | COOH | 1.0 | 1.0 | 71 | 21 |
| 6 | THF | DIPEA (1.2) | COOH | COOH | 1.0 | 1.0 | 74 | 42 |
| 7 | THF | NMM (1.2) | COOH | COOH | 1.0 | 1.0 | 77 | 56 |
| 8 | THF | NMM (1.2) | COOH | COOH | 1.5 | 1.0 | 71 | 63 |
| 9 | THF | NMM (1.2) | COOH | COOH | 1.2 | 1.0 | 47 | 52 |
| 10 | THF | NMM (1.4) | COOH | COOH | 1.0 | 1.2 | 82 | 61 |
| 11 | THF | NMM (1.7) | COOH | COOH | 1.0 | 1.5 | 98 | 70 |
| 12 | THF | NMM (2.2) | COOH | COOH | 1.0 | 2.0 | 92 | 70 |
| 13 | Toluene | NMM (1.7) | COOH | COOH | 1.0 | 1.5 | 19 | 40 |
| 14 | MeCN | NMM (1.7) | COOH | COOH | 1.0 | 1.5 | 75 | 69 |
| 15 | CH2Cl2 | NMM (1.7) | COOH | COOH | 1.0 | 1.5 | 75 | 54 |
| 16 | THF | NMM (1.7) | COOH | H | 2b (1.0) | 1.5 | no reaction | - |
| 17 | THF | NMM (1.7) | COOH | H | 3b (1.0) | 1.5 | - | no reaction |
| 18 | THF | NMM (0.2) | H | COOH | 2a (1.0) | 1b (1.5) | no reaction | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bojanowski, J.; Albrecht, A. Doubly Decarboxylative Synthesis of 4-(Pyridylmethyl)chroman-2-ones and 2-(Pyridylmethyl)chroman-4-ones under Mild Reaction Conditions. Molecules 2021, 26, 4689. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154689
Bojanowski J, Albrecht A. Doubly Decarboxylative Synthesis of 4-(Pyridylmethyl)chroman-2-ones and 2-(Pyridylmethyl)chroman-4-ones under Mild Reaction Conditions. Molecules. 2021; 26(15):4689. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154689
Chicago/Turabian StyleBojanowski, Jan, and Anna Albrecht. 2021. "Doubly Decarboxylative Synthesis of 4-(Pyridylmethyl)chroman-2-ones and 2-(Pyridylmethyl)chroman-4-ones under Mild Reaction Conditions" Molecules 26, no. 15: 4689. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154689