
Targeting Monoacylglycerol Lipase in Pursuit of Therapies for Neurological and Neurodegenerative Diseases

 , , ,
, , ,
Abstract
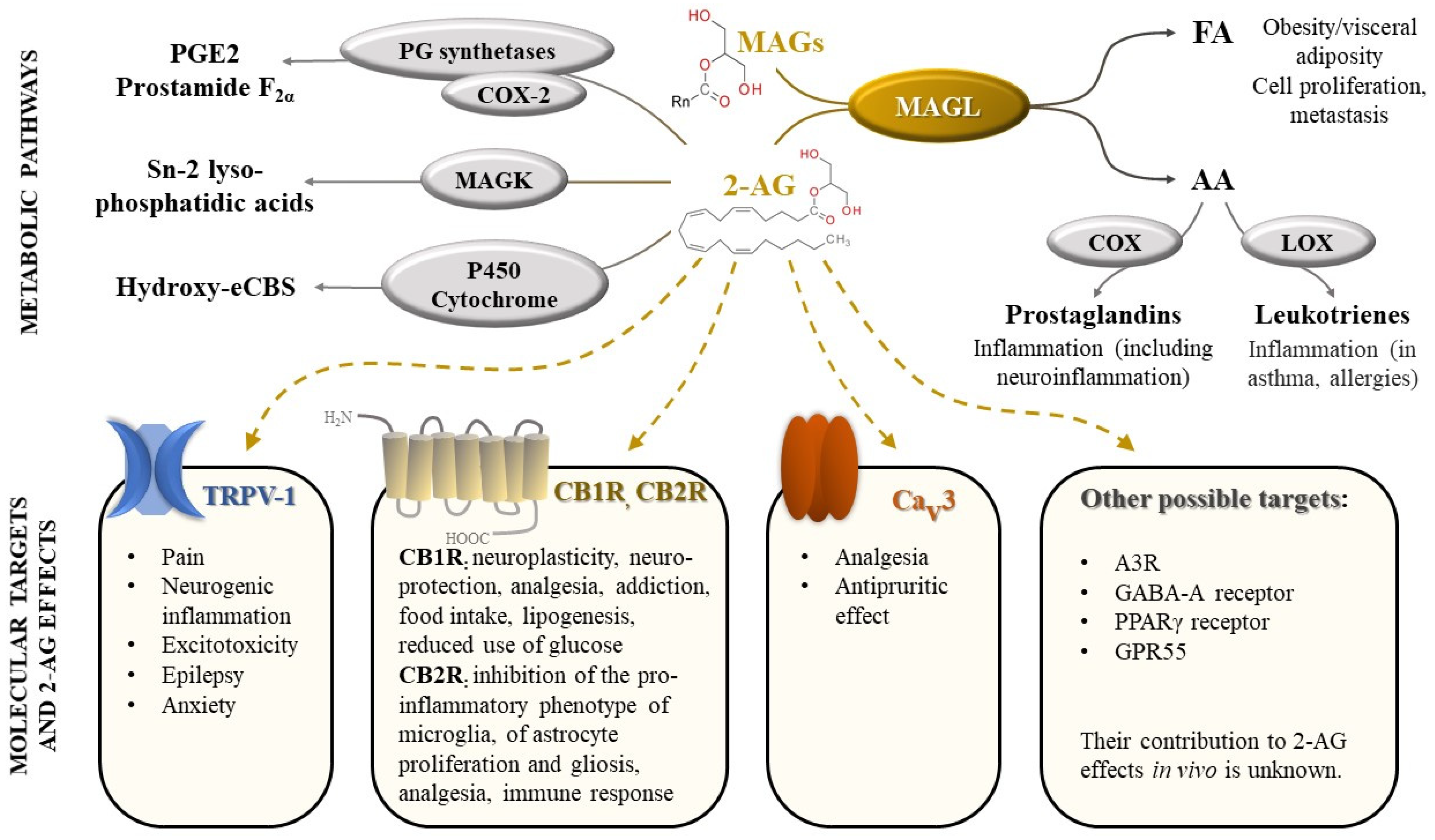
:1. Introduction
2. Molecular Characterization of MAGL and Mechanism of Catalysis
3. Development of Pharmacophore Models for MAGL Inhibitors
4. MAGL Inhibitors
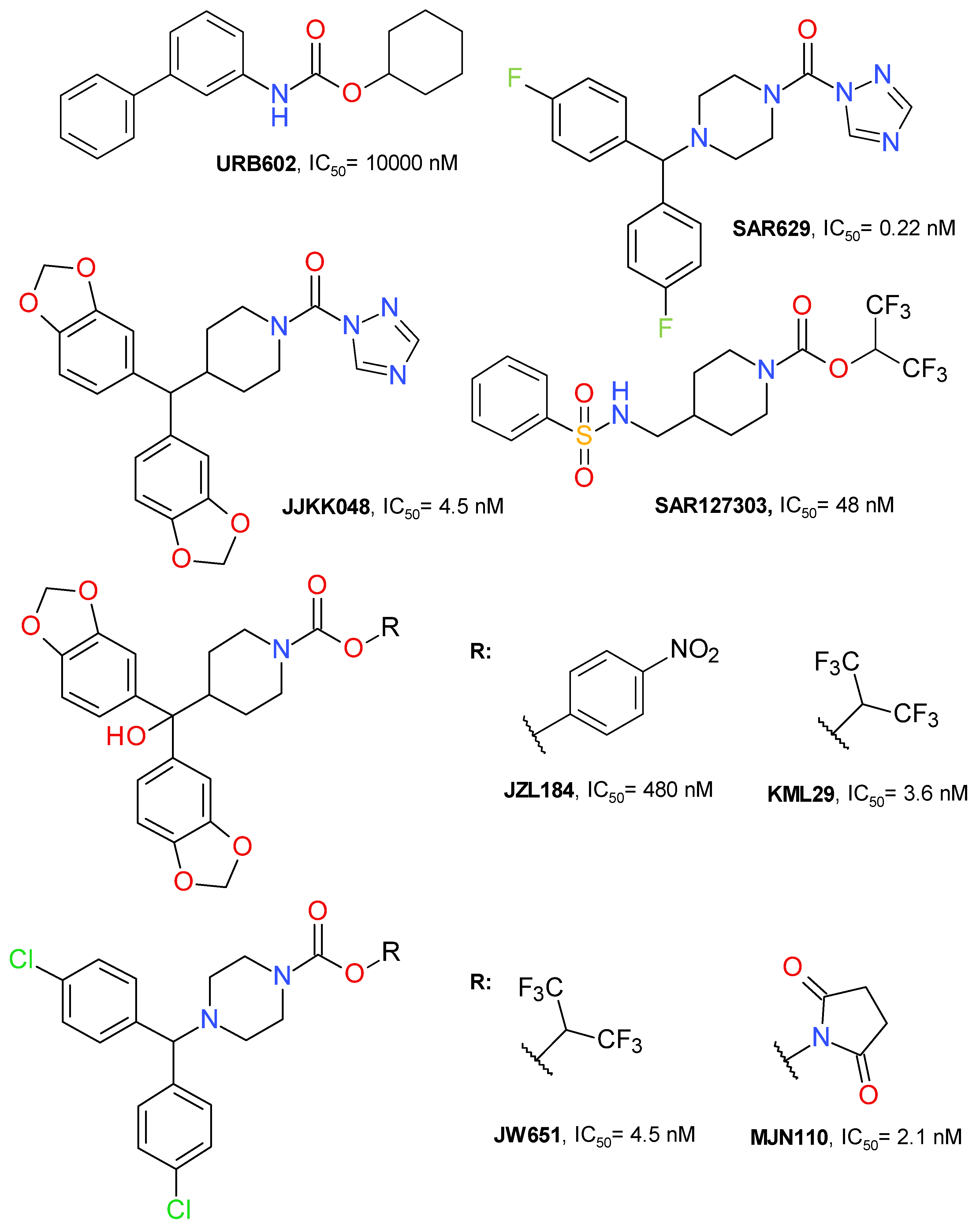
4.1. Covalent Irreversible Inhibitors
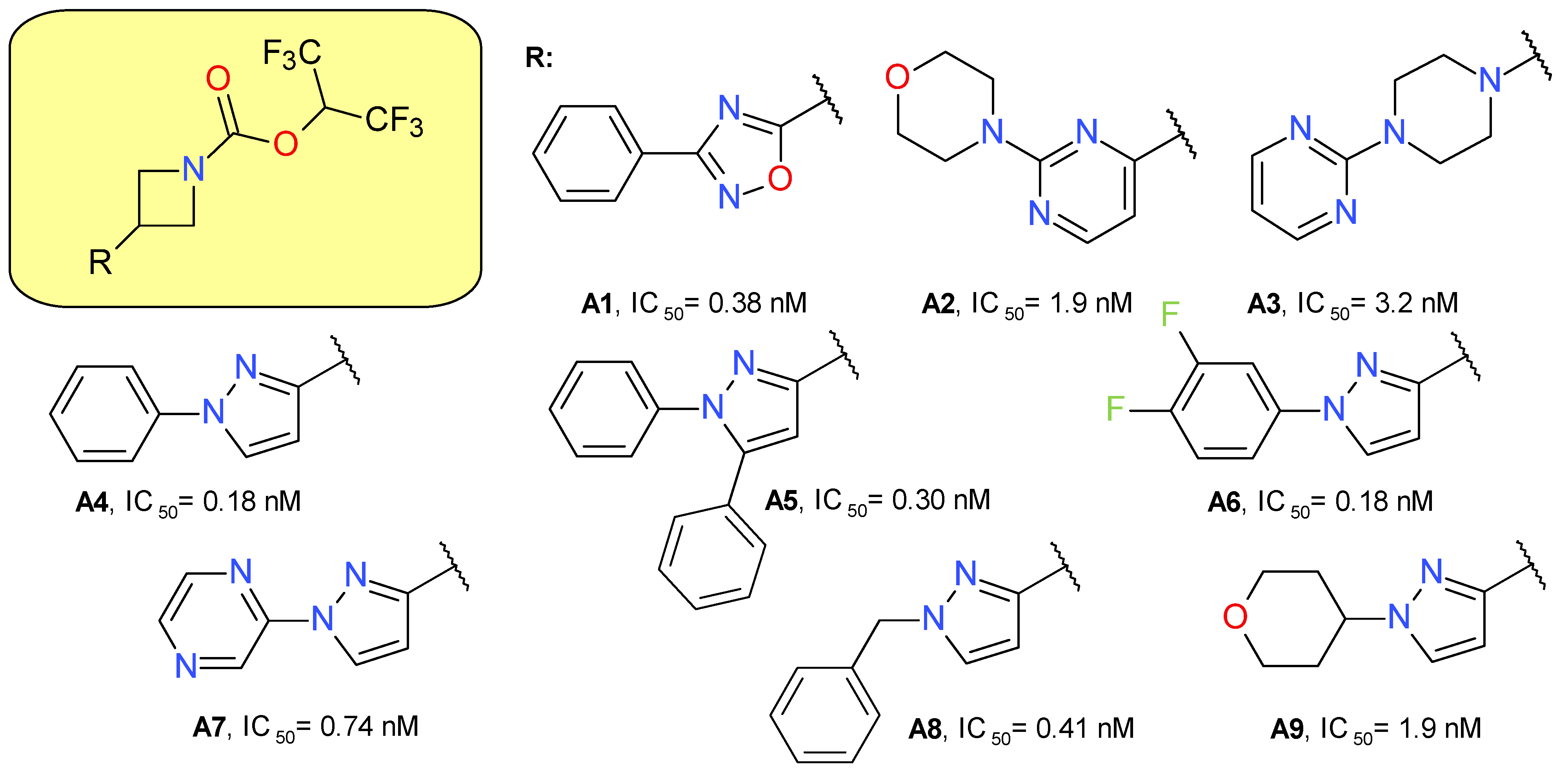
4.1.1. Azetidine HFIP Carbamates
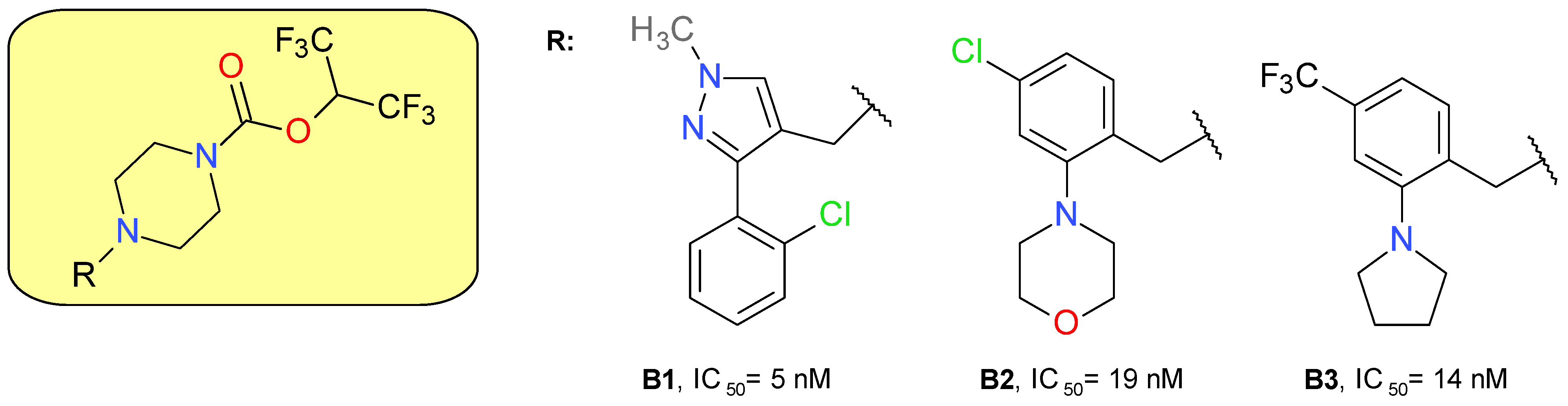
4.1.2. Piperazine HFIP Carbamates
4.1.3. Azabicyclo[3.1.0]Hexane Trifluoromethyl Glycol Carbamates
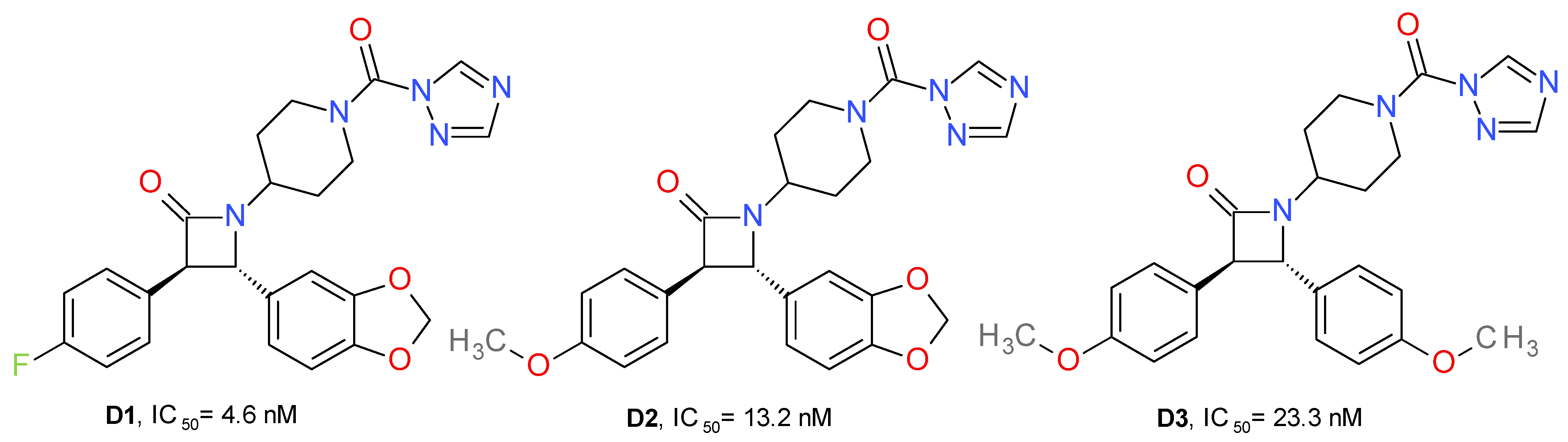
4.1.4. Azetidone Triazole Ureas
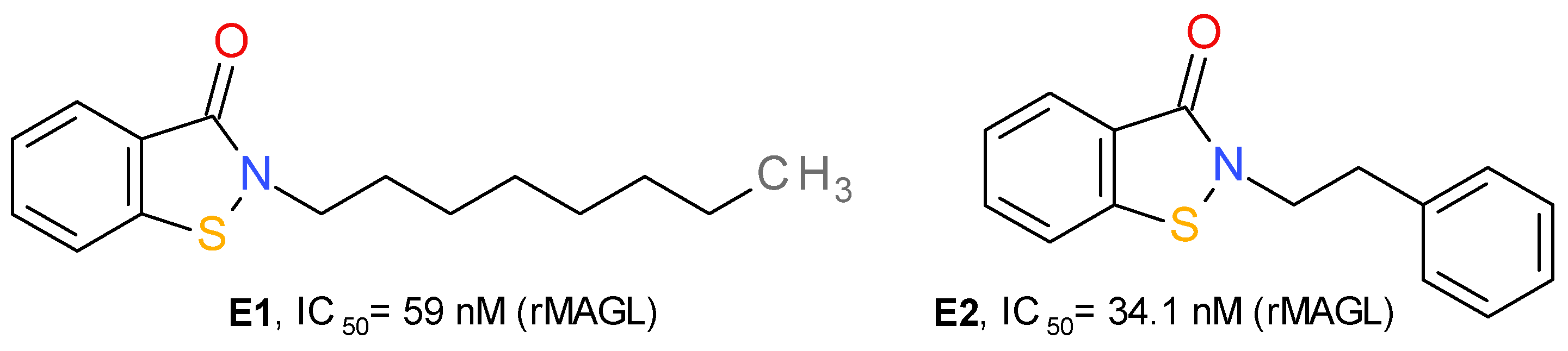
4.1.5. Benzisothiazolinone Derivatives
4.2. Reversible Inhibitors
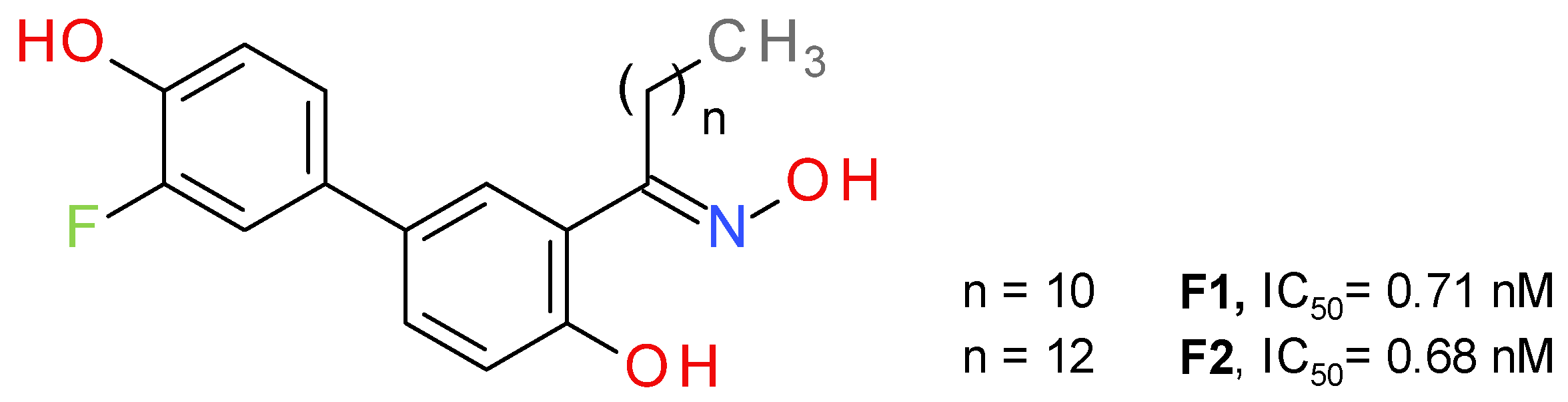
4.2.1. Salicylketoxime Derivatives
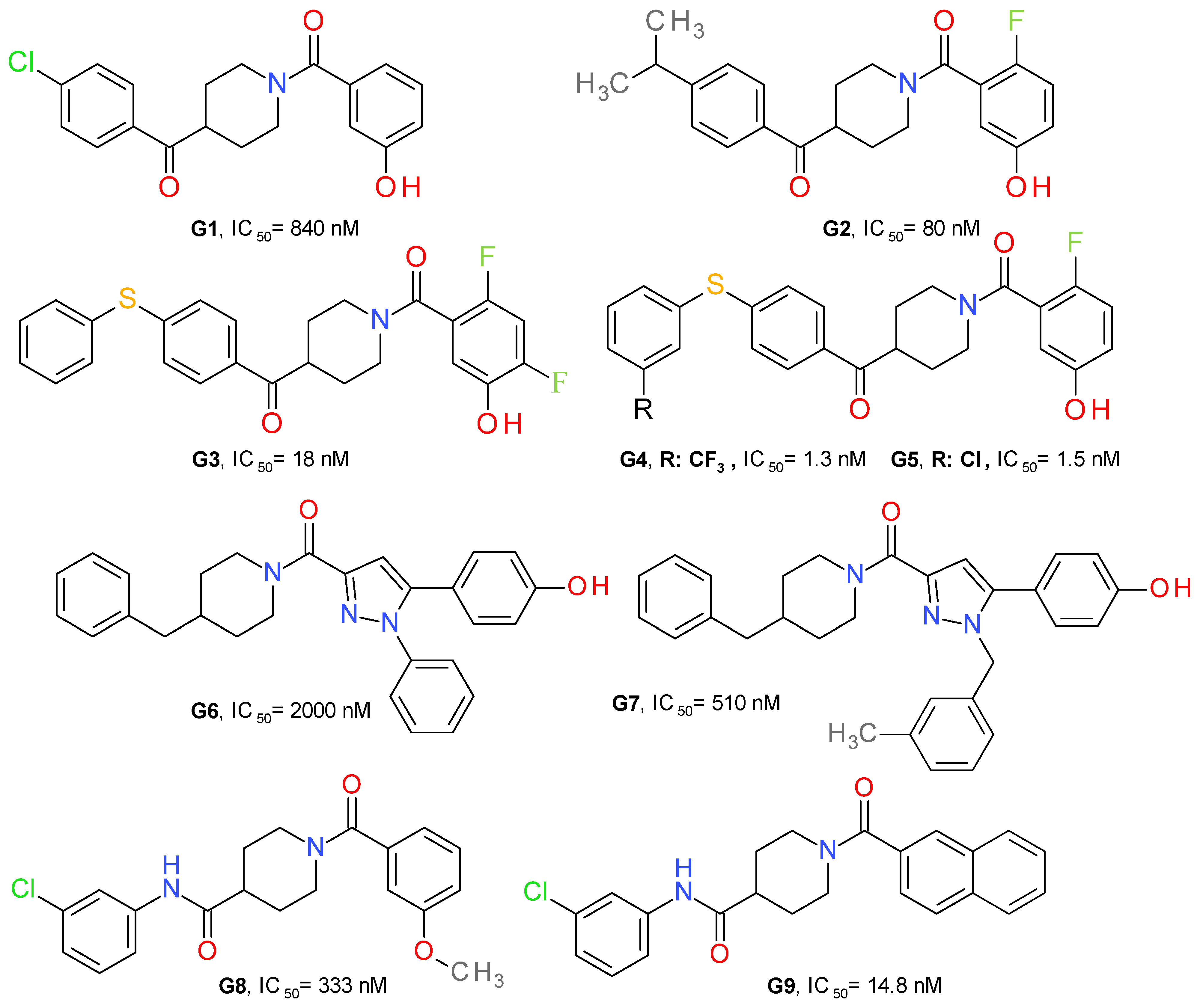
4.2.2. Piperidine Derivatives
4.2.3. Pyrrolidone Derivatives
4.2.4. Azetidinyl Amides
4.2.5. Various Structures
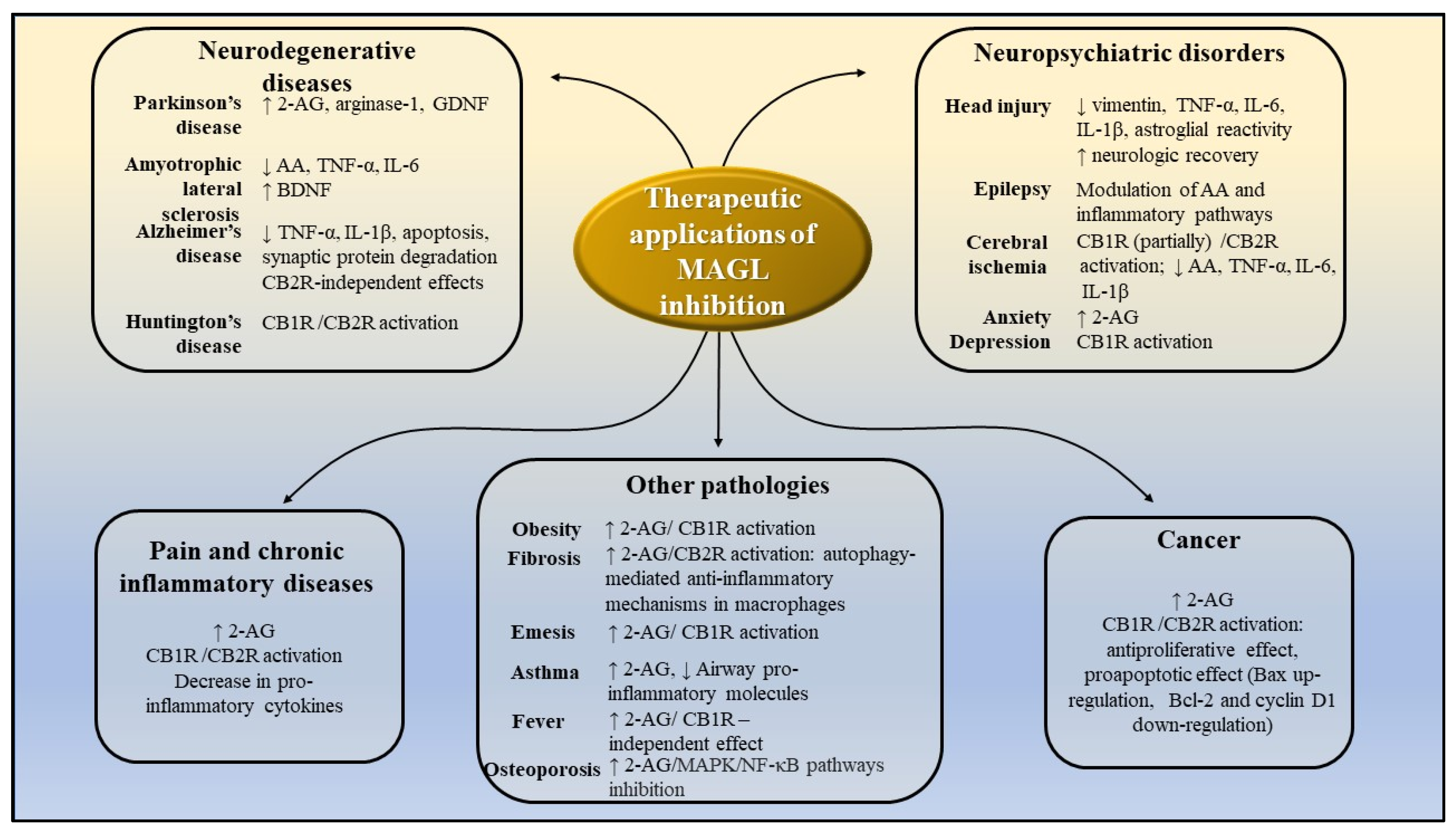
5. Potential Therapeutic Applications of MAGL Inhibition
5.1. Irreversible Pharmacological Inhibition of MAGL
5.1.1. Diseases of the Central Nervous System
5.1.2. Inflammatory Diseases
5.1.3. Other Possible Applications
Obesity and Metabolic Diseases
Neoplastic Maladies
5.2. Reversible Pharmacological Inhibition of MAGL
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Deng, H.; Li, W. Monoacylglycerol lipase inhibitors: Modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm. Sin. B 2020, 10, 582–602. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, T.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimura, A.; Yamazaki, M.; Hashimotodani, Y.; Uchigashima, M.; Kawata, S.; Abe, M.; Kita, Y.; Hashimoto, K.; Shimizu, T.; Watanabe, M.; et al. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase α mediates retrograde suppression of synaptic transmission. Neuron 2010, 65, 320–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Busquets-Garcia, A.; Bains, J.; Marsicano, G. CB1 receptor signaling in the brain: Extracting specificity from ubiquity. Neuropsychopharmacology 2018, 43, 4–20. [Google Scholar] [CrossRef]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V.; Bifulco, M.; De Petrocellis, L. The endocannabinoid system and its therapeutic exploitation. Nat. Rev. Drug Discov. 2004, 3, 771–784. [Google Scholar] [CrossRef]
- Alhouayek, M.; Masquelier, J.; Muccioli, G.G. Controlling 2-arachidonoylglycerol metabolism as an anti-inflammatory strategy. Drug Discov. Today 2014, 19, 295–304. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Marnett, L.J. Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: Cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem. Rev. 2011, 111, 5899–5921. [Google Scholar] [CrossRef]
- Nomura, D.K.; Morrison, B.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, D.K.; Hudak, C.S.; Ward, A.M.; Burston, J.J.; Issa, R.S.; Fisher, K.J.; Abood, M.E.; Wiley, J.L.; Lichtman, A.H.; Casida, J.E. Monoacylglycerol lipase regulates 2-arachidonoylglycerol action and arachidonic acid levels. Bioorganic Med. Chem. Lett. 2008, 18, 5875–5878. [Google Scholar] [CrossRef] [Green Version]
- Gil-Ordóñez, A.; Martin-Fontecha, M.; Ortega-Gutiérrez, S.; López-Rodríguez, M.L. Monoacylglycerol lipase (MAGL) as a promising therapeutic target. Biochem. Pharmacol. 2018, 157, 18–32. [Google Scholar] [CrossRef]
- Scalvini, L.; Piomelli, D.; Mor, M. Monoglyceride lipase: Structure and inhibitors. Chem. Phys. Lipids 2016, 197, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Butler, C.R.; Beck, E.M.; Harris, A.; Huang, Z.; McAllister, L.A.; Ende, C.W.A.; Fennell, K.; Foley, T.L.; Fonseca, K.; Hawrylik, S.J.; et al. Azetidine and piperidine carbamates as efficient, covalent inhibitors of monoacylglycerol lipase. J. Med. Chem. 2017, 60, 9860–9873. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Caligiuri, I.; Minutolo, F.; Rizzolio, F.; Tuccinardi, T. A patent review of monoacylglycerol lipase (MAGL) inhibitors (2013–2017). Expert Opin. Ther. Patents 2017, 27, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Bononi, G.; Poli, G.; Rizzolio, F.; Tuccinardi, T.; Macchia, M.; Minutolo, F.; Granchi, C. An updated patent review of monoacylglycerol lipase (MAGL) inhibitors (2018-present). Expert Opin. Ther. Patents 2021, 31, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Grabner, G.; Zimmermann, R.; Schicho, R.; Taschler, U. Monoglyceride lipase as a drug target: At the crossroads of arachidonic acid metabolism and endocannabinoid signaling. Pharmacol. Ther. 2017, 175, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Gutierrez, S.; Viso, A.; Cisneros, J.A. The medicinal chemistry of agents targeting monoacylglycerol lipase. Curr. Top. Med. Chem. 2008, 8, 231–246. [Google Scholar] [CrossRef]
- Tyukhtenko, S.; Ma, X.; Rajarshi, G.; Karageorgos, I.; Anderson, K.W.; Hudgens, J.W.; Guo, J.J.; Nasr, M.L.; Zvonok, N.; Vemuri, K.; et al. Conformational gating, dynamics and allostery in human monoacylglycerol lipase. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Basavarajappa, B.S. Critical enzymes involved in endocannabinoid metabolism. Protein Pept. Lett. 2007, 14, 237–246. [Google Scholar] [CrossRef] [PubMed]
- King, A.R.; Lodola, A.; Carmi, C.; Fu, J.; Mor, M.; Piomelli, D. A critical cysteine residue in monoacylglycerol lipase is targeted by a new class of isothiazolinone-based enzyme inhibitors. Br. J. Pharmacol. 2009, 157, 974–983. [Google Scholar] [CrossRef] [Green Version]
- Wouters, J.; Lambert, D. A review on the monoacylglycerol lipase: At the interface between fat and endocannabinoid signalling. Curr. Med. Chem. 2010, 17, 2588–2607. [Google Scholar] [CrossRef] [Green Version]
- Casas-Godoy, L.; Duquesne, S.; Bordes, F.; Sandoval, G.; Marty, A. Lipases: An overview. Methods Mol. Biol. 2012, 861, 3–30. [Google Scholar] [CrossRef] [PubMed]
- Bawa, S.; Afzal, O.; Kumar, S.; Kumar, R.; Jaggi, M. 3D-QSAR study of benzotriazol-1-yl carboxamide scaffold as monoacylglycerol lipase inhibitors. J. Pharm. Bioallied Sci. 2014, 6, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Afzal, O.; Kumar, S.; Kumar, R.; Firoz, A.; Jaggi, M.; Bawa, S. Docking based virtual screening and molecular dynamics study to identify potential monoacylglycerol lipase inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Brindisi, M.; Maramai, S.; Gemma, S.; Brogi, S.; Grillo, A.; Mannelli, L.D.C.; Gabellieri, E.; Lamponi, S.; Saponara, S.; Gorelli, B.; et al. Development and pharmacological characterization of selective blockers of 2-arachidonoyl glycerol degradation with efficacy in rodent models of multiple sclerosis and pain. J. Med. Chem. 2016, 59, 2612–2632. [Google Scholar] [CrossRef] [Green Version]
- Aida, J.; Fushimi, M.; Kusumoto, T.; Sugiyama, H.; Arimura, N.; Ikeda, S.; Sasaki, M.; Sogabe, S.; Aoyama, K.; Koike, T. Design, synthesis, and evaluation of piperazinyl pyrrolidin-2-ones as a novel series of reversible monoacylglycerol lipase inhibitors. J. Med. Chem. 2018, 61, 9205–9217. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Biagi, M.; Spinelli, V.; Di Stefano, M.; Macchia, M.; Minutolo, F.; Granchi, C.; Poli, G.; Tuccinardi, T. Discovery of monoacylglycerol lipase (MAGL) inhibitors based on a pharmacophore-guided virtual screening study. Molecules 2020, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Ding, X.; Zhang, H.; Luo, X.; Chen, K.; Jiang, H.; Luo, C.; Xu, H. Discovery of novel reversible monoacylglycerol lipase inhibitors via docking-based virtual screening. Bioorganic Med. Chem. Lett. 2021, 41, 127986. [Google Scholar] [CrossRef]
- Tuo, W.; Leleu-Chavain, N.; Spencer, J.; Sansook, S.; Millet, R.; Chavatte, P. Therapeutic potential of fatty acid amide hydrolase, monoacylglycerol lipase, and N-acylethanolamine acid amidase inhibitors. J. Med. Chem. 2016, 60, 4–46. [Google Scholar] [CrossRef]
- Nitulescu, G.; Margina, D.; Zanfirescu, A.; Olaru, O.; Nitulescu, G. Targeting bacterial sortases in search of anti-virulence therapies with low risk of resistance development. Pharmaceuticals 2021, 14, 415. [Google Scholar] [CrossRef]
- Martin, J.; MacKenzie, C.J.; Fletcher, D.; Gilbert, I.H. Characterising covalent warhead reactivity. Bioorganic Med. Chem. 2019, 27, 2066–2074. [Google Scholar] [CrossRef]
- Niphakis, M.J.; Cognetta, A.B., III; Chang, J.W.; Buczynski, M.W.; Parsons, L.H.; Byrne, F.; Burston, J.J.; Chapman, V.; Cravatt, B.F. Evaluation of NHS carbamates as a potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 2013, 4, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.W.; Niphakis, M.J.; Lum, K.M.; Cognetta, A.B., III; Wang, C.; Matthews, M.L.; Niessen, S.; Buczynski, M.W.; Parsons, L.H.; Cravatt, B.F. Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem. Biol. 2012, 19, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Cisar, J.S.; Weber, O.D.; Clapper, J.R.; Blankman, J.L.; Henry, C.L.; Simon, G.M.; Alexander, J.P.; Jones, T.K.; Ezekowitz, R.A.B.; O’Neill, G.P.; et al. Identification of ABX-1431, a selective inhibitor of monoacylglycerol lipase and clinical candidate for treatment of neurological disorders. J. Med. Chem. 2018, 61, 9062–9084. [Google Scholar] [CrossRef] [Green Version]
- Kharasch, E.D. Biotransformation of sevoflurane. Anesth. Analg. 1995, 81, 27S–38S. [Google Scholar] [CrossRef]
- Chang, J.W.; Cognetta, A.B., III; Niphakis, M.J.; Cravatt, B.F. Proteome-wide reactivity profiling identifies diverse carbamate chemotypes tuned for serine hydrolase inhibition. ACS Chem. Biol. 2013, 8, 1590–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Kaplan, J.; Stella, N. ABHD6: Its place in endocannabinoid signaling and beyond. Trends Pharmacol. Sci. 2019, 40, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, N.; Savinainen, J.; Ribas, C.R.; Rönkkö, J.; Kuusisto, A.; Korhonen, J.; Navia-Paldanius, D.; Häyrinen, J.; Takabe, P.; Käsnänen, H.; et al. Piperazine and piperidine triazole ureas as ultrapotent and highly selective inhibitors of monoacylglycerol lipase. Chem. Biol. 2013, 20, 379–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.Z.; Ahenkorah, S.; Vaara, M.; Staszewski, M.; Adams, Y.; Laitinen, T.; Navia-Paldanius, D.; Parkkari, T.; Savinainen, J.; Walczyński, K.; et al. Loratadine analogues as MAGL inhibitors. Bioorganic Med. Chem. Lett. 2015, 25, 1436–1442. [Google Scholar] [CrossRef]
- Jiang, M.; Van der Stelt, M. Activity-based protein profiling delivers selective drug candidate ABX-1431, a monoacylglycerol lipase inhibitor, to control lipid metabolism in neurological disorders. J. Med. Chem. 2018, 61, 9059–9061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAllister, L.A.; Butler, C.R.; Mente, S.; O’Neil, S.V.; Fonseca, K.R.; Piro, J.R.; Cianfrogna, J.A.; Foley, T.L.; Gilbert, A.M.; Harris, A.R.; et al. Discovery of trifluoromethyl glycol carbamates as potent and selective covalent monoacylglycerol lipase (MAGL) inhibitors for treatment of neuroinflammation. J. Med. Chem. 2018, 61, 3008–3026. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Jin, X.; Adibekian, A.; Li, W.; Cravatt, B.F. Characterization of tunable piperidine and piperazine carbamates as inhibitors of endocannabinoid hydrolases. J. Med. Chem. 2010, 53, 1830–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Mori, W.; Fu, H.; Schafroth, M.A.; Hatori, A.; Shao, T.; Zhang, G.; Van, R.S.; Zhang, Y.; Hu, K.; et al. Design, synthesis, and evaluation of 18F-labeled monoacylglycerol lipase inhibitors as novel positron emission tomography probes. J. Med. Chem. 2019, 62, 8866–8872. [Google Scholar] [CrossRef]
- Omran, Z. New disulfiram derivatives as MAGL-selective inhibitors. Molecules 2021, 26, 3296. [Google Scholar] [CrossRef]
- Bononi, G.; Granchi, C.; Lapillo, M.; Giannotti, M.; Nieri, D.; Fortunato, S.; El Boustani, M.; Caligiuri, I.; Poli, G.; Carlson, K.E.; et al. Discovery of long-chain salicylketoxime derivatives as monoacylglycerol lipase (MAGL) inhibitors. Eur. J. Med. Chem. 2018, 157, 817–836. [Google Scholar] [CrossRef]
- Hernandez-Torres, G.; Cipriano, M.; Heden, E.; Bjorklund, E.; Canales, A.; Zian, D.; Feliu, A.; Mecha, M.; Guaza, C.; Fowler, C.J.; et al. A reversible and selective inhibitor of monoacylglycerol lipase ameliorates multiple sclerosis. Angew. Chem. Int. Ed. Engl. 2014, 53, 13765–13770. [Google Scholar] [CrossRef]
- Tabrizi, M.A.; Baraldi, P.G.; Baraldi, S.; Ruggiero, E.; De Stefano, L.; Rizzolio, F.; Mannelli, L.D.C.; Ghelardini, C.; Chicca, A.; Lapillo, M.; et al. Discovery of 1,5-diphenylpyrazole-3-carboxamide derivatives as potent, reversible, and selective monoacylglycerol lipase (MAGL) inhibitors. J. Med. Chem. 2018, 61, 1340–1354. [Google Scholar] [CrossRef] [Green Version]
- Zhi, Z.; Zhang, W.; Yao, J.; Shang, Y.; Hao, Q.; Liu, Z.; Ren, Y.; Li, J.; Zhang, G.; Wang, J. Discovery of aryl formyl piperidine derivatives as potent, reversible, and selective monoacylglycerol lipase inhibitors. J. Med. Chem. 2020, 63, 5783–5796. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Rizzolio, F.; Palazzolo, S.; Carmignani, S.; Macchia, M.; Saccomanni, G.; Manera, C.; Martinelli, A.; Minutolo, F.; Tuccinardi, T. Structural optimization of 4-chlorobenzoylpiperidine derivatives for the development of potent, reversible, and selective monoacylglycerol lipase (MAGL) inhibitors. J. Med. Chem. 2016, 59, 10299–10314. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Lapillo, M.; Glasmacher, S.; Bononi, G.; Licari, C.; Poli, G.; el Boustani, M.; Caligiuri, I.; Rizzolio, F.; Gertsch, J.; et al. Optimization of a benzoylpiperidine class identifies a highly potent and selective reversible monoacylglycerol lipase (MAGL) inhibitor. J. Med. Chem. 2019, 62, 1932–1958. [Google Scholar] [CrossRef]
- Granchi, C.; Bononi, G.; Ferrisi, R.; Gori, E.; Mantini, G.; Glasmacher, S.; Poli, G.; Palazzolo, S.; Caligiuri, I.; Rizzolio, F.; et al. Design, synthesis and biological evaluation of second-generation benzoylpiperidine derivatives as reversible monoacylglycerol lipase (MAGL) inhibitors. Eur. J. Med. Chem. 2021, 209, 112857. [Google Scholar] [CrossRef] [PubMed]
- Altamimi, A.S.A.; Bawa, S.; Athar, F.; Hassan, Q.; Riadi, Y.; Afzal, O. Pyrrolidin-2-one linked benzofused heterocycles as novel small molecule monoacylglycerol lipase inhibitors and antinociceptive agents. Chem. Biol. Drug Des. 2020, 96, 1418–1432. [Google Scholar] [CrossRef]
- Afzal, O.; Altamimi, A.; Shahroz, M.; Sharma, H.; Riadi, Y.; Hassan, Q. Analgesic and anticancer activity of benzoxazole clubbed 2-pyrrolidinones as novel inhibitors of monoacylglycerol lipase. Molecules 2021, 26, 2389. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Connolly, P.J.; Zhang, Y.-M.; McDonnell, M.E.; Bian, H.; Lin, S.-C.; Liu, L.; Zhang, S.-P.; Chevalier, K.M.; Brandt, M.R.; et al. The discovery of azetidine-piperazine di-amides as potent, selective and reversible monoacylglycerol lipase (MAGL) inhibitors. Bioorganic Med. Chem. Lett. 2020, 30, 127243. [Google Scholar] [CrossRef]
- Dato, F.M.; Neudorfl, J.M.; Gutschow, M.; Goldfuss, B.; Pietsch, M. Omega-quinazolinonylalkyl aryl ureas as reversible inhibitors of monoacylglycerol lipase. Bioorg. Chem. 2020, 94, 103352. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, C.; Chiarlone, A.; Bellocchio, L.; Resel, E.; Pruunsild, P.; García-Rincón, D.; Sendtner, M.; Timmusk, T.; Lutz, B.; Galve-Roperh, I.; et al. The CB1 cannabinoid receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ. 2015, 22, 1618–1629. [Google Scholar] [CrossRef]
- Long, J.Z.; Nomura, D.K.; Cravatt, B.F. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem. Biol. 2009, 16, 744–753. [Google Scholar] [CrossRef] [Green Version]
- Margină, D.; Ungurianu, A.; Purdel, C.; Tsoukalas, D.; Sarandi, E.; Thanasoula, M.; Tekos, F.; Mesnage, R.; Kouretas, D.; Tsatsakis, A. Chronic inflammation in the context of everyday life: Dietary changes as mitigating factors. Int. J. Environ. Res. Public Health 2020, 17, 4135. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Díez, A.; Araque, A. Astrocytes in endocannabinoid signalling. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130599. [Google Scholar] [CrossRef]
- Costa, L.; Amaral, C.; Teixeira, N.; Correia-Da-Silva, G.; Fonseca, B.M. Cannabinoid-induced autophagy: Protective or death role? Prostagland. Other Lipid Mediat. 2016, 122, 54–63. [Google Scholar] [CrossRef]
- Choi, S.-H.; Arai, A.L.; Mou, Y.; Kang, B.; Yen, C.C.-C.; Hallenbeck, J.; Silva, A.C. Neuroprotective effects of MAGL (monoacylglycerol lipase) inhibitors in experimental ischemic stroke. Stroke 2018, 49, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.; Vitale, R. The endocannabinoid system and PPARs: Focus on their signalling crosstalk, action and transcriptional regulation. Cells 2021, 10, 586. [Google Scholar] [CrossRef] [PubMed]
- Pistis, M.; O’Sullivan, S.E. The role of nuclear hormone receptors in cannabinoid function. Cannabinoid Pharmacol. 2017, 80, 291–328. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, J.; Lehmann, C. GPR55—A putative “type 3” cannabinoid receptor in inflammation. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Calvo, A.; Bajo-Grañeras, R.; Maroto, I.B.; Zian, D.; Grabner, G.; García-Taboada, E.; Resel, E.; Zechner, R.; Zimmermann, R.; Ortega-Gutiérrez, S.; et al. Astroglial monoacylglycerol lipase controls mutant huntingtin-induced damage of striatal neurons. Neuropharmacology 2019, 150, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Benjamin, D.I.; Duerr, J.M.; Pi, Y.; Gonzales, C.; Wood, K.M.; Schwartz, J.W.; Nomura, D.K.; Samad, T.A. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep. 2012, 1, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Woodhams, S.G.; Chapman, V.; Finn, D.P.; Hohmann, A.G.; Neugebauer, V. The cannabinoid system and pain. Neuropharmacology 2017, 124, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Imperatore, R.; Morello, G.; Luongo, L.; Taschler, U.; Romano, R.; De Gregorio, D.; Belardo, C.; Maione, S.; Di Marzo, V.; Cristino, L. Genetic deletion of monoacylglycerol lipase leads to impaired cannabinoid receptor CB1 R signaling and anxiety-like behavior. J. Neurochem. 2015, 135, 799–813. [Google Scholar] [CrossRef] [Green Version]
- Navia-Paldanius, D.; Aaltonen, N.; Lehtonen, M.; Savinainen, J.R.; Taschler, U.; Radner, F.P.; Zimmermann, R.; Laitinen, J.T. Increased tonic cannabinoid CB1R activity and brain region-specific desensitization of CB1R Gi/o signaling axis in mice with global genetic knockout of monoacylglycerol lipase. Eur. J. Pharm. Sci. 2015, 77, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S.; Kunugi, H. Inhibitors of fatty acid amide hydrolase and monoacylglycerol lipase: New targets for future antidepressants. Curr. Neuropharmacol. 2015, 13, 760–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglass, J.D.; Zhou, Y.X.; Wu, A.; Zadrogra, J.A.; Gajda, A.M.; Lackey, A.I.; Lang, W.; Chevalier, K.M.; Sutton, S.W.; Zhang, S.-P.; et al. Global deletion of MGL in mice delays lipid absorption and alters energy homeostasis and diet-induced obesity. J. Lipid Res. 2015, 56, 1153–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taschler, U.; Radner, F.; Heier, C.; Schreiber, R.; Schweiger, M.; Schoiswohl, G.; Preiss-Landl, K.; Jaeger, D.; Reiter, B.; Koefeler, H.C.; et al. Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. J. Biol. Chem. 2011, 286, 17467–17477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardelli, M.; Bruschi, F.V.; Claudel, T.; Fuchs, C.; Auer, N.; Kunczer, V.; Stojakovic, T.; Scharnagl, H.; Habib, A.; Grabner, G.; et al. Lack of monoacylglycerol lipase prevents hepatic steatosis by favoring lipid storage in adipose tissue and intestinal malabsorption. J. Lipid Res. 2019, 60, 1284–1292. [Google Scholar] [CrossRef]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Baba, Y.; Funakoshi, T.; Emoto, K.; Masugi, Y.; Ekmekcioglu, S.; Amagai, M.; Mori, M.; Tanese, K. Expression of monoacylglycerol lipase as a marker of tumour invasion and progression in malignant melanoma. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 2038–2045. [Google Scholar] [CrossRef]
- Li, X.; Gao, S.; Li, W.; Liu, Z.; Shi, Z.; Qiu, C.; Jiang, J. Effect of monoacylglycerol lipase on the tumor growth in endometrial cancer. J. Obstet. Gynaecol. Res. 2019, 45, 2043–2054. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Bai, J.; Ling, Y.; Chang, W.; Xie, G.; Li, R.; Wang, G.; Tao, K. Monoacylglycerol lipase inhibitor JZL184 regulates apoptosis and migration of colorectal cancer cells. Mol. Med. Rep. 2016, 13, 2850–2856. [Google Scholar] [CrossRef] [Green Version]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K.; et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, Z.; Lian, Z.; Liao, R.; Chen, Y.; Qin, Y.; Wang, J.; Jiang, Q.; Wang, X.; Gong, J. Monoacylglycerol lipase: A novel potential therapeutic target and prognostic indicator for hepatocellular carcinoma. Sci. Rep. 2016, 6, 35784. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Zhao, Y.; Zhou, J.; Wang, X.; Pan, Q.; Zhang, N.; Wang, L.; Wang, M.; Zhan, D.; Liu, Z.; et al. Monoacylglycerol lipase promotes progression of hepatocellular carcinoma via NF-kappaB-mediated epithelial-mesenchymal transition. J. Hematol. Oncol. 2016, 9, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquarelli, N.; Engelskirchen, M.; Hanselmann, J.; Endres, S.; Porazik, C.; Bayer, H.; Buck, E.; Karsak, M.; Weydt, P.; Ferger, B.; et al. Evaluation of monoacylglycerol lipase as a therapeutic target in a transgenic mouse model of ALS. Neuropharmacology 2017, 124, 157–169. [Google Scholar] [CrossRef]
- Pihlaja, R.; Takkinen, J.; Eskola, O.; Vasara, J.; López-Picón, F.R.; Haaparanta-Solin, M.; Rinne, J.O. Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J. Neuroinflammation 2015, 12, 81. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, C. Alleviation of neuropathology by inhibition of monoacylglycerol lipase in APP transgenic mice lacking CB2 receptors. Mol. Neurobiol. 2017, 55, 4802–4810. [Google Scholar] [CrossRef]
- Hashem, J.; Hu, M.; Zhang, J.; Gao, F.; Chen, C. Inhibition of 2-arachidonoylglycerol metabolism alleviates neuropathology and improves cognitive function in a tau mouse model of Alzheimer’s disease. Mol. Neurobiol. 2021, 58, 4122–4133. [Google Scholar] [CrossRef]
- Rahmani, M.-R.; Shamsizadeh, A.; Moghadam-Ahmadi, A.; Bazmandegan, G.; Allahtavakoli, M. JZL184, as a monoacylglycerol lipase inhibitor, down-regulates inflammation in a cannabinoid pathway dependent manner. Biomed. Pharmacother. 2018, 103, 1720–1726. [Google Scholar] [CrossRef]
- Bedse, G.; Bluett, R.J.; Patrick, T.A.; Romness, N.K.; Gaulden, A.D.; Kingsley, P.J.; Plath, N.; Marnett, L.J.; Patel, S. Therapeutic endocannabinoid augmentation for mood and anxiety disorders: Comparative profiling of FAAH, MAGL and dual inhibitors. Transl. Psychiatry 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Lomazzo, E.; Bindila, L.; Remmers, F.; Lerner, R.; Schwitter, C.; Hoheisel, U.; Lutz, B. Therapeutic potential of inhibitors of endocannabinoid degradation for the treatment of stress-related hyperalgesia in an animal model of chronic pain. Neuropsychopharmacology 2014, 40, 488–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alteba, S.; Zer-Aviv, T.M.; Tenenhaus, A.; Ben David, G.; Adelman, J.; Hillard, C.J.; Doron, R.; Akirav, I. Antidepressant-like effects of URB597 and JZL184 in male and female rats exposed to early life stress. Eur. Neuropsychopharmacol. 2020, 39, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Worley, N.B.; Varela, J.A.; Gaillardetz, G.P.; Hill, M.N.; Christianson, J.P. Monoacylglycerol lipase alpha inhibition alters prefrontal cortex excitability and blunts the consequences of traumatic stress in rat. Neuropharmacology 2020, 166, 107964. [Google Scholar] [CrossRef]
- Nie, X.; Kitaoka, S.; Shinohara, M.; Kakizuka, A.; Narumiya, S.; Furuyashiki, T. Roles of toll-like receptor 2/4, monoacylglycerol lipase, and cyclooxygenase in social defeat stress-induced prostaglandin E2 synthesis in the brain and their behavioral relevance. Sci. Rep. 2019, 9, 17548. [Google Scholar] [CrossRef]
- Wang, Y.; Gu, N.; Duan, T.; Kesner, P.; Blaskovits, F.; Liu, J.; Lu, Y.; Tong, L.; Gao, F.; Harris, C.; et al. Monoacylglycerol lipase inhibitors produce pro- or antidepressant responses via. hippocampal CA1 GABAergic synapses. Mol. Psychiatry 2017, 22, 215–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, X.; Yang, C.; Zhao, S. Effect of monoacylglycerol lipase inhibition on intestinal permeability in chronic stress model. Biochem. Biophys. Res. Commun. 2020, 525, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Terrone, G.; Pauletti, A.; Salamone, A.; Rizzi, M.; Villa, B.R.; Porcu, L.; Sheehan, M.J.; Guilmette, E.; Butler, C.R.; Piro, J.R.; et al. Inhibition of monoacylglycerol lipase terminates diazepam-resistant status epilepticus in mice and its effects are potentiated by a ketogenic diet. Epilepsia 2017, 59, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Griebel, G.; Pichat, P.; Beeské, S.; Leroy, T.; Redon, N.; Jacquet, A.; Françon, D.; Bert, L.; Even, L.; Lopez-Grancha, M.; et al. Selective blockade of the hydrolysis of the endocannabinoid 2-arachidonoylglycerol impairs learning and memory performance while producing antinociceptive activity in rodents. Sci. Rep. 2015, 5, 7642. [Google Scholar] [CrossRef]
- Muldoon, P.P.; Chen, J.; Harenza, J.L.; Abdullah, R.A.; Sim-Selley, L.J.; Cravatt, B.F.; Miles, M.F.; Chen, X.; Lichtman, A.H.; Damaj, M.I. Inhibition of monoacylglycerol lipase reduces nicotine withdrawal. Br. J. Pharmacol. 2015, 172, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Crowe, M.S.; Wilson, C.D.; Leishman, E.; Prather, P.L.; Bradshaw, H.B.; Banks, M.L.; Kinsey, S.G. The monoacylglycerol lipase inhibitor KML29 with gabapentin synergistically produces analgesia in mice. Br. J. Pharmacol. 2017, 174, 4523–4539. [Google Scholar] [CrossRef]
- Philpott, H.T.; McDougall, J.J. Combatting joint pain and inflammation by dual inhibition of monoacylglycerol lipase and cyclooxygenase-2 in a rat model of osteoarthritis. Arthritis Res. 2020, 22, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nass, S.R.; Steele, F.F.; Ware, T.B.; Libby, A.H.; Hsu, K.-L.; Kinsey, S.G. Monoacylglycerol lipase inhibition using JZL184 attenuates paw inflammation and functional deficits in a mouse model of inflammatory arthritis. Cannabis Cannabinoid Res. 2021, 6, 233–241. [Google Scholar] [CrossRef]
- Sakin, Y.S.; Dogrul, A.; Ilkaya, F.; Seyrek, M.; Ulas, U.H.; Gülşen, M.; Bagci, S. The effect of FAAH, MAGL, and dual FAAH/MAGL inhibition on inflammatory and colorectal distension-induced visceral pain models in rodents. Neurogastroenterol. Motil. 2015, 27, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Ignatowska-Jankowska, B.; Wilkerson, J.L.; Mustafa, M.; Abdullah, R.; Niphakis, M.; Wiley, J.L.; Cravatt, B.F.; Lichtman, A.H. Selective monoacylglycerol lipase inhibitors: Antinociceptive versus cannabimimetic effects in mice. J. Pharmacol. Exp. Ther. 2015, 353, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Kamimura, R.; Hossain, M.Z.; Unno, S.; Ando, H.; Masuda, Y.; Takahashi, K.; Otake, M.; Saito, I.; Kitagawa, J. Inhibition of 2-arachydonoylgycerol degradation attenuates orofacial neuropathic pain in trigeminal nerve-injured mice. J. Oral Sci. 2018, 60, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Curry, Z.; Wilkerson, J.L.; Bagdas, D.; Kyte, S.L.; Patel, N.; Donvito, G.; Mustafa, M.A.; Poklis, J.L.; Niphakis, M.J.; Hsu, K.-L.; et al. Monoacylglycerol lipase inhibitors reverse paclitaxel-induced nociceptive behavior and proinflammatory markers in a mouse model of chemotherapy-induced neuropathy. J. Pharmacol. Exp. Ther. 2018, 366, 169–183. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Okine, B.N.; Finn, D.P.; Masocha, W. Peripheral deficiency and antiallodynic effects of 2-arachidonoyl glycerol in a mouse model of paclitaxel-induced neuropathic pain. Biomed. Pharmacother. 2020, 129, 110456. [Google Scholar] [CrossRef]
- Burston, J.J.; Mapp, P.I.; Sarmad, S.; Barrett, D.; Niphakis, M.J.; Cravatt, B.F.; Walsh, D.; Chapman, V. Robust anti-nociceptive effects of monoacylglycerol lipase inhibition in a model of osteoarthritis pain. Br. J. Pharmacol. 2016, 173, 3134–3144. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Suidan, G.L.; Quan, J.; Pi, Y.; O’Neill, S.M.; Ilardi, M.; Pozdnyakov, N.; Lanz, T.A.; Xi, H.; Bell, R.D.; et al. Inhibition of 2-AG hydrolysis differentially regulates blood brain barrier permeability after injury. J. Neuroinflamm. 2018, 15, 142. [Google Scholar] [CrossRef]
- Martínez-Torres, S.; Cutando, L.; Pastor, A.; Kato, A.; Sakimura, K.; De la Torre, R.; Valjent, E.; Maldonado, R.; Kano, M.; Ozaita, A. Monoacylglycerol lipase blockade impairs fine motor coordination and triggers cerebellar neuroinflammation through cyclooxygenase-2. Brain Behav. Immun. 2019, 81, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alavez, M.; Nguyen, W.; Mori, S.; Moroncini, G.; Viader, A.; Nomura, D.K.; Cravatt, B.F.; Conti, B. Monoacylglycerol lipase regulates fever response. PLoS ONE 2015, 10, e0134437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Mulvihill, M.M.; Mukhopadhyay, P.; Xu, H.; Erdélyi, K.; Hao, E.; Holovac, E.; Haskó, G.; Cravatt, B.F.; Nomura, D.K.; et al. Monoacylglycerol lipase controls endocannabinoid and eicosanoid signaling and hepatic injury in mice. Gastroenterology 2013, 144, 808–817.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, A.; Chokr, D.; Wan, J.; Hegde, P.; Mabire, M.; Siebert, M.; Ribeiro-Parenti, L.; Le Gall, M.; Lettéron, P.; Pilard, N.; et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut 2018, 68, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.-K.; Zhang, M.; Tian, Z.-L.; Wang, M.; Zhao, R.; Wang, L.-L.; Li, S.-S.; Liu, M.; Li, J.-Y.; Zhang, M.-Z.; et al. The monoacylglycerol lipase inhibitor JZL184 decreases inflammatory response in skeletal muscle contusion in rats. Eur. J. Pharmacol. 2015, 761, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yao, H.; Cheng, Y.; Gong, D.; Liao, X.; Wang, R. Effects of monoacylglycerol lipase inhibitor URB602 on lung ischemia-reperfusion injury in mice. Biochem. Biophys. Res. Commun. 2018, 506, 578–584. [Google Scholar] [CrossRef]
- Abohalaka, R.; Bozkurt, T.E.; Nemutlu, E.; Onder, S.C.; Sahin-Erdemli, I. The effects of fatty acid amide hydrolase and monoacylglycerol lipase inhibitor treatments on lipopolysaccharide-induced airway inflammation in mice. Pulm. Pharmacol. Ther. 2020, 62, 101920. [Google Scholar] [CrossRef] [PubMed]
- Sticht, M.A.; Lau, D.J.; Keenan, C.M.; Cavin, J.-B.; Morena, M.; Vemuri, V.K.; Makriyannis, A.; Cravatt, B.F.; Sharkey, K.A.; Hill, M.N. Endocannabinoid regulation of homeostatic feeding and stress-induced alterations in food intake in male rats. Br. J. Pharmacol. 2019, 176, 1524–1540. [Google Scholar] [CrossRef] [PubMed]
- Gradinaru, D.; Margina, D.; Borsa, C.; Ionescu, C.; Ilie, M.; Costache, M.; Dinischiotu, A.; Prada, G.-I. Adiponectin: Possible link between metabolic stress and oxidative stress in the elderly. Aging Clin. Exp. Res. 2016, 29, 621–629. [Google Scholar] [CrossRef]
- Elmazoglu, Z.; Rangel-Lopez, E.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Tunez, I.; Aschner, M.; Santamaria, A.; Karasu, C. Cannabinoid-profiled agents improve cell survival via. reduction of oxidative stress and inflammation, and Nrf2 activation in a toxic model combining hyperglycemia+Abeta1-42 peptide in rat hippocampal neurons. Neurochem. Int. 2020, 140, 104817. [Google Scholar] [CrossRef]
- Marino, S.; de Ridder, D.; Bishop, R.T.; Renema, N.; Ponzetti, M.; Sophocleous, A.; Capulli, M.; Aljeffery, A.; Carrasco, G.; Gens, M.D.; et al. Paradoxical effects of JZL184, an inhibitor of monoacylglycerol lipase, on bone remodelling in healthy and cancer-bearing mice. EBioMedicine 2019, 44, 452–466. [Google Scholar] [CrossRef]
- Pasquarelli, N.; Porazik, C.; Bayer, H.; Buck, E.; Schildknecht, S.; Weydt, P.; Witting, A.; Ferger, B. Contrasting effects of selective MAGL and FAAH inhibition on dopamine depletion and GDNF expression in a chronic MPTP mouse model of Parkinson’s disease. Neurochem. Int. 2017, 110, 14–24. [Google Scholar] [CrossRef]
- Covey, D.P.; Dantrassy, H.M.; Yohn, S.E.; Castro, A.; Conn, P.J.; Mateo, Y.; Cheer, J.F. Inhibition of endocannabinoid degradation rectifies motivational and dopaminergic deficits in the Q175 mouse model of Huntington’s disease. Neuropsychopharmacology 2018, 43, 2056–2063. [Google Scholar] [CrossRef]
- Bernal-Chico, A.; Canedo, M.; Manterola, A.; Victoria Sanchez-Gomez, M.; Perez-Samartin, A.; Rodriguez-Puertas, R.; Matute, C.; Mato, S. Blockade of monoacylglycerol lipase inhibits oligodendrocyte excitotoxicity and prevents demyelination In Vivo. Glia 2015, 63, 163–176. [Google Scholar] [CrossRef]
- Zhang, J.; Teng, Z.; Song, Y.; Hu, M.; Chen, C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J. Cereb. Blood Flow. Metab. 2015, 35, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Pagano, E.; Borrelli, F.; Orlando, P.; Romano, B.; Monti, M.; Morbidelli, L.; Aviello, G.; Imperatore, R.; Capasso, R.; Piscitelli, F.; et al. Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol. Res. 2017, 119, 227–236. [Google Scholar] [CrossRef]
- Parker, L.A.; Niphakis, M.J.; Downey, R.; Limebeer, C.L.; Rock, E.M.; Sticht, M.A.; Morris, H.; Abdullah, R.A.; Lichtman, A.H.; Cravatt, B.F. Effect of selective inhibition of monoacylglycerol lipase (MAGL) on acute nausea, anticipatory nausea, and vomiting in rats and Suncus murinus. Psychopharmacology 2015, 232, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.A.; Limebeer, C.L.; Rock, E.M.; Sticht, M.A.; Ward, J.; Turvey, G.; Benchama, O.; Rajarshi, G.; Wood, J.T.; Alapafuja, S.O.; et al. A comparison of novel, selective fatty acid amide hydrolase (FAAH), monoacyglycerol lipase (MAGL) or dual FAAH/MAGL inhibitors to suppress acute and anticipatory nausea in rat models. Psychopharmacology 2016, 233, 2265–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhou, C.; Qi, D.; Gao, Y.; Zhu, M.; Tao, T.; Sun, X.; Xiao, J. Inhibiting monoacylglycerol lipase suppresses RANKL-induced osteoclastogenesis and alleviates ovariectomy-induced bone loss. Front. Cell Dev. Biol. 2021, 9, 640867. [Google Scholar] [CrossRef]
- Muller-Vahl, K.R.; Fremer, C.; Beals, C.; Ivkovic, J.; Loft, H.; Schindler, C. Monoacylglycerol lipase inhibition in tourette syndrome: A 12-week, randomized, controlled study. Mov. Disord. 2021, 36. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
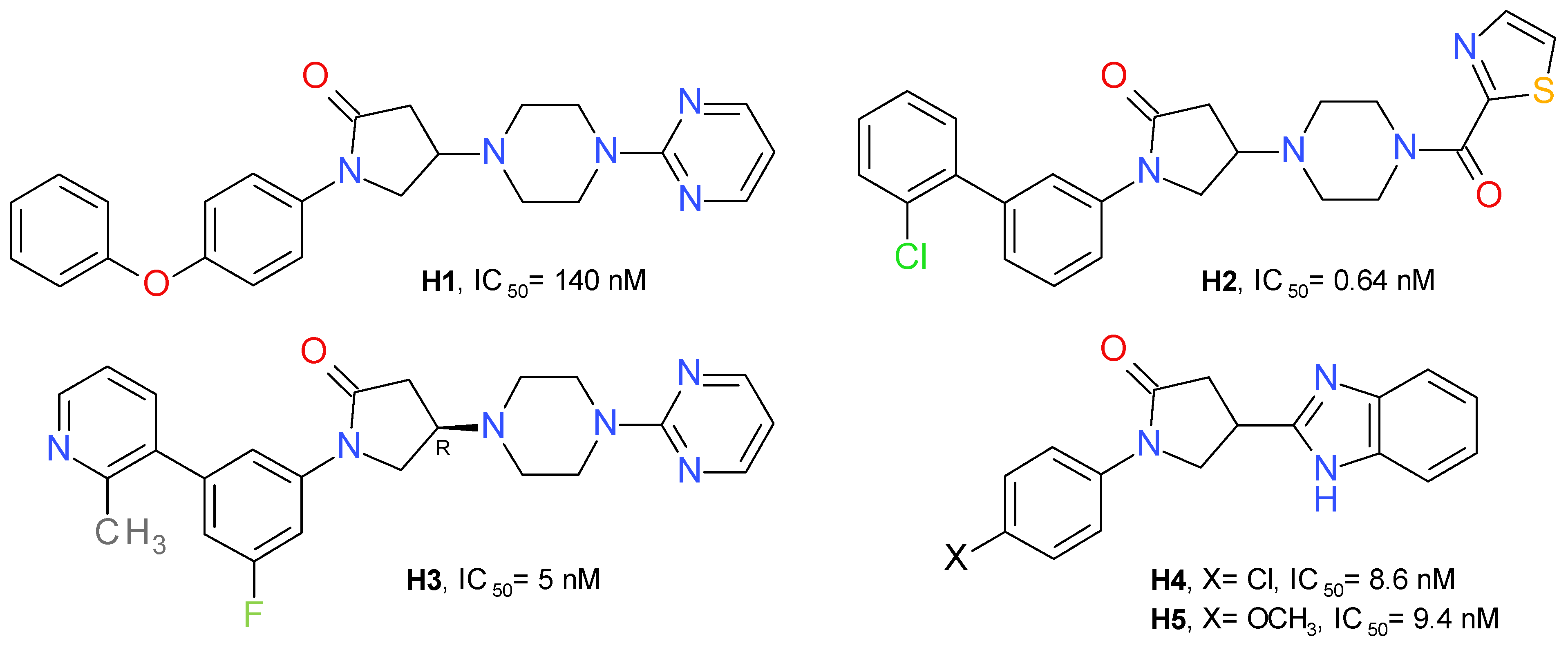{kind=link}
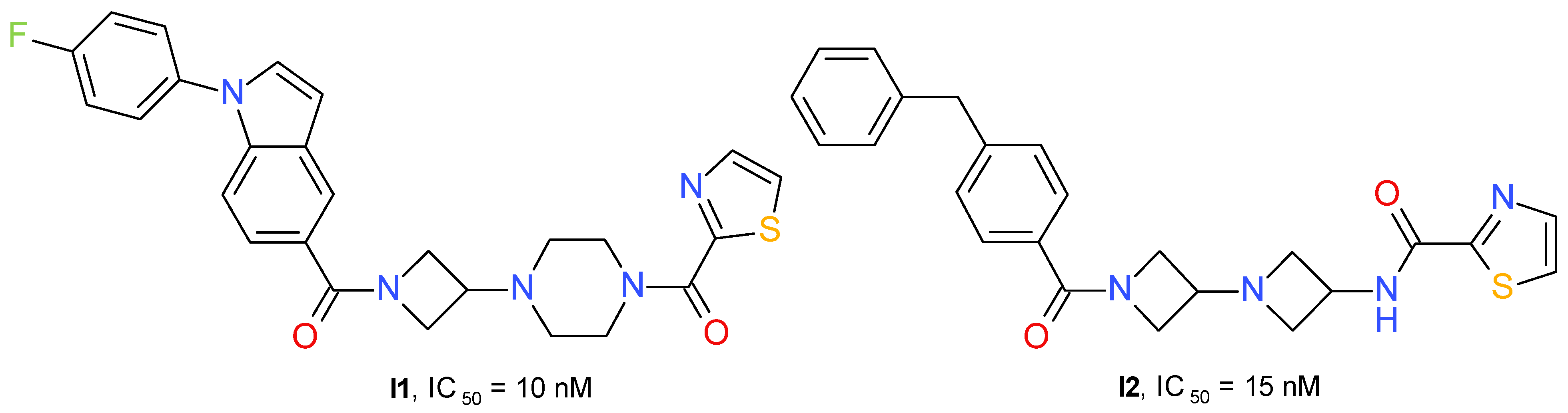{kind=link}
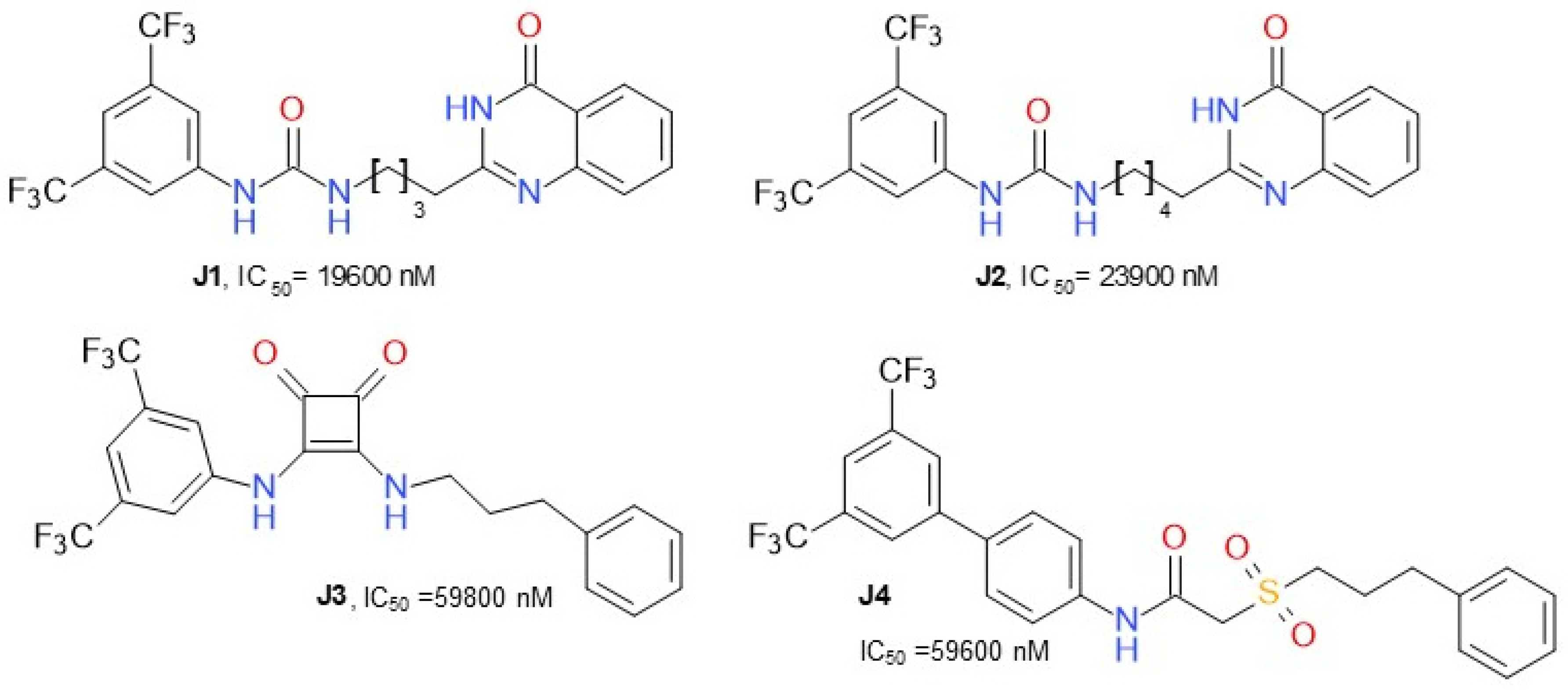{kind=link}
{kind=link}
| Therapeutical Application | Compound | Animal Model | Animal Species | Results | Alleged Mechanism | Reference |
|---|---|---|---|---|---|---|
| Pain and inflammatory disorders | ||||||
| Neuropathic pain | JZL184 | Neuropathic pain induced by trigeminal nerve injury | C57BL/6J mice | Attenuates orofacial neuropathic pain. | Not mentioned | [97] |
| MJN110 JZL184 | Chronic constriction injury model | C57BL/6J mice | Reduced chronic constriction injury-induced mechanical allodynia and thermal hyperalgesia. | CB1R/CB2R activation Decreased whole-brain arachidonic acid levels, no change in AEA, OEA, or PEA levels | [101] | |
| JZL184 | Neuropathic pain induced by trigeminal nerve injury | C57Bl/6J mice | Attenuates orofacial neuropathic pain. | Not mentioned | [102] | |
| MJN110 | Paclitaxel-induced neuropathic pain | C57BL/6J mice | Reverse paclitaxel-induced mechanical allodynia. Prevents increased expression of MCP-1 and p-p38 MAPK in dorsal root ganglia as well as MCP-1 in spinal dorsal horn. | CB1R/CB2R activation | [103] | |
| JZL184 | Paclitaxel-induced neuropathic pain | C57BL/6J/ BALB/c mice | Reverse paclitaxel-induced mechanical allodynia. | CB1R/CB2R activation | [104] | |
| Inflammatory pain/other inflammatory disorders | KML29 | Monoiodoacetate-induced osteoarthritis | Wistar rats | Analgesic effect—reduces secondary allodynia. Anti-inflammatory effect characterized by a decrease in rolling and adherent leukocytes. | CB1R/CB2R activation | [98] |
| SAR127303 | Formalin-induced and phenylbenzoquinone -induced pain | CB1R7 SCID mice | Antinociceptive effects Alters learning performance in several assays related to episodic, working and spatial memory. | CB1R (visceral pain). None reported for inflammatory pain Elevates hippocampal levels of 2-AG in mice, not AEA, PEA and OEA levels | [95] | |
| MJN 110 | Monoiodoacetate-induced osteoarthritis | Sprague Dawley rats | Analgesic effect—reduces secondary allodynia. Anti-inflammatory effect. | CB1R/CB2R activation | [105] | |
| JZL184 | Colonic distension model/acetic acid-induced pain | BALB/c mice | Alleviate pain-related behaviors. | Not mentioned | [100] | |
| JZL184 | Collagen-induced arthritis | DB1A mice | Reduced paw inflammation and pain-depressed behavioral signs. Dose-dependently attenuated grip strength and balance beam deficits caused by arthritis. | CB2R activation | [99] | |
| B3 (Figure 4) | Formalin-induced pain | Sprague-Dawley rats | Dose-dependent reduction of pain response in both acute and late phases. No reduction in motor activity. | 2-AG increase | [34] | |
| JZL184 | Collagen-induced arthritis | DB1A mice | Reduced paw inflammation and pain-depressed behavioral signs. Dose-dependently attenuated grip strength and balance beam deficits caused by arthritis. | CB2R activation | [99] | |
| B3 (Figure 4) | Formalin-induced pain | Sprague-Dawley rats | Dose-dependent reduction of pain response in both acute and late phases. No reduction in motor activity. | 2-AG increase | [34] | |
| Neurodegenerative diseases | ||||||
| Parkinson disease | KML29 | MPTP/probenecid-induced Parkinson | C57BL/6J mice | Attenuated striatal dopamine depletion. | Striatal 2-AG, arginase-1 and GDNF increase | [118] |
| Amyotrophic lateral sclerosis | KML29 | Genetic model | Low-copy SOD1G93A mice | Slows down onset, progression, increases survival. Delays the decrease in body weight and in motor activity Neurotrophic and anti-inflammatory effects. | AA, TNFα and IL6 decrease BDNF increase in spinal cord Effects are not PG-related | [82] |
| Alzheimer’s disease | JZL184 | Genetic model | APdE9 mice | Marked decrease in total Aβ burden in the temporal and parietal cortex and, to some extent, in the hippocampus. Decreased the pro-inflammatory reactions of microglia. | Not mentioned | [83] |
| JZL184 | Genetic model | 5XFAD APP/5XFAD APP-CB2R-KO transgenic mice | Reduces neuroinflammation and neurodegeneration. Improvements in spatial learning and memory decrease in the expression of APP and β-secretase as well as production of total Aβ and Aβ42. | Not mediated via CB2R. Other receptors. Prevents deterioration in expression of synaptic proteins (PSD95, AMPA receptor subunits GluA1 and GluA2, and NMDA receptor subunit GluN2B). | [84] | |
| JZL184 | Genetic model | tau P301S/PS19 transgenic (TG) mice | Suppressed inflammatory responses in astrocytes and reactive microglial cells in the cortex and hippocampus. Decreased tau neuronal loss. | Decreased hippocampal IL-1β and TNFα. Prevented the increase in p-GSK3β, P35/25, p-NF-kB. expression and the decrease in expression of PPARγ. Inhibited apoptosis through a caspase-3-dependent signaling pathway. | [85] | |
| Huntington disease | JZL184 | Knock-in mouse model | Q175 mice | Prevents motivational deficit. Increases dopamine release during a progressive-ratio task. | CB1R/CB2R activation | [119] |
| Autoimmune encephalomyelitis/demyelination | JZL184 D1 (Figure 6) C5 (Figure 5) | Immunization with myelin oligodendrocyte glycoprotein in incomplete Freund’s adjuvant, lipopolysaccharide (LPS) or cuprizone-induced demyelination. | C57BL/6J mice | Protects oligodendrocytes from excitotoxicity, thus protecting white matter. Attenuates neurological deficits and/or prevents myelin loss. | 2-AG increase. CB1R activation. Other receptors Decrease in AMPA-induced cytosolic calcium overload, mitochondrial membrane depolarization, and production of reactive oxygen species. Prevented LPS-induced increase in TNFα, PGE2, IL-1β. | [26,42,120] |
| Neuropsychiatric disorders | ||||||
| Head injury | JZL184 | Repetitively Mild Closed Head Injury model | C57BL/6 mice | Reduces chronic traumatic encephalopathy-like neuropathologic changes (impairments in basal synaptic transmission, long-term synaptic plasticity, and spatial learning) and promotes neurologic recovery. Decreases expression of APP and the enzymes that synthesize Aβ, production of Aβ, neurodegeneration, aggregation of TDP-43 protein and phosphorylation of tau. | Pro-inflammatory markers vimentin, IL-1β, IL-6, and TNFα) decrease and reactivation of astroglial cells inhibition | [121] |
| Focal cerebral ischemia model | JZL184 | Endothelin-1-induced, transient or non-transient occlusion of the middle cerebral artery ischemia | Wistar-Kyoto rats | Attenuated infarct volume and hemispheric swelling, sensorimotor deficits, inflammatory response, and decreased the number of degenerating neurons. Decrease in microglial activation and neuroinflammatory response. | CB2R activation/Partially CB1R activation (sensory impairment) Other receptors Significant TNF-α microglial reduction | [62] |
| Stroke | JZL184 | Permanent cerebralischemia model | Mice Strain not specified | Lowered brain infarction, reduced brain edema, improvement of behavioral functions. | CB1R activation 2-AG, IL-10 increase AA, MMP9, TNF-α decrease | [86] |
| Inflammatory and ischemic blood-brain barrier disruption | CPD-4645 | LPS-induced and ischemic-induced blood-brain barrier disruption | C57BL/6 mice | Reduced blood-brain barrier damage in both models. Prevented neuroinflammation. | CB1R/CB2R activation, in ischemic model Other receptors 2-AG increase AA, IL1β and IL6 decrease | [106] |
| Epilepsy—status epilepticus (SE) | CPD-4645 | Diazepam-resistant SE model | C57BL6N mice/(Cnr1−/−) mice | Reduces benzodiazepine-refractory SE and prevents cell loss and cognitive deficits. | Independent on CB1R receptors Modulation of arachidonic acid and inflammatory pathways | [94] |
| Epilepsy—focal seizures | SAR127303 | Corneal kindling-induced seizures | CB1R7 SCID mice | Inhibits seizure initiation and protects against focal seizure activity. | Elevates hippocampal levels of 2-AG in mice, not AEA, PEA and OEA levels | [95] |
| Anxiety/depression | KML29 | Chronic corticosterone-induced stress | CD1 or C57BL/6 mice | Antidepressant effects on acute stress-exposed mice, through astrocyte-mediated glutamatergic synaptic long-term depression (low dose), rapid and long-lasting antidepressant responses in chronically stressed mice likely through disinhibition of GABAergic synapses (high dose). | 2-AG increase, consequent CB1R activation | [92] |
| JZL184 | Early life stress model | Sprague Dawley pups | Prevented depression- and anxiety-like behavior and the impairment in social behavior and neuronal Plasticity. Prevented induced alterations in BDNF hippocampus and in nucleus accumbens. | Partially via CB1R activation | [89] | |
| JZL184 | Non-stress and stress (foot-shock, restraint)-induced anxiety | ICR (CD-1) mice | Prevents anxiety-like behavior in rodents. | Not mentioned. | [87] | |
| JZL184 | Chronic unpredictable stress-model | C57BL/6J mice | Reduce chronic unpredictable stress-induced anxiety and thermal hyperalgesia. | Increase in 2-AG | [88] | |
| KML29 | Stress-induced anxiety | Fischer-344 rats | MAGL inhibition in the ventromedial prefrontal cortex augments the output of neurons that project to brainstem and limbic structures that mediate stress responses, preventing stress-induced anxiety. | Increase in 2-AG | [90] | |
| Cancer | ||||||
| Breast and prostate cancers Osteosarcoma | JZL184 | Genetic model | C57BL/6 J and BALB/c-nu/nu mice | Decrease in cancer-related bone damage (osteoprotective effect), reduced skeletal tumor growth and metastasis, suppressed cachexia, prolonged survival. | 2-AG increase consequent CB1R/CB2R activation and inflammatory markers decrease | [117] |
| Colon cancer | Xenograft and azoxymethane-induced colon cancer | ICR mice | Attenuated azoxymethane-induced preneoplastic lesions, polyps and tumors and reduced xenograft tumor volume. Antiangiogenic effect. | Down-regulation of VEGF and FGF-2, reduction in the number of vessels and down-regulation of cyclin D1 | [122] | |
| Other pathologies | ||||||
| Fibrosis | MJN110 | Carbon tetrachloride-induced liver fibrosis | C57BL/6J/(MAGL−/−) mice | Reduced hepatic macrophage number, inflammatory gene expression and slowed down fibrosis progression. Accelerated fibrosis regression. | Reduces the production of IL-1α, IL-1β, PGE2 and TXA2 from macrophages, via an autophagy-dependent pathway (independently of CB2R receptors) | [110] |
| Emesis | MJN110 | Taste reactivity model—LiCl-induced acute vomiting and contextually elicited anticipatory gaping | Sprague– Dawley rats | Suppressed both acute and anticipatory nausea. | 2-AG increase consequent CB1R activation | [123] |
| Emesis | AM4301 | Taste reactivity model | Sprague-Dawley rats | Suppressed acute nausea, when delivered systemically or into the interoceptive insular cortex. | CB1R-mediated | [124] |
| Asthma | JZL184 | LPS-induced airway inflammation | CD1 mice | Prevents increased serotonin-induced contractions and reduces peribronchial and parenchymal inflammation | 2-AG increase Reduces airway TNF-α, IL-1β | [113] |
| Lung ischemia | URB602 | Lung ischemia-reperfusion injury model | C57BL/6 mice | Preventive or therapeutic regimen reduced lung injury score while increased oxygenation. | 2-AG increase Reduces airway pro-inflammatory mediators AA, PGI2, TXB2, LTB4 and inflammatory citokines IL-6, TNF-α | [112] |
| Fever | JZL184 | Centrally and peripherally-administered LPS or IL-1β-induced fever models | MAGL −/− and MAGL +/+ mice | Reduces fever (but it does not suppress it). Does not alter normal temperature. | 2-AG increase Not mediated via CB1R | [108] |
| Osteoporosis | JZL184 | Ovariectomized mouse model | C57BL/6 mice | Ameliorated bone loss. Suppressed osteoclast differentiation, bone resorption, and osteoclast-specific gene expression. | MAPK and NF-κB inhibition | [125] |
| Muscle contusion | JZL184 | Sprague Dawley rats | Rat muscle contusion model | Decreases inflammatory response in skeletal muscle contusion in rats: decreased neutrophil and macrophage infiltration and pro-inflammatory cytokine expression. | CB1R/CB2R activation 2-AG increase Decreased arachidonic acid levels and pro-inflammatory cytokines | [111] |
| Therapeutical Application | Compound | Animal Model | Animal Species | Results | Alleged Mechanism | Reference |
|---|---|---|---|---|---|---|
| Pain and inflammatory disorders | ||||||
| Neuropathic pain | G7 (Figure 9) | Oxaliplatin-induced neuropathic pain | 4r5CD-1 mice | Reverse oxaliplatin-induced cold allodynia. | Does not increase 2-AG in brain or spinal cord. Presumably, modulation of 2-AG levels in the peripheral nervous system (not proven) | [47] |
| Inflammatory pain/other inflammatory disorders | I1 (Figure 11) | Complete Freund’s adjuvant-induced inflammation | Sprague-Dawley rats | Anti-hyperalgesic efficacy correlated with the dose-dependent elevation of brain 2-AG levels. | 2-AG increase | [55] |
| H5 (Figure 10) | Formalin-induced pain | Wistar rats | Dose-dependent reduction of pain response in both acute and late phases, indicating its peripheral and central effects. | Not mentioned | [53] | |
| Neuropsychiatric disorders | ||||||
| Depression | G9 (Figure 9) | Reserpine-induced depression | ICR mice | Significantly improved the results of the cumulative immobility time in the forced swim test and tail suspension test induced by reserpine, | Increase in 2-AG | [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanfirescu, A.; Ungurianu, A.; Mihai, D.P.; Radulescu, D.; Nitulescu, G.M. Targeting Monoacylglycerol Lipase in Pursuit of Therapies for Neurological and Neurodegenerative Diseases. Molecules 2021, 26, 5668. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185668
Zanfirescu A, Ungurianu A, Mihai DP, Radulescu D, Nitulescu GM. Targeting Monoacylglycerol Lipase in Pursuit of Therapies for Neurological and Neurodegenerative Diseases. Molecules. 2021; 26(18):5668. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185668
Chicago/Turabian StyleZanfirescu, Anca, Anca Ungurianu, Dragos Paul Mihai, Denise Radulescu, and George Mihai Nitulescu. 2021. "Targeting Monoacylglycerol Lipase in Pursuit of Therapies for Neurological and Neurodegenerative Diseases" Molecules 26, no. 18: 5668. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185668