
A Simple Synthesis Route for Selectively Methylated β-Cyclodextrin Using a Copper Complex Sandwich Protecting Strategy

 ,
,  ,
,  ,
,
Abstract
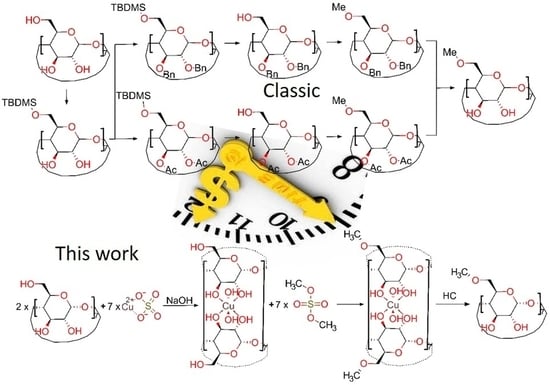
:
1. Introduction and Current Status of the Subject
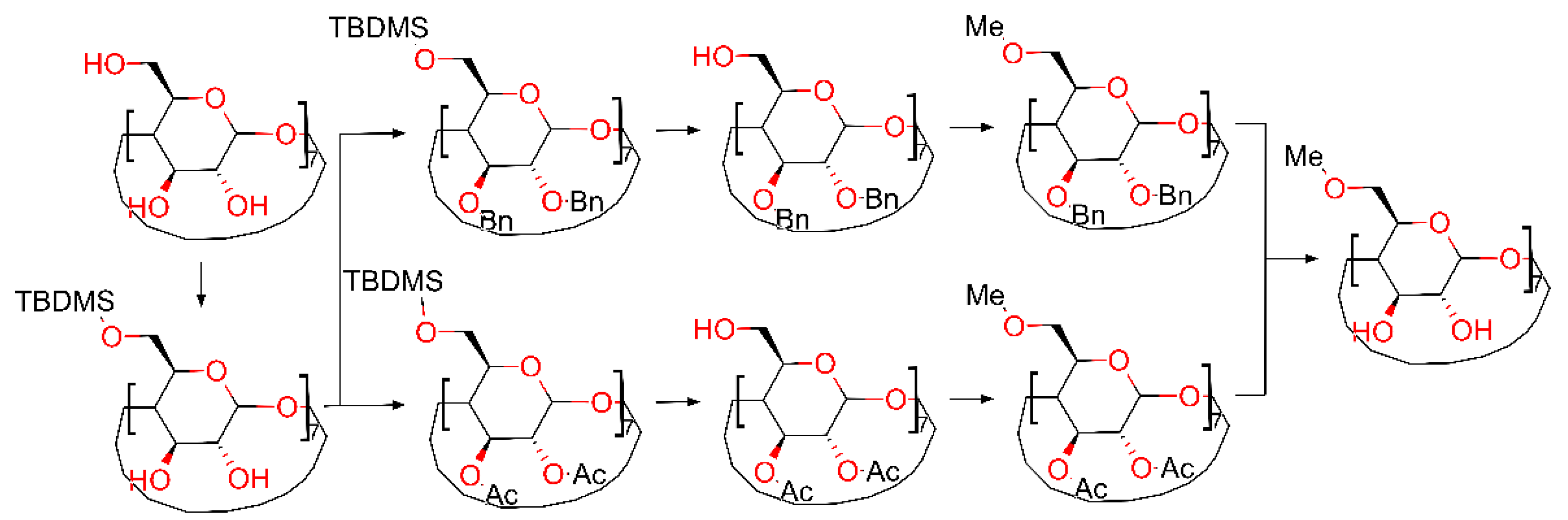
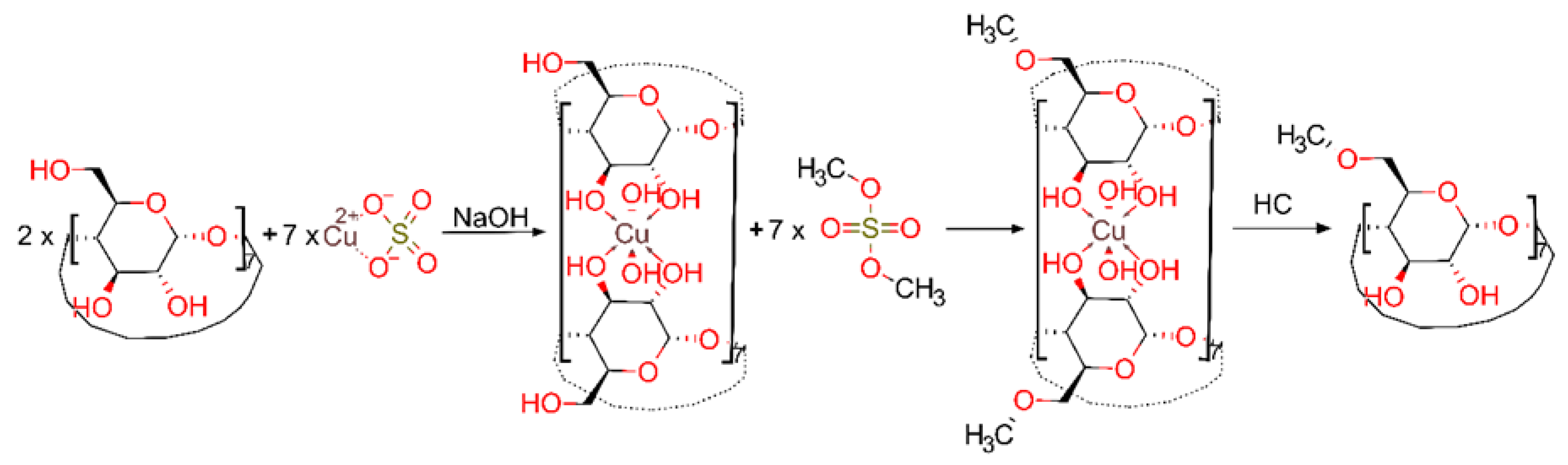
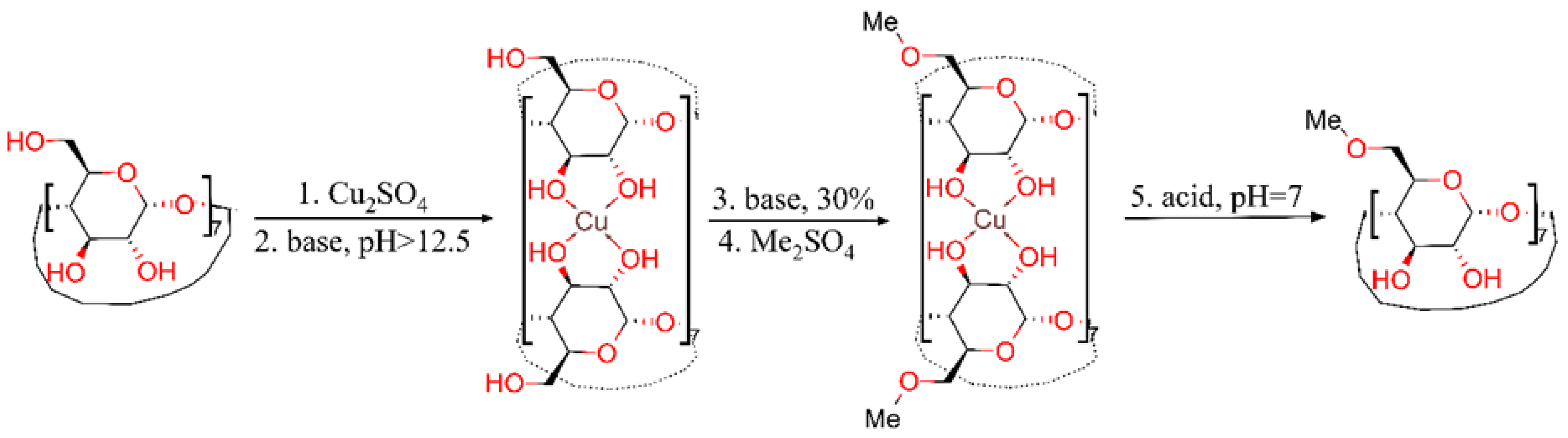
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wenz, G. Influence of intramolecular hydrogen bonds on the binding potential of methylated β-cyclodextrin derivatives. Beilstein J. Org. Chem. 2012, 8, 1890–1895. [Google Scholar] [CrossRef] [Green Version]
- Szejtli, J.; Lipták, A.; Jodál, I.; Fügedi, P.; Nánási, P.; Neszmélyi, A. Synthesis and 13C-NMR spectroscopy of methylated beta-cyclodextrins. Starch Stärke 1980, 32, 165–169. [Google Scholar] [CrossRef]
- Nakazono, K.; Takashima, T.; Arai, T.; Koyama, Y.; Takata, T. High-yield one-pot synthesis of permethylated alpha-cyclodextrin-based polyrotaxane in hydrocarbon solvent through an efficient heterogeneous reaction. Macromolecules 2010, 43, 691–696. [Google Scholar] [CrossRef]
- Stefanache, A.; Silion, M.; Stoica, I.; Fifere, A.; Harabagiu, V.; Farcas, A. Poly[2,7-(9,9-dioctylfluorene)-alt-(5,5′-bithiophene/permethylated beta-cyclodextrin)] main-chain polyrotaxane: Synthesis, characterization and surface morphology. Eur. Polym. J. 2014, 50, 223–234. [Google Scholar] [CrossRef]
- Trotta, F.; Costa, L.; Giancarlo, C. A new easy access to enantiomerically pure 2,2′-dihydroxy-1,1′-binaphthyl. J. Incl. Phenom. Macrocycl. Chem. 2002, 44, 341–344. [Google Scholar] [CrossRef]
- Ashton, P.R.; Königer, R.; Stoddart, J.F.; Alker, D.; Harding, V.D. Amino acid derivatives of β-cyclodextrin. J. Org. Chem. 1996, 61, 903–908. [Google Scholar] [CrossRef]
- Bom, A.; Bradley, M.; Cameron, K.; Clark, J.K.; van Egmond, J.; Feilden, H.; MacLean, E.J.; Muir, A.W.; Palin, R.; Rees, D.C.; et al. A novel concept of reversing neuromuscular block: Chemical encapsulation of rocuronium bromide by a cyclodextrin-based synthetic host. Angew. Chem. Int. Ed. 2002, 41, 265–270. [Google Scholar] [CrossRef]
- Takeo, K.; Mitoh, H.; Uemura, K. Selective chemical modification of cyclomalto- oligosaccharides via tert-butyldimethylsilylation. Carbohydr. Res. 1989, 187, 203–221. [Google Scholar] [CrossRef]
- Uccello-Barretta, G.; Sicoli, G.; Balzano, F.; Salvadori, P. NMR spectroscopy: A powerful tool for detecting the conformational features of symmetrical persubstituted mixed cyclomaltoheptaoses (β-cyclodextrins). Carbohydr. Res. 2005, 340, 271–281. [Google Scholar] [CrossRef]
- Bálint, M.; Darcsi, A.; Benkovics, G.; Varga, E.; Malanga, M.; Béni, S. Synthesis of the chiral selector heptakis(6-O-methyl)-β-cyclodextrin by phase-transfer catalysis and hydrazine-mediated transfer-hydrogenation. Electrophoresis 2019, 40, 1941–1950. [Google Scholar] [CrossRef] [Green Version]
- Estrada, R.I.I.I.; Vigh, G. Comparison of charge state distribution in commercially available sulfated cyclodextrins used as chiral resolving agents in capillary electrophoresis. J. Chromatogr. A 2012, 1226, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Bakó, P.; Fenichel, L.; Töke, L.; Szente, L.; Szejtli, J. Methylation of cyclodextrins by phase-transfer catalysis. J. Incl. Phenom. Mol. Recognit. Chem. 1994, 18, 307–314. [Google Scholar] [CrossRef]
- Wimmer, T. Process for the Preparation of Alkylated Cyclodextrin Derivatives, the Methylated Cyclodextrin Derivatives Obtainable from This Process and the Use of the Products. Patent DE4333598 (A1), 5 April 1995. [Google Scholar]
- Cui, Y.; Wang, C.; Mao, J.; Yu, Y. A facile and practical approach to randomly methylated betacyclodextrin. J. Chem. Technol. Biotechnol. 2010, 85, 248–251. [Google Scholar] [CrossRef]
- Gan, Y.; Zhang, Y.; Xiao, C.; Zhou, C.; Zhao, Y. A novel preparation of methyl-beta-cyclodextrin from dimethyl carbonate and beta-cyclodextrin. Carbohydr. Res. 2011, 346, 389–392. [Google Scholar] [CrossRef]
- Pitha, J.; Milecki, J.; Fales, H.; Pannell, L.; Uekama, K. Hydroxypropyl-beta-cyclodextrin-preparation and characterization-effects on solubility of drugs. Int. J. Pharm. 1986, 29, 73–82. [Google Scholar] [CrossRef]
- Rao, C.T.; Pitha, J.; Lindberg, B.; Lindberg, J. Distribution of substituents in O-(2-hydroxypropyl) derivatives of cyclomalto-oligosaccharides (cyclodextrins): Influence of increasing substitution, of the base used in the preparation, and of macrocyclic size. Carbohydr. Res. 1992, 223, 99–107. [Google Scholar] [CrossRef]
- Yuan, C.; Liu, B.; Liu, H. Characterization of hydroxypropyl-beta-cyclodextrins with different substitution patterns via FTIR, GC-MS, and TG-DTA. Carbohydr. Polym. 2015, 118, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Patil, G. Aminoglycoside antibiotics-A method for selective N-acylation based on the temporary protection of amino alcohol functions as copper chelates. Tetrahedron. Lett. 1978, 12, 1035–1038. [Google Scholar] [CrossRef]
- Bellia, F.; La Mendola, D.; Pedone, C.; Rizzarelli, E.; Saviano, M.; Vecchio, G. Selectively functionalized cyclodextrins and their metal complexes. Chem. Soc. Rev. 2009, 38, 2756–2781. [Google Scholar] [CrossRef]
- Masurier, N.; Lafont, O.; Le Provost, R.; Lesur, D.; Masson, P.; Djedaïni-Pilard, F.; Estour, F. Regioselective access to 3I-O-substituted-β-cyclodextrin derivatives. Chem. Commun. 2009, 5, 589–591. [Google Scholar] [CrossRef]
- Law, H.; Benito, J.M.; Fernández, J.M.G.; Jicsinszky, L.; Crouzy, S.; Defaye, J. Copper(II)-Complex Directed Regioselective Mono-p-Toluenesulfonylation of Cyclomaltoheptaose at a Primary Hydroxyl Group Position: An NMR and Molecular Dynamics-Aided Design. J. Phys. Chem. B 2011, 115, 7524–7532. [Google Scholar] [CrossRef]
- Fuchs, R.; Habermann, N.; Klufers, P. Multinuclear Sandwich-type Complexes of Deprotonated β-Cyclodextrin and Copper(II) Ions. Angew. Chem. Int. Ed. Engl. 1993, 32, 852–854. [Google Scholar] [CrossRef]
- Bagabas, A.A.; Frasconi, M.; Iehl, J.; Hauser, B.; Farha, O.K.; Hupp, J.T.; Hartlieb, K.J.; Botros, Y.Y.; Stoddart, J.F. γ-Cyclodextrin Cuprate Sandwich-Type Complexes. Inorg. Chem. 2013, 52, 2854–2861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz, T.E.; Jankowski, C.K.; Hocquelet, C.; del Rio, P.F.; Barrios, H. The unambiguous assignment of NMR spectra of per-O-methylated 6-mono and 6,6-diamino-β-cyclodextrins. Can. J. Chem. 2008, 86, 726–736. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2-OH | 3-OH | H1 | 6-OH | 3-O-Me | 2-O-Me | 6-O-Me | |
|---|---|---|---|---|---|---|---|
| β-CD | 5.73 | 5.69 | 4.80 | 4.51 | - | - | - |
| MβCD | 5.82 | 5.76 | 4.77 | 4.51 | - | - | 3.24 |
| MβCD [9] | 5.80 | 5.72 | 4.77 | - | 3.59 * | 3.47 * | 3.24 |
| Reference sample | 5.83 | 5.74 | 4.77 | 4.46 | 3.69 | 3.40 | 3.23 |
| TRIMEB | - | - | 5.04 | - | 3.50 | 3.38 | 3.23 |
| TRIMEB [25] | - | - | 5.08 | - | 3.64 | 3.50 | 3.32 |
| DIMEB [25] | - | N/S | 4.95 | - | - | 3.6 | 3.4 |
| C1 | C4 | C3 | C2 | C5 | C6 | Me3 | Me2 | Me6 | |
|---|---|---|---|---|---|---|---|---|---|
| β-CD | 102.14 | 81.73 | 73.27 | 72.58 | 72.24 | 60.15 | - | - | - |
| MβCD | 102.24 | 82.26 | 73.06 | 72.44 | 70.31 | 70.94 | - | - | 52.87 |
| MβCD [9] | 97.5 | 77.6 | 68.3 | 67.6 | 65.6 | 66.2 | 61.3 * | 58.6 * | 53.3 |
| Reference sample | 102.30 | 82.27 | 73.10 | 72.37 | 70.40 | 70.99 | 63.44 | 59.30 | 58.15 |
| TRIMEB [25] | 98.4 | 79.7 | 82.4 | 81.6 | 70.5 | 71.0 | 60.9 | 58.0 | 58.4 |
| DIMEB [25] | 101.3 | 82.1 | 73.1 | 83.6 | 70.9 | 71.4 | - | 60.3 | 58.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bucur, S.; Niculaua, M.; Ciobanu, C.I.; Lungu, N.C.; Mangalagiu, I. A Simple Synthesis Route for Selectively Methylated β-Cyclodextrin Using a Copper Complex Sandwich Protecting Strategy. Molecules 2021, 26, 5669. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185669
Bucur S, Niculaua M, Ciobanu CI, Lungu NC, Mangalagiu I. A Simple Synthesis Route for Selectively Methylated β-Cyclodextrin Using a Copper Complex Sandwich Protecting Strategy. Molecules. 2021; 26(18):5669. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185669
Chicago/Turabian StyleBucur, Stefan, Marius Niculaua, Catalina Ionica Ciobanu, Neculai Catalin Lungu, and Ionel Mangalagiu. 2021. "A Simple Synthesis Route for Selectively Methylated β-Cyclodextrin Using a Copper Complex Sandwich Protecting Strategy" Molecules 26, no. 18: 5669. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185669