
Simultaneous Determination of Tetracyclines and Fluoroquinolones in Poultry Eggs by UPLC Integrated with Dual-Channel-Fluorescence Detection Method

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Stock and Working Solutions
2.3. UPLC-FLD Analysis
2.4. Sample Collection and Pretreatment
2.5. Method Validation
2.5.1. Linearity
2.5.2. Recovery
2.5.3. Precision
2.5.4. Sensitivity
3. Results and Discussion
3.1. Optimization of the Sample Pretreatment
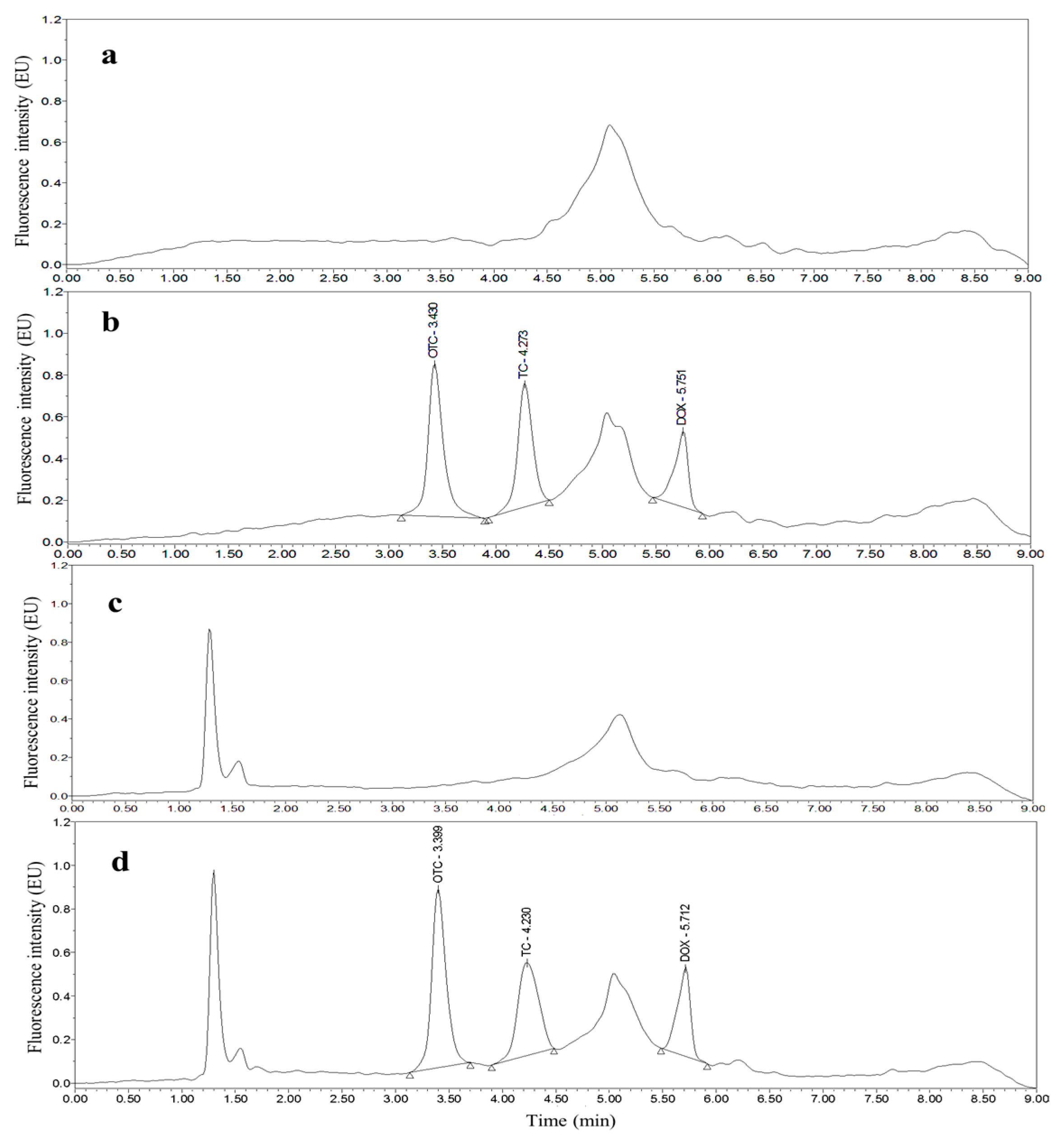
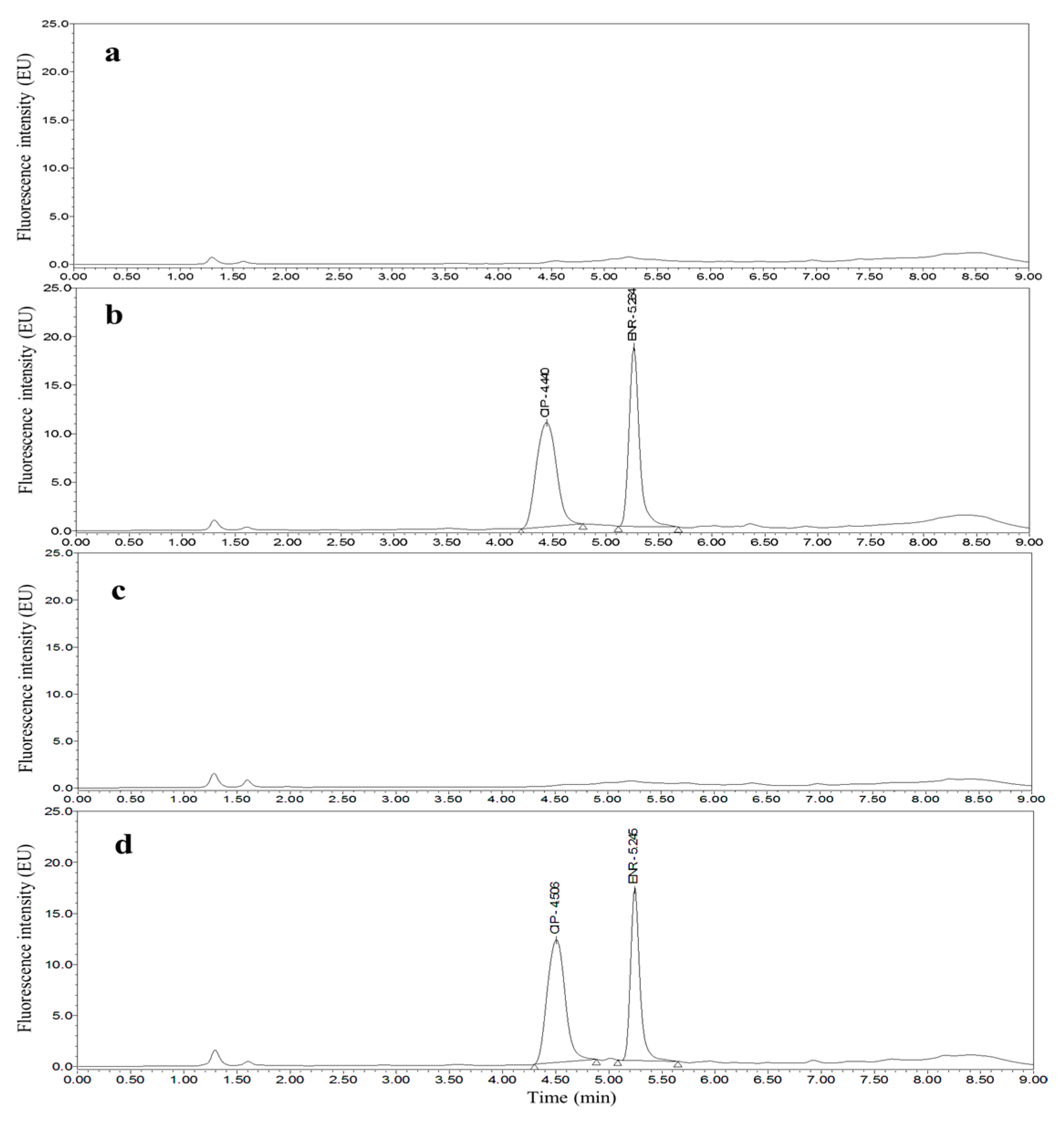
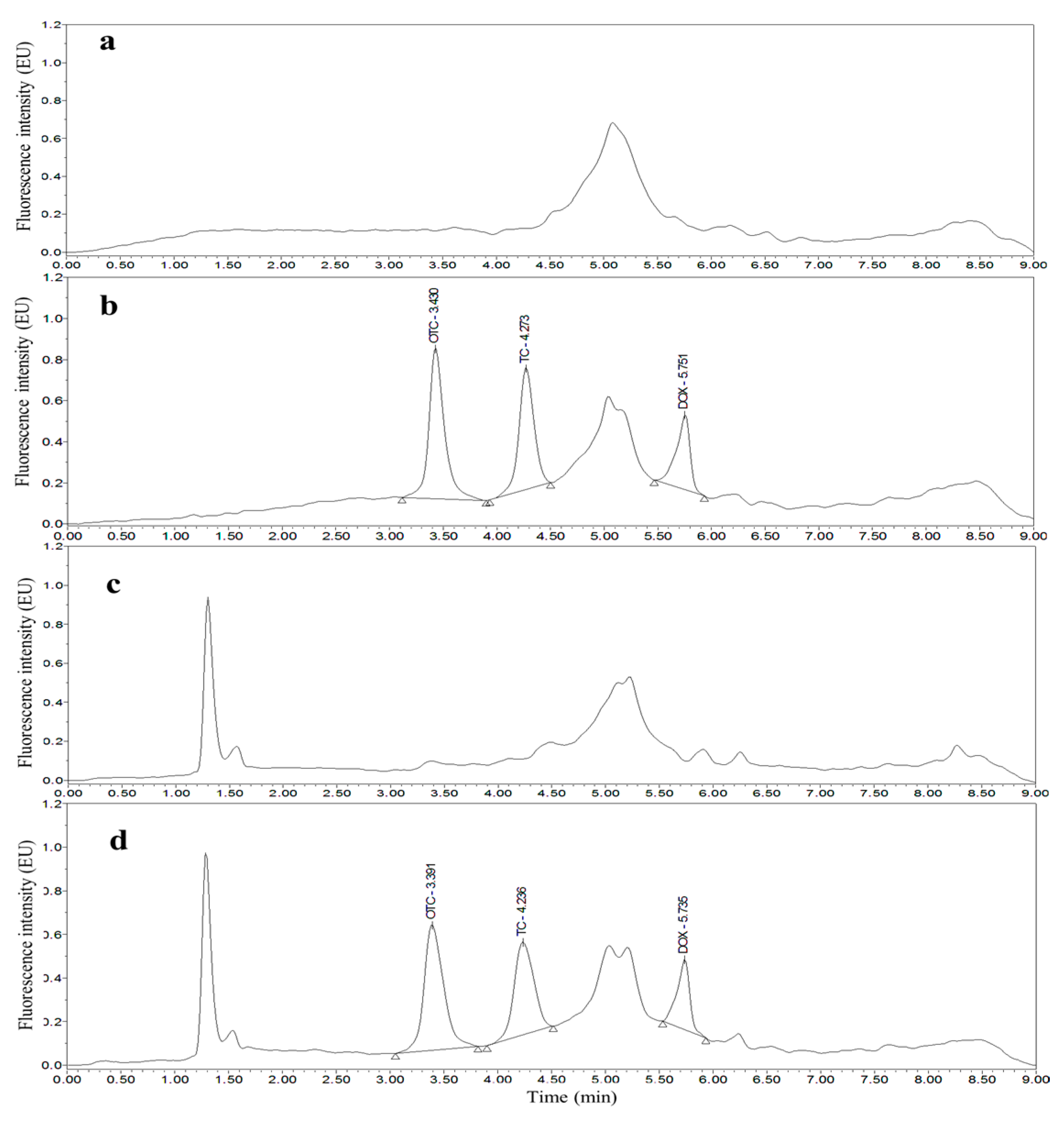
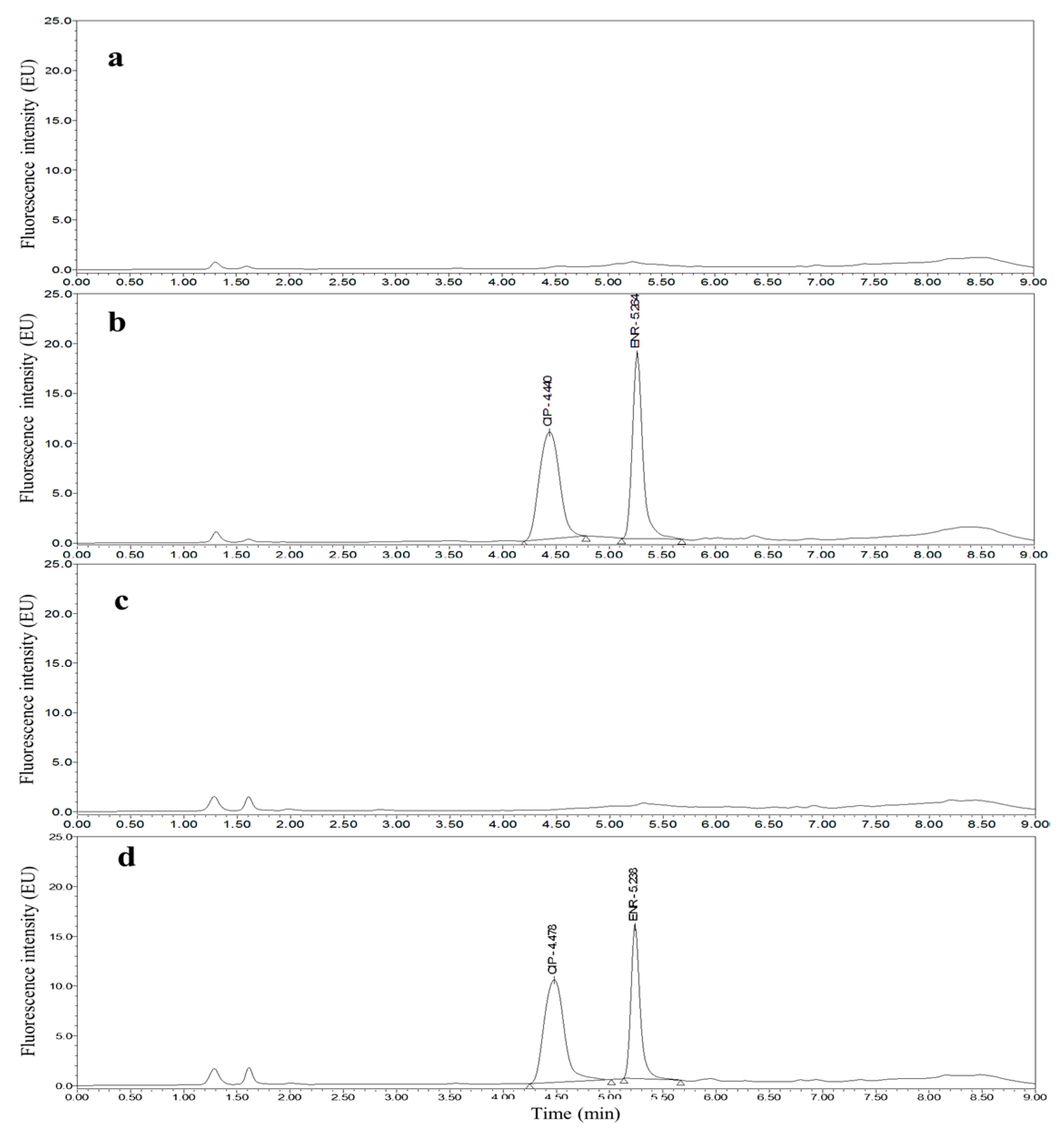
3.2. Optimization of UPLC Separation and FLD
3.3. Validation
3.4. Applications
3.5. Comparison with Previously Reported Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- European Commission. Agriculture and Rural development. European Commisision Press Release Database. Available online: https://ec.europa.eu/agriculture/eggs_en (accessed on 24 June 2019).
- Slana, M.V.; Pahor, L.; Cvitkovič Maričič, L.; Sollner-Dolenc, M. Excretion pattern of enrofloxacin after oral treatment of chicken broilers. J. Vet. Pharmacol. Ther. 2014, 37, 611–614. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Ban on antibiotics as growth promoters in animal feed enters into effect. European Commisision Press Release Database. Available online: http://europa.eu/rapid/press-releaseIP-05-1687en.htm (accessed on 22 December 2005).
- AccessScience Editors. U.S. Bans Antibiotics Use for Enhancing Growth in Livestock; McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Dutil, L.; Irwin, R.; Finley, R.; Ng, L.K.; Avery, B.; Boerlin, P.; Bourgault, A.M.; Cole, L.; Daignault, D.; Desruisseau, A.; et al. Ceftiofur resistance in Salmonella enterica serovar Heidelberg from chicken meat and humans, Canada. Emerg. Infect. Dis. 2010, 16, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xia, S.B.; Bu, F.Y.; Qi, J.; Liu, Y.; Xu, H. Identification of integrons and phylogenetic groups of drug-resistant Escherichia coli from broiler carcasses in China. Int. J. Food Microbiol. 2015, 211, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A. Fluoroquinolones in animal health. J. Vet. Pharmacol. Ther. 1996, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Agriculture of the People’s Republic of China. Maxium Residue Level of Veterinary Drugs in Food of Animal Origin; Notice no. 235 (Appendix 4); Ministry of Agriculture of the People’s Republic of China: Beijing, China, 2002. [Google Scholar]
- The European Medicines Agency. Commission Regulation (EU) No. 37/2010 of 22 December 2009 on Pharmacologically Active Substances and Their Classification Regarding Maximum Residue Limits in Foodstuffs of Animal Origin; The European Medicines Agency: Amsterdam, the Netherlands, 2010. [Google Scholar]
- Castillo-García, M.L.; Aguilar-Caballos, M.P.; Gómez-Hens, A. A europium- and terbium-coated magnetic nanocomposite as sorbent in dispersive solid phase extraction coupled with ultra-high performance liquid chromatography for antibiotic determination in meat samples. J. Chromatogr. A 2015, 1425, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Whittow, G.C. Sturkie’s Avian Physiology. Sturkie’s Avian Physiology, 5th ed.; Academic Press: London, UK, 2000. [Google Scholar]
- Standardization Administration of the People’s Republic of China. Water for Analytical Laboratory Use-Specification and Test Methods; Standards Press of China: Beijing, China, 2008. Available online: https://www.sac.gov.cn (accessed on 15 May 2008).
- Frenich, A.G.; Aguilera-Luiz, M.; Vidal, J.; Romero-González, R. Comparison of several extraction techniques for multiclass analysis of veterinary drugs in eggs using ultra-high pressure liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2010, 661, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Heller, D.N.; Nochetto, C.B.; Rummel, N.G.; Thomas, M.H. Development of multiclass methods for drug residues in eggs: Hydrophilic solid-phase extraction cleanup and liquid chromatography/tandem mass spectrometry analysis of tetracycline, fluoroquinolone, sulfonamide, and beta-lactam residues. J. Agric. Food Chem. 2006, 54, 5267–5278. [Google Scholar] [CrossRef] [PubMed]
- Piatkowska, M.; Jedziniak, P.; Zmudzki, J. Multiresidue method for the simultaneous determination of veterinary medicinal products, feed additives and illegal dyes in eggs using liquid chromatography-tandem mass spectrometry. Food Chem. 2016, 197, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, N. A simple and small-scale sample preparation technique to determine canthaxanthin in hen egg yolk. Food Chem. 2011, 124, 1643–1646. [Google Scholar] [CrossRef]
- Peres, G.T.; Rath, S.; Reyes, F. A HPLC with fluorescence detection method for the determination of tetracyclines residues and evaluation of their stability in honey. Food Control. 2010, 21, 620–625. [Google Scholar] [CrossRef]
- Bani-Yaseen, A.D.; Hammad, F.; Ghanem, B. On the photophysicochemical properties of selected fluoroquinolones: Solvatochromic and fluorescence spectroscopy study. J. Fluoresc. 2013, 23, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.J.; Darwish, A.M.; Freeman, D.W. Simultaneous multiresidue determination of tetracyclines and fluoroquinolones in catfish muscle using high performance liquid chromatography with fluorescence detection. Anal. Chim. Acta 2007, 586, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.J.; Braden, S.E.; Reyes-Herrera, I.; Donoghue, D. Simultaneous determination of fluoroquinolones and tetracyclines in chicken muscle using HPLC with fluorescence detection. J. Chromatogr. B 2007, 846, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Moudgil, P.; Bedi, J.S.; Aulakh, R.S.; Gill, J.; Kumar, A. Validation of HPLC multi-residue method for determination of fluoroquinolones, tetracycline, sulphonamides and chloramphenicol residues in bovine milk. Food Anal. Method. 2018, 12, 338–346. [Google Scholar] [CrossRef]
- The European Communities. Commission Decision 2002/657/EC of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results; Official Journal of European Communities: Brussels, Belgium, 2002. [Google Scholar]
- U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research; Center for Veterinary Medicine. Guidance for Industry: Bioanalytical Method Validation; U.S. Department of Health and Human Services: Washington, DC, USA, 2001. [Google Scholar]
- Wei, H.; Ma, L.; Guo, C.; Sha, J.; Zhu, X.; Wang, Y. Simultaneous extraction and determination of fluoroquinolones, tetracyclines and sulfonamides antibiotics in soils using optimised solid phase extraction chromatography-tandem mass spectrometry. Int. J. Environ. Anal. Chem. 2012, 92, 698–713. [Google Scholar] [CrossRef]
- Lillenberg, M.; Yurchenko, S.; Kipper, K.; Herodes, K.; Pihl, V.; Sepp, K.; Lõhmus, R.; Nei, L. Simultaneous determination of fluoroquinolones, sulfonamides and tetracyclines in sewage sludge by pressurized liquid extraction and liquid chromatography electrospray ionization-mass spectrometry. J. Chromatogr. A 2009, 1216, 5949–5954. [Google Scholar] [CrossRef] [PubMed]
- BŁĄDEK, T.; Posyniak, A.; Gajda, A.; Gbylik, M.; Zmudzki, J. Multi-class procedure for analysis of antibacterial compounds in eggs by liquid chromatography-tandem mass spectrometry. J. Vet. Res. 2013, 56, 321–327. [Google Scholar] [CrossRef]
- Martins, M.T.; Melo, J.; Barreto, F.; Hoff, R.B.; Jank, L.; Bittencourt, M.S.; Arsand, J.B.; Schapoval, E.E.S. A simple, fast and cheap non-SPE screening method for antibacterial residue analysis in milk and liver using liquid chromatography-tandem mass spectrometry. Talanta 2014, 129, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.N.; Zhang, L.; Song, Y.P.; Liu, J.; Wang, J. Application of molecularly imprinted polymer based matrix solid phase dispersion for determination of fluoroquinolones, tetracyclines and sulfonamides in meat. J. Chromatogr. B 2017, 1065, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Mu, H.; Hu, Y.M. Determination of fluoroquinolones, sulfonamides, and tetracyclines multiresidues simultaneously in porcine tissue by MSPD and HPLC-DAD. J. Pharm. Anal. 2012, 2, 76–81. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Analyte | Linearity Range (µg/kg) | Linear Regression Equation | Determination Coefficient (R2) |
|---|---|---|---|---|
| Blended whole chicken egg | OTC | 19.2–1000.0 | y = 425.51x − 2034.3 | 0.9998 |
| TC | 31.2–1000.0 | y = 360.26x + 1118.4 | 0.9998 | |
| DOX | 35.3–300.0 | y = 248.98x + 7889.8 | 0.9993 | |
| CIP | 0.6–300.0 | y = 35325x − 9347 | 0.9998 | |
| ENR | 0.4–300.0 | y = 39161x + 282018 | 0.9998 | |
| Chicken albumen | OTC | 17.4–1000.0 | y = 418.55x − 1723.3 | 0.9997 |
| TC | 28.8–1000.0 | y = 396.38x − 3708.4 | 0.9994 | |
| DOX | 31.9–300.0 | y = 281.71x + 3259.9 | 0.9992 | |
| CIP | 0.7–300.0 | y = 33285x + 10819 | 0.9996 | |
| ENR | 0.3–300.0 | y = 37248x + 398456 | 0.9996 | |
| Chicken yolk | OTC | 25.6–1000.0 | y = 449.03x − 6847.2 | 0.9994 |
| TC | 36.0–1000.0 | y = 363.79x − 4823.8 | 0.9993 | |
| DOX | 40.1–300.0 | y = 286.29x − 71.871 | 0.9998 | |
| CIP | 1.2–300.0 | y = 35999x + 30938 | 0.9996 | |
| ENR | 0.3–300.0 | y = 43943x + 166512 | 0.9994 | |
| Blended whole duck egg | OTC | 20.1–1000.0 | y = 476.11x − 5044.9 | 0.9999 |
| TC | 32.1–1000.0 | y = 359.6x + 1599.6 | 0.9998 | |
| DOX | 34.7–300.0 | y = 302.9x − 2627.5 | 0.9995 | |
| CIP | 1.1–300.0 | y = 36124x − 20065 | 0.9997 | |
| ENR | 0.5–300.0 | y = 44043x + 124503 | 0.9997 | |
| Duck albumen | OTC | 19.2–1000.0 | y = 444.68x − 968.85 | 0.9996 |
| TC | 27.3–1000.0 | y = 394.95x − 7178.2 | 0.9994 | |
| DOX | 32.5–300.0 | y = 296.54x − 815.06 | 0.9994 | |
| CIP | 0.7–300.0 | y = 35712x + 126377 | 0.9998 | |
| ENR | 0.4–300.0 | y = 39981x + 118026 | 0.9995 | |
| Duck yolk | OTC | 21.6–1000.0 | y = 413.89x + 538.36 | 0.9997 |
| TC | 38.3–1000.0 | y = 333.34x − 3420.4 | 0.9998 | |
| DOX | 37.2–300.0 | y = 281.82x + 29.831 | 0.9995 | |
| CIP | 1.5–300.0 | y = 40759x + 79433 | 0.9998 | |
| ENR | 0.3–300.0 | y = 39750x + 220046 | 0.9993 |
| Analyte | Spiked Level (µg/kg) | Recovery (%) | RSD (%) | Intraday RSD (%) | Interday RSD (%) |
|---|---|---|---|---|---|
| OTC | 19.2 | 86.93 ± 1.39 | 1.60 | 2.59 | 2.78 |
| 200 | 86.90 ± 2.56 | 2.95 | 3.71 | 3.83 | |
| 400 α | 89.43 ± 1.63 | 1.82 | 2.03 | 2.32 | |
| 800 | 91.15 ± 3.21 | 3.52 | 4.52 | 4.72 | |
| TC | 31.2 | 86.58 ± 2.88 | 3.32 | 4.14 | 4.26 |
| 200 | 87.40 ± 1.70 | 1.95 | 3.85 | 3.57 | |
| 400 α | 89.28 ± 2.48 | 2.78 | 2.85 | 3.89 | |
| 800 | 90.20 ± 1.38 | 1.53 | 2.33 | 2.58 | |
| DOX | 35.3 | 86.33 ± 2.25 | 2.60 | 3.61 | 3.13 |
| 50 | 85.10 ± 3.01 | 3.54 | 3.10 | 3.94 | |
| 100 α | 85.15 ± 1.87 | 2.19 | 3.17 | 3.70 | |
| 200 | 84.65 ± 3.72 | 4.40 | 3.98 | 4.59 | |
| CIP | 0.6 | 87.65 ± 2.29 | 2.60 | 3.49 | 3.90 |
| 50 | 89.88 ± 2.23 | 2.48 | 3.62 | 3.41 | |
| 100 α | 92.35 ± 2.16 | 2.34 | 2.46 | 3.99 | |
| 200 | 92.88 ± 2.41 | 2.60 | 2.05 | 3.27 | |
| ENR | 0.4 | 94.05 ± 1.88 | 1.99 | 3.07 | 4.17 |
| 50 | 88.63 ± 2.49 | 2.80 | 2.93 | 3.61 | |
| 100 α | 95.58 ± 3.68 | 3.85 | 3.41 | 3.90 | |
| 200 | 92.63 ± 2.57 | 2.78 | 3.15 | 3.74 |
| Analyte | Spiked Level (µg/kg) | Recovery (%) | RSD (%) | Intraday RSD (%) | Interday RSD (%) |
|---|---|---|---|---|---|
| OTC | 17.4 | 87.28 ± 1.76 | 2.01 | 2.57 | 3.96 |
| 200 | 86.33 ± 1.07 | 1.24 | 2.04 | 2.86 | |
| 400 α | 89.70 ± 2.60 | 2.90 | 3.15 | 3.09 | |
| 800 | 89.03 ± 3.30 | 3.71 | 4.28 | 4.32 | |
| TC | 28.8 | 85.60 ± 3.15 | 3.69 | 3.82 | 3.95 |
| 200 | 85.20 ± 1.53 | 1.79 | 2.24 | 3.40 | |
| 400 α | 90.05 ± 3.40 | 3.77 | 2.68 | 4.84 | |
| 800 | 89.28 ± 2.42 | 2.70 | 2.67 | 3.64 | |
| DOX | 31.9 | 86.20 ± 2.15 | 2.50 | 3.41 | 3.57 |
| 50 | 86.40 ± 1.54 | 1.79 | 2.23 | 2.37 | |
| 100 α | 87.53 ± 2.00 | 2.29 | 2.99 | 3.07 | |
| 200 | 89.58 ± 1.67 | 1.86 | 2.13 | 2.44 | |
| CIP | 0.7 | 88.45 ± 2.65 | 2.99 | 2.59 | 3.69 |
| 50 | 88.13 ± 1.72 | 1.96 | 2.32 | 2.66 | |
| 100 α | 90.15 ± 1.22 | 1.35 | 1.99 | 2.10 | |
| 200 | 90.78 ± 1.96 | 2.16 | 2.69 | 2.54 | |
| ENR | 0.3 | 86.15 ± 3.52 | 4.09 | 4.67 | 6.24 |
| 50 | 91.48 ± 1.78 | 1.94 | 2.36 | 4.18 | |
| 100 α | 89.58 ± 1.51 | 1.69 | 2.65 | 2.81 | |
| 200 | 91.45 ± 3.36 | 3.67 | 4.31 | 4.94 |
| Analyte | Spiked Level (µg/kg) | Recovery (%) | RSD (%) | Intraday RSD (%) | Interday RSD (%) |
|---|---|---|---|---|---|
| OTC | 25.6 | 85.30 ± 2.17 | 2.54 | 2.95 | 4.10 |
| 200 | 86.53 ± 1.35 | 1.56 | 3.62 | 3.91 | |
| 400 α | 87.93 ± 2.40 | 2.73 | 2.94 | 4.92 | |
| 800 | 88.85 ± 2.96 | 3.33 | 3.35 | 4.12 | |
| TC | 36.0 | 86.58 ± 2.88 | 3.32 | 3.55 | 3.97 |
| 200 | 86.70 ± 1.16 | 1.34 | 3.29 | 3.11 | |
| 400 α | 88.05 ± 2.00 | 2.27 | 2.56 | 3.99 | |
| 800 | 89.15 ± 1.07 | 1.20 | 2.77 | 3.63 | |
| DOX | 40.1 | 86.08 ± 1.83 | 2.12 | 2.35 | 3.97 |
| 50 | 86.03 ± 2.70 | 3.15 | 3.63 | 4.71 | |
| 100 α | 85.15 ± 1.87 | 2.20 | 3.17 | 3.77 | |
| 200 | 85.13 ± 2.79 | 3.28 | 3.89 | 4.28 | |
| CIP | 1.2 | 87.38 ± 1.89 | 2.17 | 2.38 | 3.89 |
| 50 | 89.63 ± 1.99 | 2.22 | 3.03 | 3.82 | |
| 100 α | 91.85 ± 2.45 | 2.66 | 3.19 | 3.66 | |
| 200 | 92.88 ± 2.41 | 2.60 | 3.05 | 3.27 | |
| ENR | 0.3 | 90.73 ± 1.70 | 1.88 | 2.07 | 3.37 |
| 50 | 90.38 ± 2.00 | 2.20 | 2.74 | 3.30 | |
| 100 α | 95.58 ± 3.68 | 3.85 | 4.11 | 5.60 | |
| 200 | 93.88 ± 2.66 | 2.83 | 3.31 | 3.45 |
| Analyte | Spiked Level (µg/kg) | Recovery (%) | RSD (%) | Intraday RSD (%) | Interday RSD (%) |
|---|---|---|---|---|---|
| OTC | 20.1 | 85.33 ± 2.87 | 3.36 | 3.50 | 4.55 |
| 200 | 85.78 ± 1.60 | 1.86 | 2.66 | 3.30 | |
| 400 α | 88.38 ± 1.88 | 2.13 | 2.91 | 3.11 | |
| 800 | 89.40 ± 2.50 | 2.81 | 3.85 | 4.19 | |
| TC | 32.1 | 87.30 ± 2.15 | 2.46 | 3.61 | 4.48 |
| 200 | 87.58 ± 2.18 | 2.49 | 3.20 | 3.55 | |
| 400 α | 88.35 ± 2.50 | 2.83 | 3.38 | 3.40 | |
| 800 | 89.18 ± 1.72 | 1.93 | 2.57 | 3.14 | |
| DOX | 34.7 | 84.33 ± 2.68 | 3.18 | 3.62 | 4.51 |
| 50 | 83.50 ± 1.10 | 1.32 | 3.12 | 3.31 | |
| 100 α | 86.15 ± 3.33 | 3.87 | 3.22 | 4.28 | |
| 200 | 84.83 ± 1.26 | 1.49 | 3.58 | 3.46 | |
| CIP | 1.1 | 85.53 ± 2.05 | 2.40 | 3.51 | 4.17 |
| 50 | 90.13 ± 2.66 | 2.95 | 3.68 | 4.14 | |
| 100 α | 91.80 ± 1.94 | 2.11 | 2.91 | 3.39 | |
| 200 | 92.03 ± 2.96 | 3.21 | 3.60 | 4.45 | |
| ENR | 0.5 | 87.98 ± 3.12 | 3.55 | 3.46 | 4.01 |
| 50 | 89.13 ± 2.65 | 2.97 | 3.29 | 3.04 | |
| 100 α | 94.35 ± 2.80 | 2.97 | 3.93 | 4.30 | |
| 200 | 92.63 ± 2.57 | 2.78 | 2.11 | 3.24 |
| Analyte | Spiked Level (µg/kg) | Recovery (%) | RSD (%) | Intraday RSD (%) | Interday RSD (%) |
|---|---|---|---|---|---|
| OTC | 19.2 | 87.03 ± 2.14 | 2.46 | 3.57 | 4.22 |
| 200 | 87.58 ± 1.82 | 2.08 | 3.83 | 3.54 | |
| 400 α | 88.48 ± 2.11 | 2.39 | 2.80 | 3.12 | |
| 800 | 88.18 ± 2.20 | 2.49 | 3.03 | 4.96 | |
| TC | 27.3 | 84.10 ± 3.50 | 4.17 | 4.91 | 5.55 |
| 200 | 86.33 ± 2.91 | 3.37 | 4.25 | 4.68 | |
| 400 α | 88.90 ± 3.66 | 4.12 | 4.06 | 5.28 | |
| 800 | 89.23 ± 2.12 | 2.38 | 3.86 | 3.37 | |
| DOX | 32.5 | 84.45 ± 2.18 | 2.58 | 3.51 | 4.78 |
| 50 | 85.88 ± 1.52 | 1.77 | 2.35 | 3.17 | |
| 100 α | 86.68 ± 3.40 | 3.92 | 3.96 | 5.80 | |
| 200 | 88.88 ± 1.79 | 2.02 | 3.76 | 3.69 | |
| CIP | 0.7 | 88.45 ± 2.65 | 2.99 | 3.55 | 4.11 |
| 50 | 87.85 ± 1.22 | 1.39 | 3.32 | 3.16 | |
| 100 α | 88.90 ± 3.53 | 3.98 | 4.70 | 5.33 | |
| 200 | 89.08 ± 1.60 | 1.79 | 2.07 | 3.88 | |
| ENR | 0.4 | 87.90 ± 3.26 | 3.70 | 3.95 | 4.42 |
| 50 | 90.98 ± 1.71 | 1.88 | 3.20 | 3.23 | |
| 100 α | 89.85 ± 2.05 | 2.28 | 3.23 | 4.77 | |
| 200 | 91.23 ± 3.50 | 3.84 | 2.88 | 4.27 |
| Analyte | Spiked Level (µg/kg) | Recovery (%) | RSD (%) | Intraday RSD (%) | Interday RSD (%) |
|---|---|---|---|---|---|
| OTC | 21.6 | 86.93 ± 1.39 | 1.60 | 3.54 | 3.59 |
| 200 | 86.85 ± 2.56 | 2.95 | 3.71 | 4.83 | |
| 400 α | 89.43 ± 1.63 | 1.82 | 2.13 | 3.32 | |
| 800 | 91.15 ± 3.21 | 3.52 | 3.86 | 4.72 | |
| TC | 38.3 | 86.58 ± 2.88 | 3.32 | 3.14 | 4.26 |
| 200 | 87.40 ± 1.70 | 1.95 | 2.95 | 2.57 | |
| 400 α | 89.28 ± 2.48 | 2.78 | 2.87 | 3.26 | |
| 800 | 90.20 ± 1.38 | 1.53 | 3.58 | 3.33 | |
| DOX | 37.2 | 86.33 ± 2.25 | 2.60 | 3.25 | 4.13 |
| 50 | 85.10 ± 3.01 | 3.54 | 3.48 | 5.10 | |
| 100 α | 87.65 ± 1.60 | 1.84 | 2.37 | 3.19 | |
| 200 | 90.90 ± 2.40 | 2.64 | 3.66 | 3.23 | |
| CIP | 1.5 | 88.65 ± 3.30 | 3.72 | 4.11 | 4.52 |
| 50 | 89.88 ± 2.23 | 2.48 | 2.62 | 3.41 | |
| 100 α | 92.35 ± 2.16 | 2.34 | 3.46 | 3.99 | |
| 200 | 92.63 ± 2.03 | 2.19 | 3.09 | 4.53 | |
| ENR | 0.3 | 89.73 ± 1.42 | 1.59 | 2.40 | 3.66 |
| 50 | 90.48 ± 1.60 | 1.78 | 2.82 | 3.86 | |
| 100 α | 92.51 ± 3.68 | 3.98 | 3.11 | 4.26 | |
| 200 | 92.63 ± 2.57 | 2.78 | 3.17 | 3.24 |
| Analyte | Matrix | LODs (µg/kg) | LOQs (µg/kg) |
|---|---|---|---|
| OTC | Blended whole chicken egg | 5.5 | 19.2 |
| Chicken albumen | 5.2 | 17.4 | |
| Chicken Yolk | 7.7 | 25.6 | |
| Blended whole duck egg | 6.0 | 20.1 | |
| Duck albumen | 5.4 | 19.2 | |
| Duck yolk | 6.5 | 21.6 | |
| TC | Blended whole chicken egg | 9.1 | 31.2 |
| Chicken albumen | 9.5 | 28.8 | |
| Chicken yolk | 10.6 | 36.0 | |
| Blended whole duck egg | 9.3 | 32.1 | |
| Duck albumen | 8.9 | 27.3 | |
| Duck yolk | 11.8 | 38.3 | |
| DOX | Blended whole chicken egg | 10.5 | 35.3 |
| Chicken albumen | 9.7 | 31.9 | |
| Chicken yolk | 13.4 | 40.1 | |
| Blended whole duck egg | 10.4 | 34.7 | |
| Duck albumen | 9.6 | 32.5 | |
| Duck yolk | 10.6 | 37.2 | |
| CIP | Blended whole chicken egg | 0.2 | 0.6 |
| Chicken albumen | 0.2 | 0.7 | |
| Chicken yolk | 0.4 | 1.2 | |
| Blended whole duck egg | 0.3 | 1.1 | |
| Duck albumen | 0.2 | 0.7 | |
| Duck yolk | 0.5 | 1.5 | |
| ENR | Blended whole chicken egg | 0.1 | 0.4 |
| Chicken albumen | 0.1 | 0.3 | |
| Chicken yolk | 0.1 | 0.3 | |
| Blended whole duck egg | 0.1 | 0.5 | |
| Duck albumen | 0.1 | 0.4 | |
| Duck yolk | 0.1 | 0.3 |
| Detection Method | Sample Pretreatment Method | Analyte | Matrix | Analysis Time (min) | LOD (µg/kg) | LOQ (µg/kg) | Recovery (%) |
|---|---|---|---|---|---|---|---|
| UPLC-FLD (this study) | LLE-SPE | Tetracyclines and fluoroquinolones | Poultry eggs | 9 | 0.1–13.4 | 0.3–40.1 | 83.50–95.58 |
| UPLC-FLD [10] | Dispersive SPE | Tetracyclines and acidic quinolones | Pork and chicken muscle | 6 | 0.25–3.8 | - | 61.5–102.6 |
| HPLC-FLD [19] | LLE | Tetracyclines and fluoroquinolones | Catfish muscle | >25 | - | 0.15–1.5 | 60–92 |
| HPLC-FLD [20] | LLE | Tetracyclines and fluoroquinolones | Chicken muscle | >25 | - | 0.5–5 | 63–95 |
| UPLC-DAD [28] | Matrix solid-phase dispersion | Tetracyclines, fluoroquinolones and sulfonamides | Pork | 14 | 0.5–3.0 | - | 74.5–102.7 |
| HPLC-DAD [29] | Matrix solid-phase dispersion | Tetracyclines and fluoroquinolones | Porcine tissues | 12 | 2–10 | 7–34 | 80.6–99.2 |
| HPLC-DAD [21] | LLE-SPE | Tetracyclines, fluoroquinolones, sulfonamides and chloramphenicols | Milk | 40 | 17.2–24.9 | 51.5–68.1 | 83.3–111.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; He, Z.; Chen, J.; Chen, L.; Xie, K.; Zhang, T.; Zhang, G.; Dai, G. Simultaneous Determination of Tetracyclines and Fluoroquinolones in Poultry Eggs by UPLC Integrated with Dual-Channel-Fluorescence Detection Method. Molecules 2021, 26, 5684. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185684
Guo Y, He Z, Chen J, Chen L, Xie K, Zhang T, Zhang G, Dai G. Simultaneous Determination of Tetracyclines and Fluoroquinolones in Poultry Eggs by UPLC Integrated with Dual-Channel-Fluorescence Detection Method. Molecules. 2021; 26(18):5684. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185684
Chicago/Turabian StyleGuo, Yawen, Zhaoyuan He, Jinyuan Chen, Lan Chen, Kaizhou Xie, Tao Zhang, Genxi Zhang, and Guojun Dai. 2021. "Simultaneous Determination of Tetracyclines and Fluoroquinolones in Poultry Eggs by UPLC Integrated with Dual-Channel-Fluorescence Detection Method" Molecules 26, no. 18: 5684. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185684