
Simulation of the Interactions of Arginine with Wild-Type GALT Enzyme and the Classic Galactosemia-Related Mutant p.Q188R by a Computational Approach

 , ,
, , 
Abstract
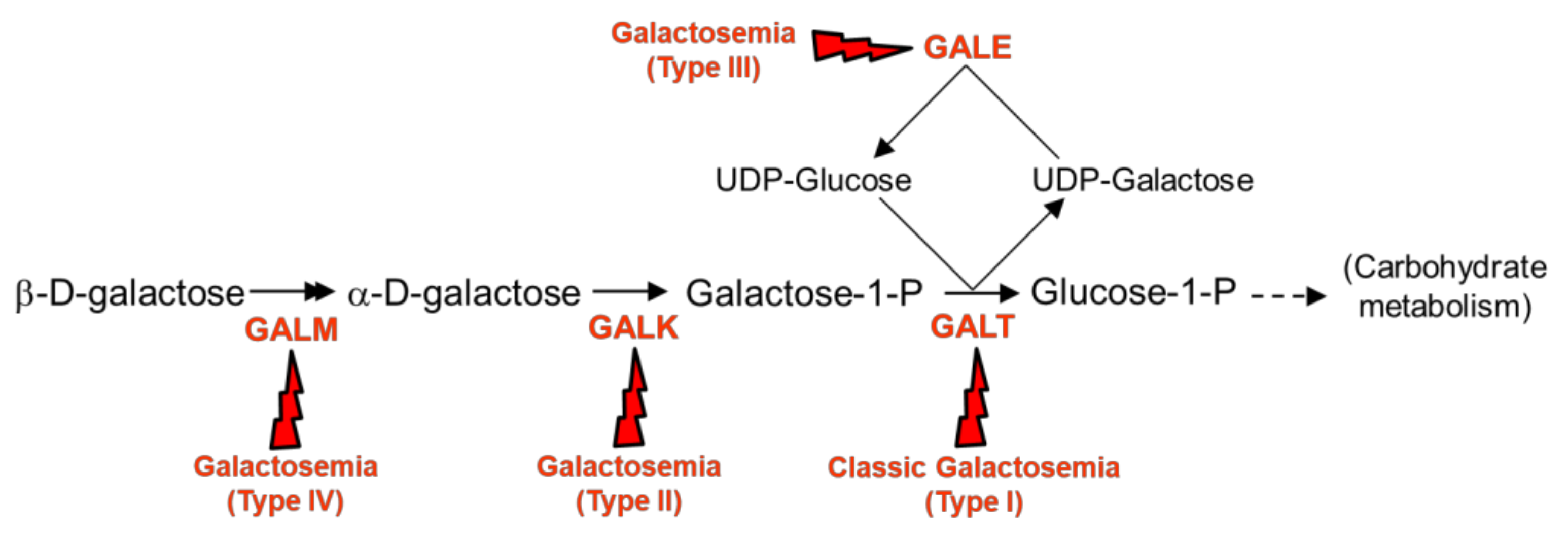
:1. Introduction
2. Results and Discussion
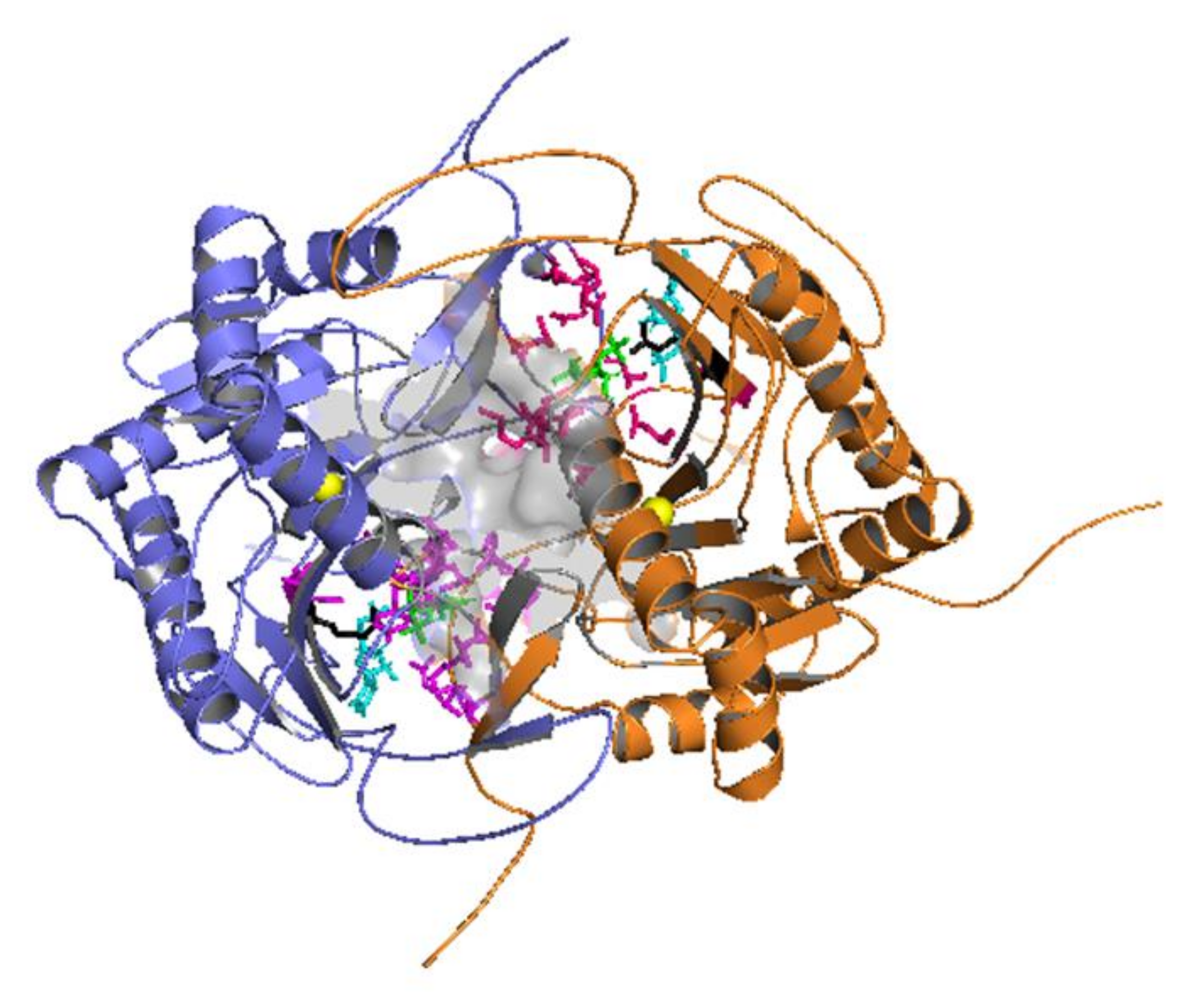
2.1. Docking Simulations
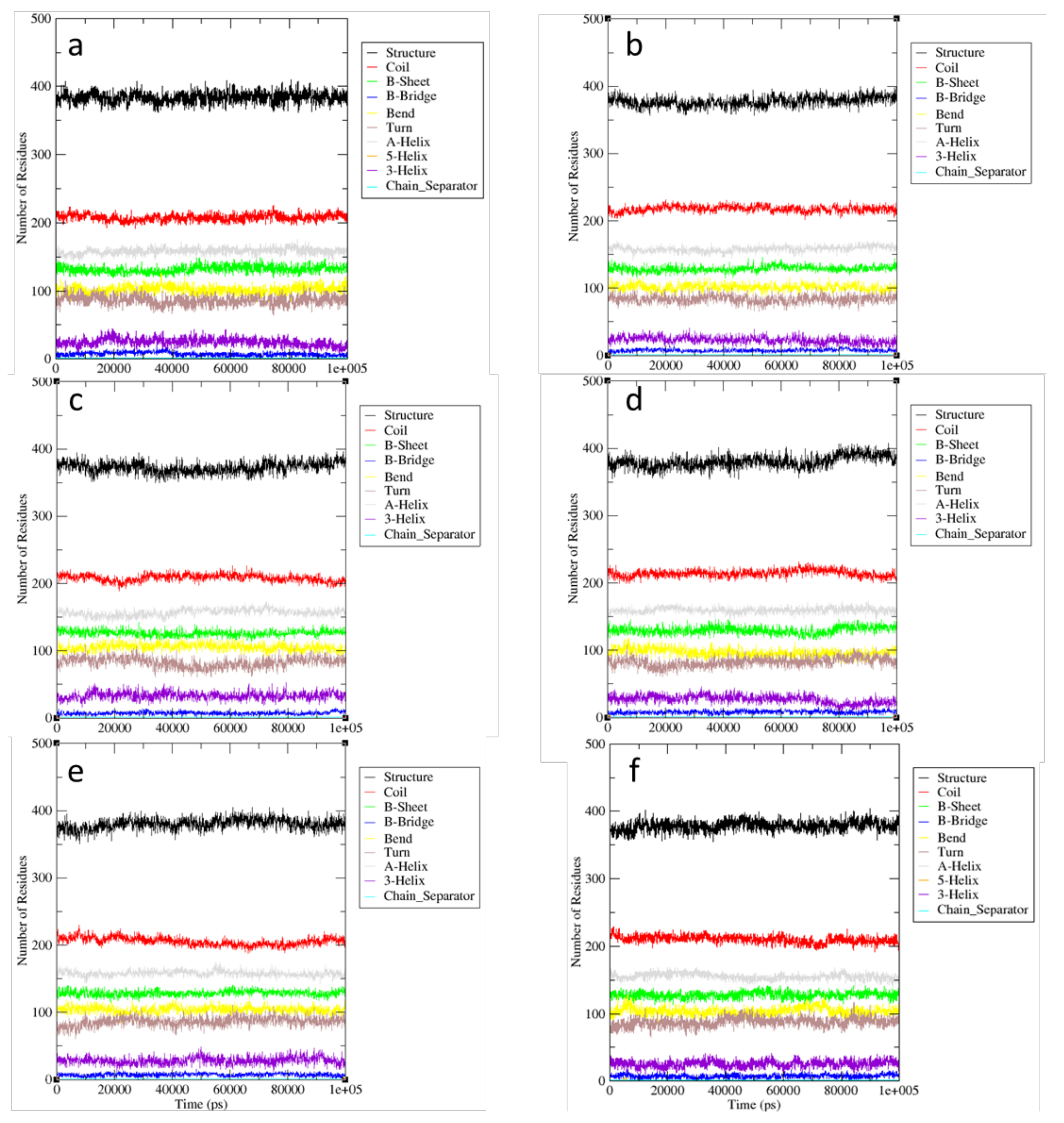
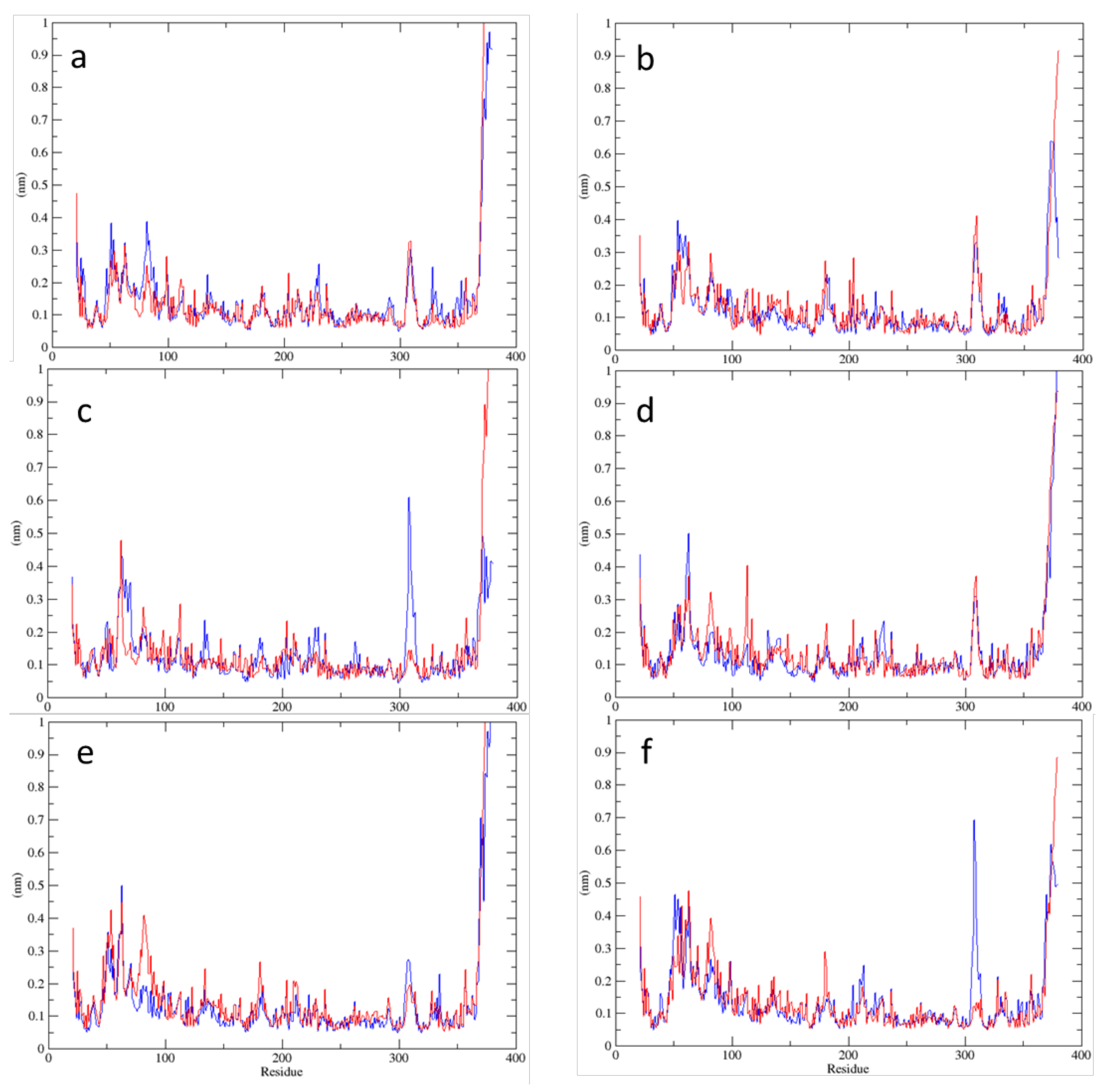
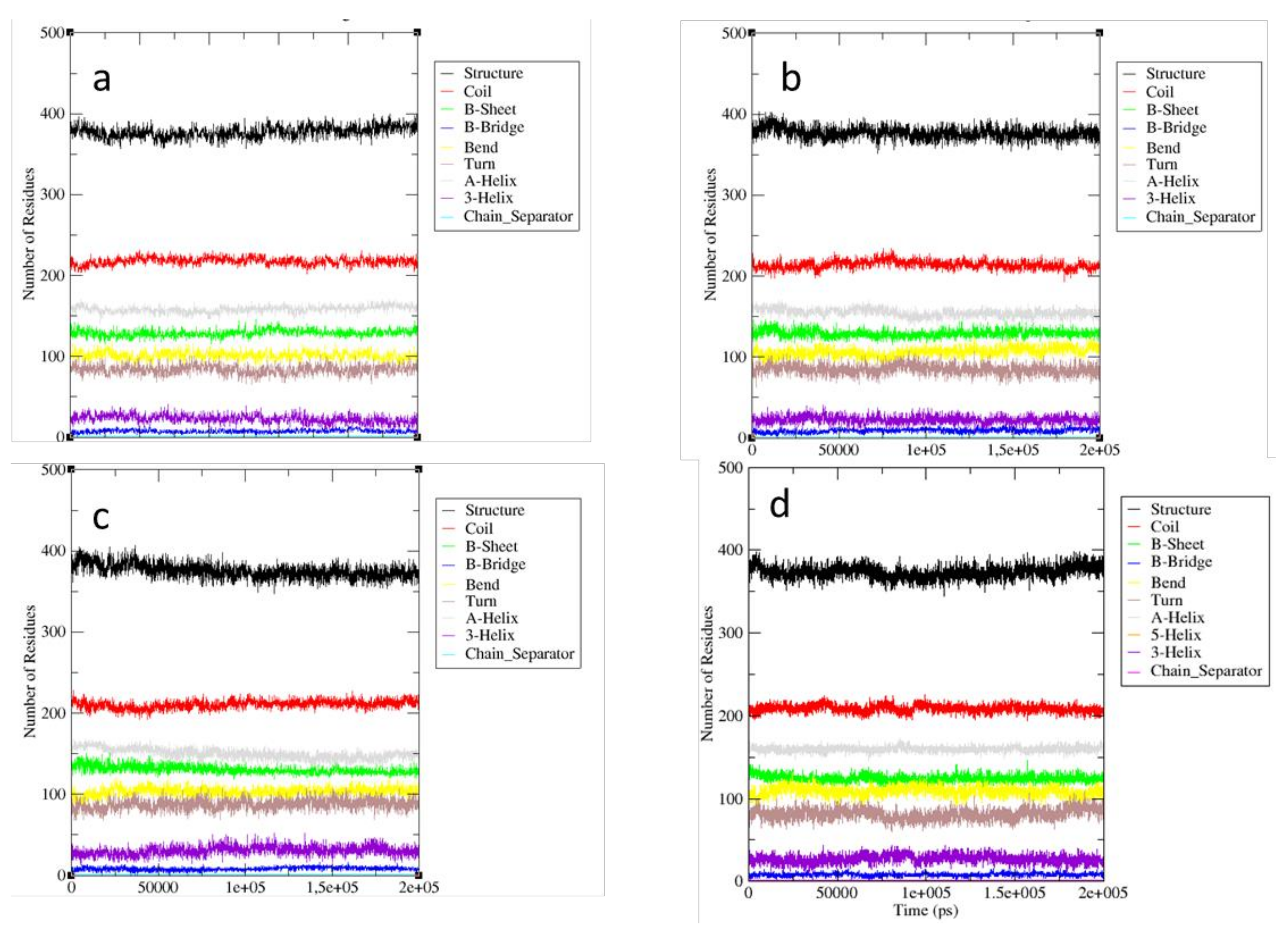
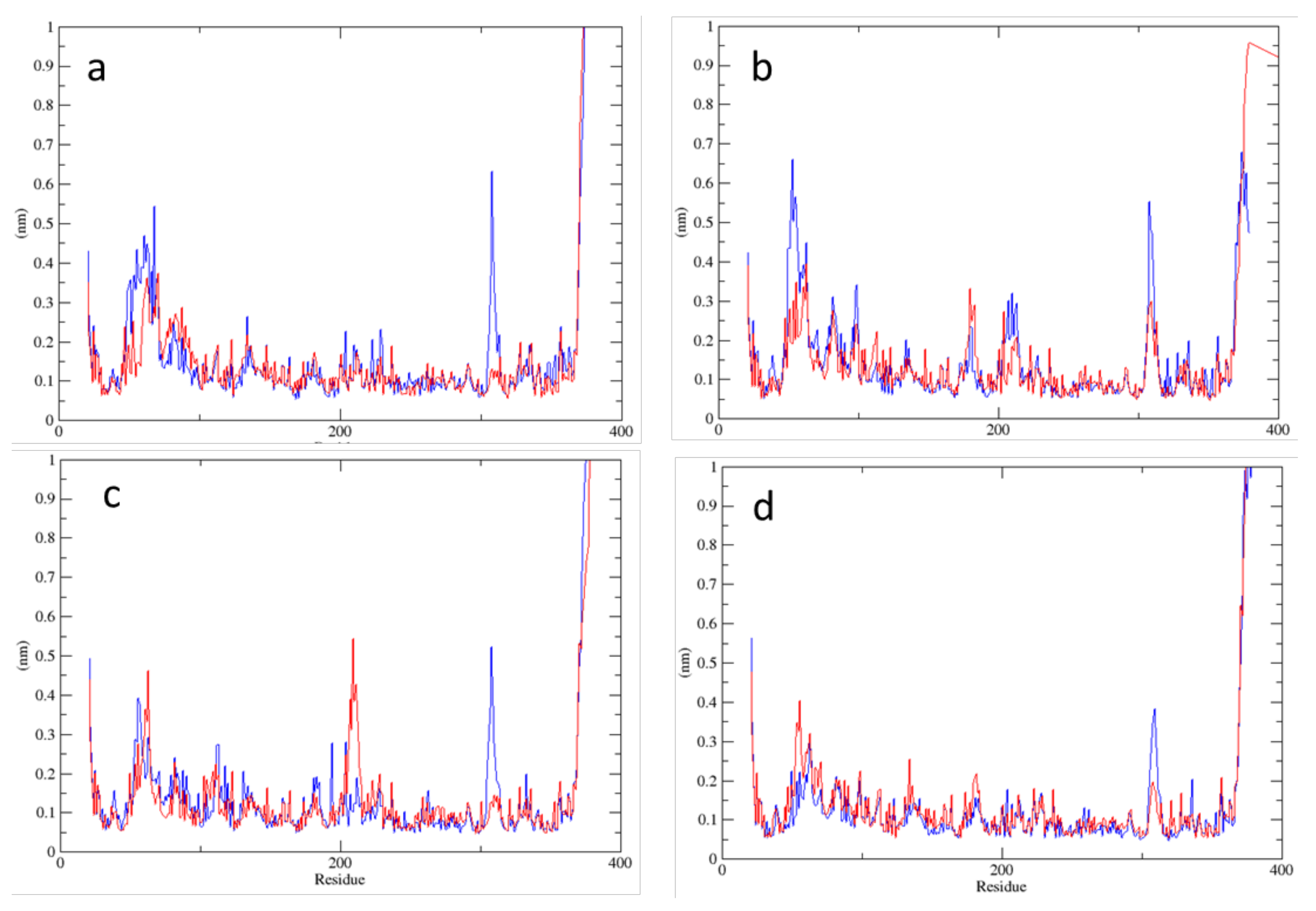
2.2. MD Simulations—Arginine in the Active Site
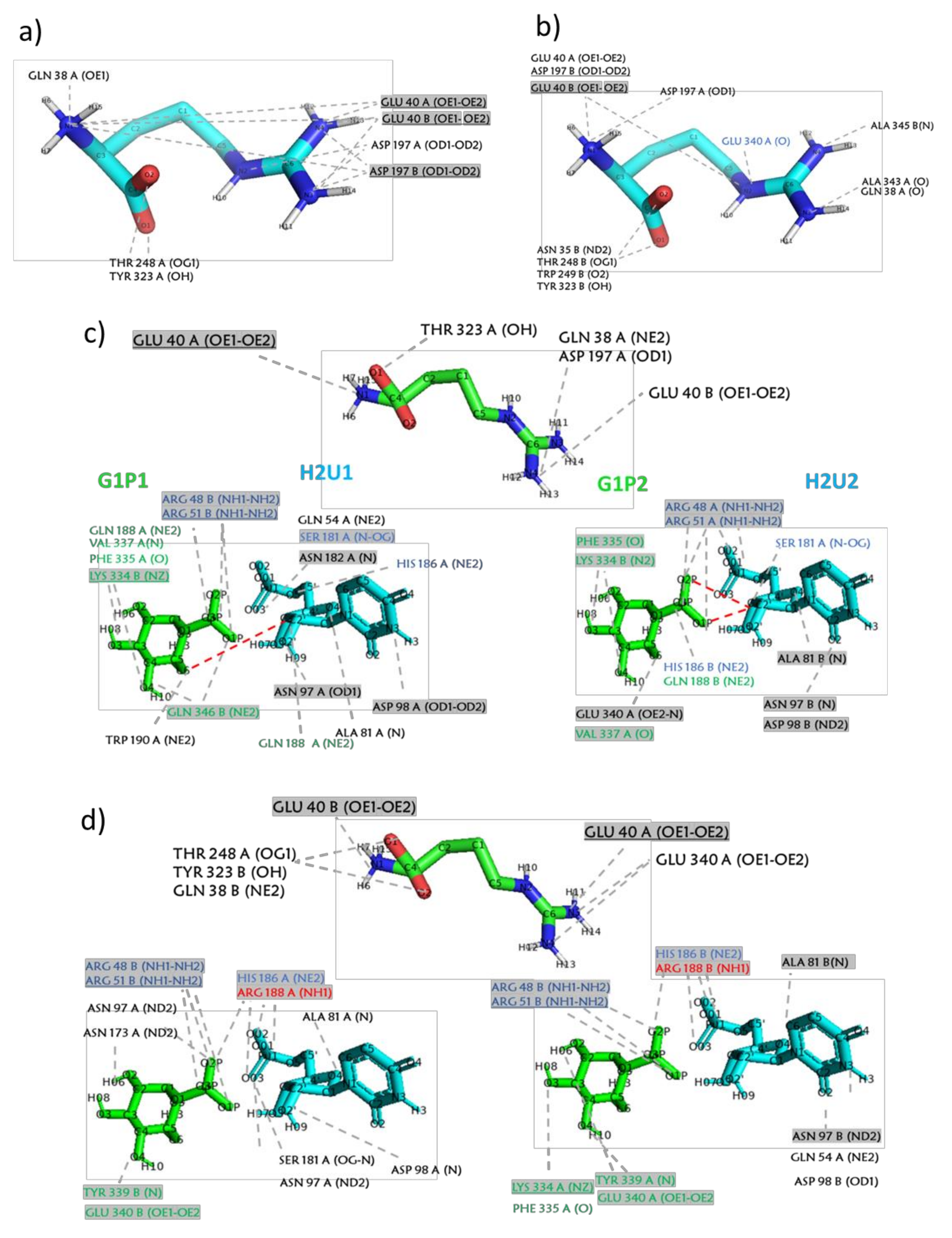
2.3. MD Simulations—Arginine in the Central Cavity
2.4. Comparison of the Results of Simulations in the Absence of Arginine
3. Materials and Methods
3.1. Starting Structures
3.2. Docking Simulations
3.3. MD Simulations and Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ringe, D.; Petsko, G.A. What are pharmacological chaperones and why are they interesting? J. Biol. 2009, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological chaperones: A therapeutic approach for diseases caused by destabilizing missense mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef] [Green Version]
- Serapian, S.A.; Sanchez-Martin, C.; Moroni, E.; Rasola, A.; Colombo, G. Targeting the mithocondrial chaperone TRAP1: Strategies and therapeutic perspectives. Trends Pharm. Sci. 2021, 42, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Leloir, L.F. Two decades of research on the biosynthesis of saccharides. Science 1971, 172, 1299–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, G.T. Classic galactosemia and clinical variant galactosemia. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993 2020. [Google Scholar]
- McCorvie, T.J.; Gleason, T.J.; Fridovich-Keil, J.L.; Timson, D.J. Misfolding of galactose 1-phosphate uridylyltransferase can result in type I galactosemia. Biochim. Biophys. Acta 2013, 1832, 1279–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- d’Acierno, A.; Scafuri, B.; Facchiano, A.; Marabotti, A. The evolution of a Web resource: The Galactosemia Proteins Database 2.0. Hum. Mutat. 2018, 39, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Timson, D.J. The molecular basis of galactosemia-Past, present and future. Gene 2016, 589, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, A.I.; Trabuco, M.; Ramos, R.; Silva, M.J.; Tavares de Almeida, I.; Leandro, P.; Rivera, I.; Vicente, J.B. Functional and structural impact of the most prevalent missense mutations in classic galactosemia. Mol. Genet. Genomic Med. 2014, 2, 484–496. [Google Scholar] [CrossRef]
- Baynes, B.M.; Wang, D.I.C.; Trout, B.L. Role of arginine in the stabilization of proteins against aggregation. Biochemistry 2005, 44, 4919–4925. [Google Scholar] [CrossRef]
- Coelho, A.I.; Trabuco, M.; Silva, M.J.; de Almeida, I.T.; Leandro, P.; Rivera, I.; Vicente, J.B. Arginine functionally improves clinically relevant human galactose-1-phosphate uridylyltransferase (GALT) variants expressed in a prokaryotic model. JIMD Rep. 2015, 23, 1–6. [Google Scholar]
- Haskovic, M.; Derks, B.; van der Ploeg, L.; Trommelen, J.; Nyakayiru, J.; van Loon, L.J.C.; Mackinnon, S.; Yue, W.W.; Peake, R.W.A.; Zha, L.; et al. Arginine does not rescue p.Q188R mutation deleterious effect in classic galactosemia. Orphanet. J. Rare Dis. 2018, 13, 212. [Google Scholar] [CrossRef] [Green Version]
- Marabotti, A.; Facchiano, A.M. Homology modeling studies on human galactose-1-phosphate uridylyltransferase and on its galactosemia-related mutant Q188R provide an explanation of molecular effects of the mutation on homo- and heterodimers. J. Med. Chem. 2005, 48, 773–779. [Google Scholar] [CrossRef]
- Facchiano, A.; Marabotti, A. Analysis of galactosemia-linked mutations of GALT enzyme using a computational biology approach. Protein Eng Des. Sel. 2010, 23, 103–113. [Google Scholar] [CrossRef] [Green Version]
- d’Acierno, A.; Facchiano, A.; Marabotti, A. GALT protein database, a bioinformatics resource for the management and analysis of structural features of a galactosemia-related protein and its mutants. Genom. Proteom. Bioinform. 2009, 7, 71–76. [Google Scholar] [CrossRef] [Green Version]
- d’Acierno, A.; Facchiano, A.; Marabotti, A. GALT protein database: Querying structural and functional features of GALT enzyme. Hum. Mutat. 2014, 35, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Verdino, A.; D’Urso, G.; Tammone, C.; Scafuri, B.; Marabotti, A. Analysis of the structure-function-dynamics relationships of GALT enzyme and of its pathogenic mutant p.Q188R: A molecular dynamics simulation study in different experimental conditions. Molecules 2021, 26, 5941. [Google Scholar]
- Chiappori, F.; Merelli, I.; Milanesi, L.; Marabotti, A. Static and dynamic interactions between GALK enzyme and known inhibitors: Guidelines to design new drugs for galactosemic patients. Eur J. Med. Chem. 2013, 63, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.; Boxer, M.B.; Marabotti, A. GALK inhibitors for classic galactosemia. Future Med. Chem. 2014, 6, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- McCorvie, T.J.; Kopec, J.; Pey, A.L.; Fitzpatrick, F.; Patel, D.; Chalk, R.; Shrestha, L.; Yue, W.W. Molecular basis of classic galactosemia from the structure of human galactose 1-phosphate uridylyltransferase. Hum. Mol. Genet. 2016, 25, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15–Ligand discovery for everyone. J. Chem. Inf. Model 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, J. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Castrignanò, T.; Gioiosa, S.; Flati, T.; Cestari, M.; Picardi, E.; Chiara, M.; Fratelli, M.; Amente, S.; Cirilli, M.; Tangaro, M.A.; et al. ELIXIR-IT HPC@CINECA: High performance computing resources for the bioinformatics community. BMC Bioinform. 2020, 21, 352. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Prot. Chem. 2003, 66, 27–85. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE-AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Abascal, J.L.F.; Vega, C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Berendsen, H.J.C. A leap-frog algorithm for stochastic dynamics. Mol. Simul. 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comp. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Verlet, L. Computer “experiments” on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Binding Energy of the Representative Pose | Number of Poses in the Cluster | Predicted Interactions with Residues |
|---|---|---|---|
| wtGALT + arginine (active site) | −5.25 | 35 | R48, R51, R333, K334, F335, V337, E340, D348 |
| p.Gln188Arg + arginine (active site) | −5.26 | 36 | R48, R51, R188, R333, K334, F335, V337, E340, D348 |
| wtGALT + G1P + arginine (active site) | −5.55 | 87 | R48, N97, D98, G1P |
| p.Gln188Arg + G1P + arginine (active site) | −5.22 | 84 | R48, N97, D98, F99, R188, G1P |
| wtGALT + H2U + arginine (active site) | −6.96 | 72 | R48, R51, G179, S181, K334, F335, V337, E340, Q346, H2U |
| p.Gln188Arg + H2U + arginine (active site) | −6.76 | 90 | R48, R51, N173, G179, R188, K334, F335, V337, G338, Y339, E340, H2U |
| wtGALT + arginine (central cavity) | −4.73 | 26 | Q38, E40, D197, R201 |
| p.Gln188Arg + arginine (central cavity) | −5.00 | 15 | Q38, E40, T248, Y323, M341 |
| wtGALT + G1P + H2U + arginine (central cavity) | −5.19 | 20 | Q38, E40, W41, D197, R201 |
| p.Gln188Arg + G1P + H2U + arginine (central cavity) | −5.09 | 19 | Q38, E40, M341, Q344, A345 |
| wtGALT + Arginine | p.Gln188Arg + Arginine | wtGALT + G1P + Arginine | p.Gln188Arg + G1P + Arginine | wtGALT + H2U + Arginine | p.Gln188Arg + H2U + Arginine |
|---|---|---|---|---|---|
| Average Number of H-Bonds per Timeframe: 26/26 | Average Number of H-Bonds per Timeframe: 25/25 | Average Number of H-Bonds per Timeframe: 26/22 | Average Number of H-Bonds per Timeframe: 24/27 | Average Number of H-Bonds per Timeframe: 29/27 | Average Number of H-Bonds per Timeframe: 23/26 |
| ILE32A-LYS120B | ILE32A-LYS120B | ILE32A-LYS120B | ILE32A-LYS120B | ILE32A-LYS120B | ILE32A-LYS120B |
| ILE32B-LYS120A | ILE32B-LYS120A | ILE32B-LYS120A | ILE32B-LYS120A | ILE32B-LYS120A | ILE32B-LYS120A |
| TYR34A-GLN118B | TYR34A-GLN118B | TYR34A-GLN118B | TYR34A-GLN118B | TYR34A-GLN118B | TYR34A-GLN118B |
| TYR34B-GLN118A | TYR34B-GLN118A | TYR34B-GLN118A | TYR34B-GLN118A | TYR34B-GLN118A | TYR34B-GLN118A |
| ILE198A-ALA343B | ILE198A-ALA343B | ILE198A-ALA343B | ILE198A-ALA343B | ILE198A-ALA343B | ILE198A-ALA343B |
| ILE198B-ALA343A | ILE198B-ALA343A | ILE198B-ALA343A | ILE198B-ALA343A | ILE198B-ALA343A | ILE198B-ALA343A |
| ASP197A-GLN344B | ASP197A-GLN344B | ASP197A-GLN344B | ASP197B-GLN344A | ARG48A-PHE99B | ASP197B-GLN344A |
| ARG48A-PHE99B | ASP197B-GLN344A | HIS301A-LEU342B | HIS301A-LEU342B | HIS301B-LEU342A | TRP41A-ASP197B |
| TRP41A-ASP197B | ARG48A-PHE99B | TRP41A-ASP197B | HIS301B-LEU342A | TRP41A-ASP197B | TRP41B-ASP197A |
| TRP41B-ASP197A | SER45A-ALA101B | TRP41B-ASP197A | TRP41A-ASP197B | TRP41B-ASP197A | ARG228A-ASP113B |
| ARG228A-ASP113B | HIS47A-PRO100B | ARG228B-ASP113A | TRP41B-ASP197A | ARG228A-ASP113B | ARG228B-ASP113A |
| ARG228B-ASP113A | HIS301B-LEU342A | GLN30A-ALA101B | ARG228B-ASP113A | ARG228B-ASP113A | ARG201B-ASP39A |
| ARG204A-ASP39B | TRP41A-ASP197B | GLY338A-SER297B | GLN30A-GLN103B | GLN30A-GLN103B | ARG204A-ASP39B |
| ARG201A-ASP39B | TRP41B-ASP197A | GLY338B-SER297A | GLN30A-ALA122B | GLN30A-ALA122B | GLY338B-SER297A |
| ARG333B-GLU58A | ARG228A-ASP113B | GLN56A-GLU58B | ARG48B-PRO100A | ARG201B-ASP39A | GLY338A-SER297B |
| ARG48B-PHE99A | ARG228B-ASP113A | ARG51A-ASP98B | TYR339A-SER192B | GLN103A-GLN30B | GLN30A-GLN103B |
| GLY338A-SER297B | ARG48B-PRO100A | ARG201A-ASP39B | SER45B-ALA101A | ARG201A-ASP39B | SER45A-ALA101B |
| ALA122A-GLN30B | GLN30A-ALA122B | ARG333A-GLU58B | ARG204A-ASP39B | GLN30B-ALA122A | ARG48A-PHE99B |
| GLN30A-GLN103B | ARG48B-PHE99A | ARG228A-ASP113B | ARG48B-PHE99A | ARG201A-ASP39B | |
| GLY338A-SER297B | SER45B-ALA101A | GLY338A-SER297B | ARG51B-ASP98A | GLN30B-GLN103A | |
| ARG204A-ASP39B | SER45A-ALA101B | HIS114B-GLU220A | GLY338A-SER297B | ||
| GLN169B-ILE32A | GLN103A-GLN30B | ARG201B-GLN38A | SER45A-ALA101B | ||
| TYR339A-SER192B | SER45B-ALA101A | ||||
| GLN30B-ALA122A | ASP197A-GLN344B | ||||
| ARG204A-ASP39B | |||||
| HIS301A-LEU342B | |||||
| ARG228B-ASP113A |
| wtGALT + Arginine | p.Gln188Arg + Arginine | wtGALT + G1P + Arginine | p.Gln188Arg + G1P + Arginine | wtGALT + H2U + Arginine | p.Gln188Arg + H2U + Arginine |
|---|---|---|---|---|---|
| GLU58B-ARG333A | |||||
| ASP113B-ARG228A | ASP113B-ARG228A | ASP113B-ARG228A | ASP113B-ARG228A | ASP113B-ARG228A | |
| ASP113A-ARG228B | ASP113A-ARG228B | ASP113A-ARG228B | ASP113A-ARG228B | ASP113A-ARG228B | |
| ARG204A-ASP39B | ARG204A-ASP39B | ASP39A-ARG201B |
| wtGALT + Arginine | p.Gln188Arg + Arginine | wtGALT+ Ligands + Arginine | p.Gln188Arg + Ligands + Arginine |
|---|---|---|---|
| Average Number of H-Bonds per Timeframe: 26 | Average Number of H-Bonds per Timeframe: 26 | Average Number of H-Bonds per timeframe: 25 | Average Number of H-Bonds per Timeframe: 30 |
| ILE32A-LYS120B | ILE32A-LYS120B | ILE32A-LYS120B | ILE32A-LYS120B |
| ILE32B-LYS120A | ILE32B-LYS120A | ILE32B-LYS120A | ILE32B-LYS120A |
| TYR34A-GLN118B | TYR34A-GLN118B | TYR34A-GLN118B | TYR34A-GLN118B |
| TYR34B-GLN118A | TYR34B-GLN118A | TYR34B-GLN118A | TYR34B-GLN118A |
| ILE198A-ALA343B | ILE198B-ALA343A | ILE198B-ALA343A | ILE198B-ALA343A |
| ASP197A-GLN344B | ASP197B-GLN344A | ILE198A-ALA343B | ILE198A-ALA343B |
| ARG48A-PHE99B | HIS301A-LEU342B | ASP197B-GLN344A | HIS301B-LEU342A |
| HIS301A-LEU342B | SER297A-VAL337B | ARG48A-PHE99B | TRP41B-ASP197A |
| HIS114B-GLU220A | SER45A-ALA101B | HIS301A-LEU342B | ARG201A-ASP39B |
| HIS47A-PRO100B | TRP41A-ASP197B | TRP41A-ASP197B | TRP41A-ASP197B |
| GLY338A-SER297B | GLY338A-ASN173B | TRP41B-ASP197A | ARG228B-ASP113A |
| GLN30A-ALA101B | GLN30A-ALA122B | ARG201A-ASP39B | GLN30A-GLN103B |
| GLN103A-GLN30B | ARG48A-PRO100B | ARG228B-ASP113A | GLN30A-ALA122B |
| GLN30A-GLN103B | ARG204A-ASP39B | SER45B-ALA101A | HIS47A-PHE99B |
| ARG333A-GLN58B | ARG228B-ASP113A | TRP167B-TYR339A | ARG204A-ASP39B |
| ARG201A-ASP39B | ARG201A-GLU40B | ARG228A-ASP113B | SER297A-VAL337B |
| ARG228B-ASP113A | GLN103A-GLN30B | GLN346A-ALA101B | |
| TRP41A-ASP197B | GLN30A-GLN103B | GLN30B-ALA101A | |
| TRP41B-ASP197A | ARG228A-ASP113B | ARG48B-PHE99A | |
| GLY338B-SER297A | ARG201B-ASP39A | ARG51B-ASP98A | |
| ARG48B-PHE99A | ARG201B-ASP39A | ||
| GLN224A-HIS114B | ARG228A-ASP113B | ||
| ARG333B-GLU58A |
| wtGALT + Arginine | p.Gln188Arg + Arginine | wtGALT + Ligands + Arginine | p.Gln188Arg + Ligands + Arginine |
|---|---|---|---|
| GLU58B-ARG333A | |||
| ASP113B-ARG228A | ASP113B-ARG228A | ASP113B-ARG228A | |
| ASP113A-ARG228B | ASP113A-ARG228B | ASP113A-ARG228B | |
| ASP39B-ARG201A | ASP39B-ARG201A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdino, A.; D’Urso, G.; Tammone, C.; Scafuri, B.; Catapano, L.; Marabotti, A. Simulation of the Interactions of Arginine with Wild-Type GALT Enzyme and the Classic Galactosemia-Related Mutant p.Q188R by a Computational Approach. Molecules 2021, 26, 6061. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26196061
Verdino A, D’Urso G, Tammone C, Scafuri B, Catapano L, Marabotti A. Simulation of the Interactions of Arginine with Wild-Type GALT Enzyme and the Classic Galactosemia-Related Mutant p.Q188R by a Computational Approach. Molecules. 2021; 26(19):6061. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26196061
Chicago/Turabian StyleVerdino, Anna, Gaetano D’Urso, Carmen Tammone, Bernardina Scafuri, Lucrezia Catapano, and Anna Marabotti. 2021. "Simulation of the Interactions of Arginine with Wild-Type GALT Enzyme and the Classic Galactosemia-Related Mutant p.Q188R by a Computational Approach" Molecules 26, no. 19: 6061. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26196061