
Comparing Dimerization Free Energies and Binding Modes of Small Aromatic Molecules with Different Force Fields


Abstract
: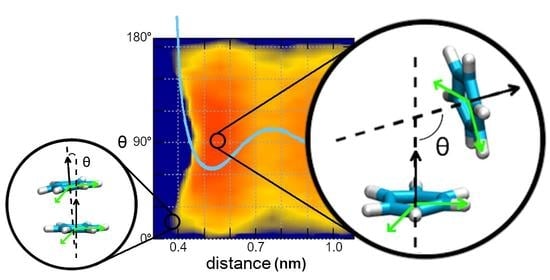
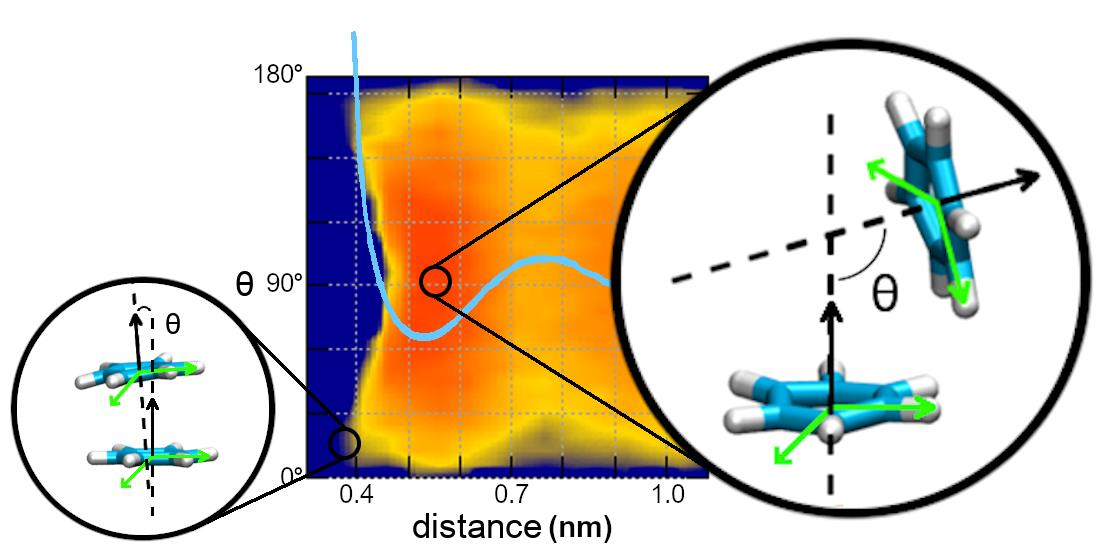{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
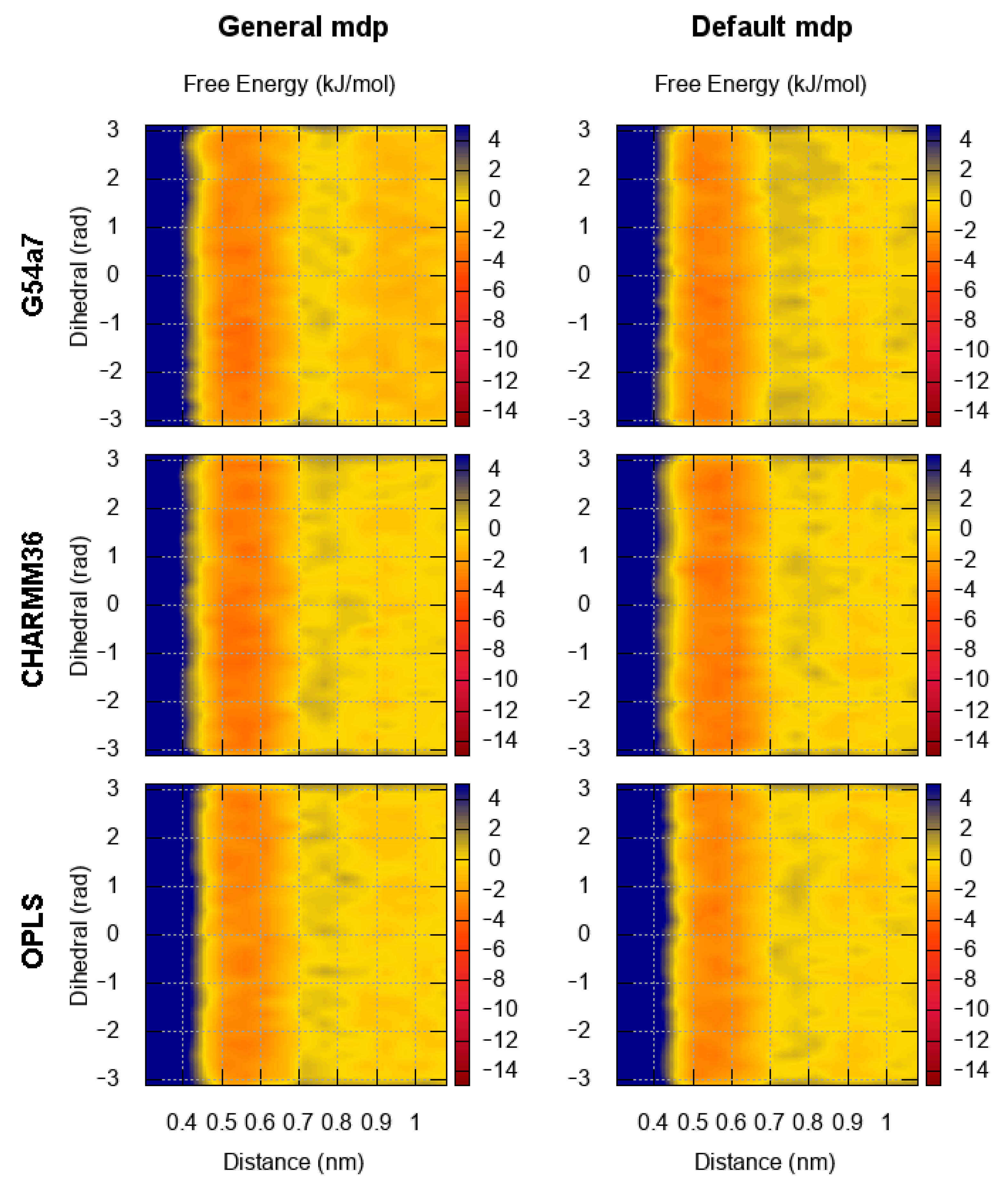
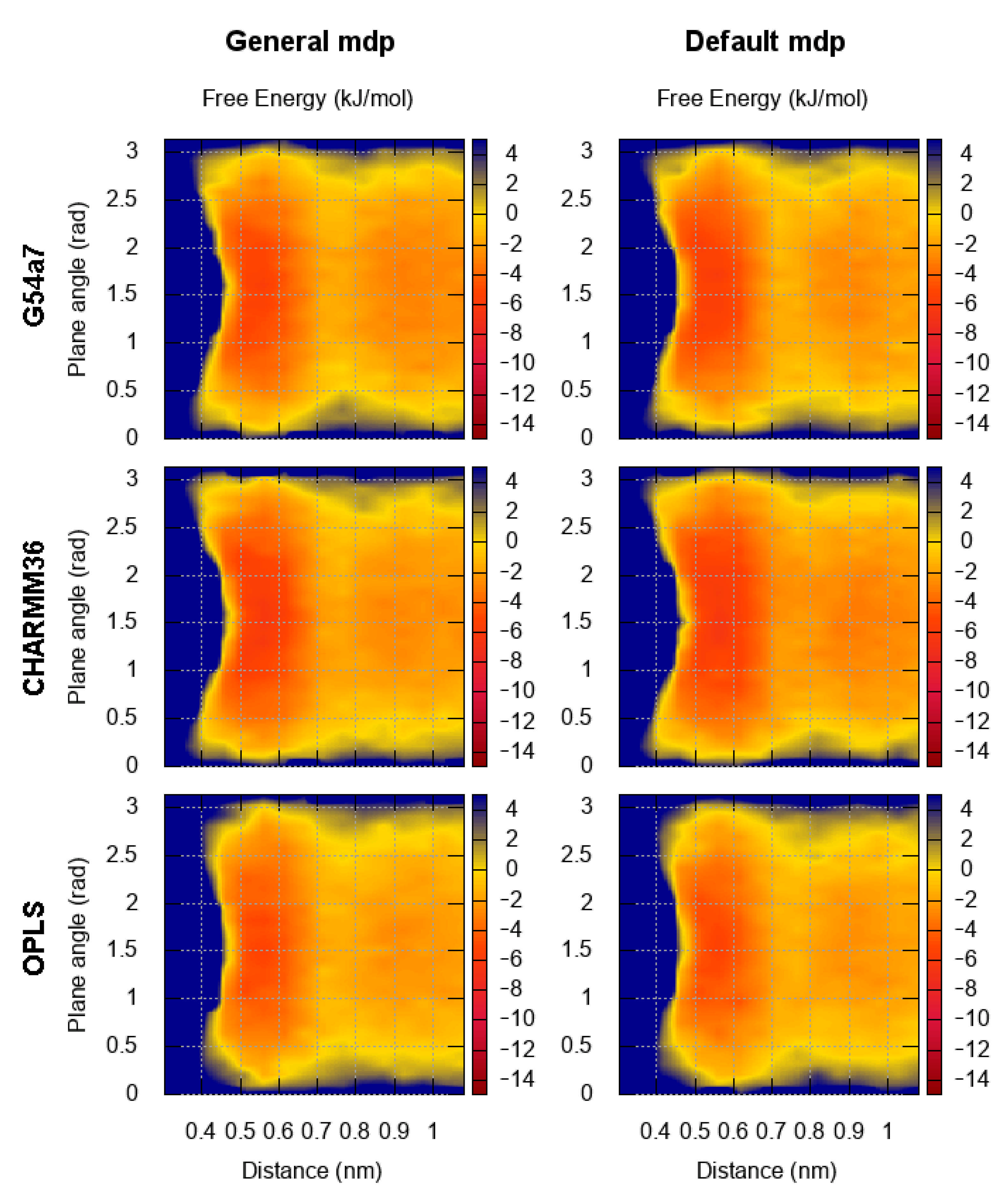
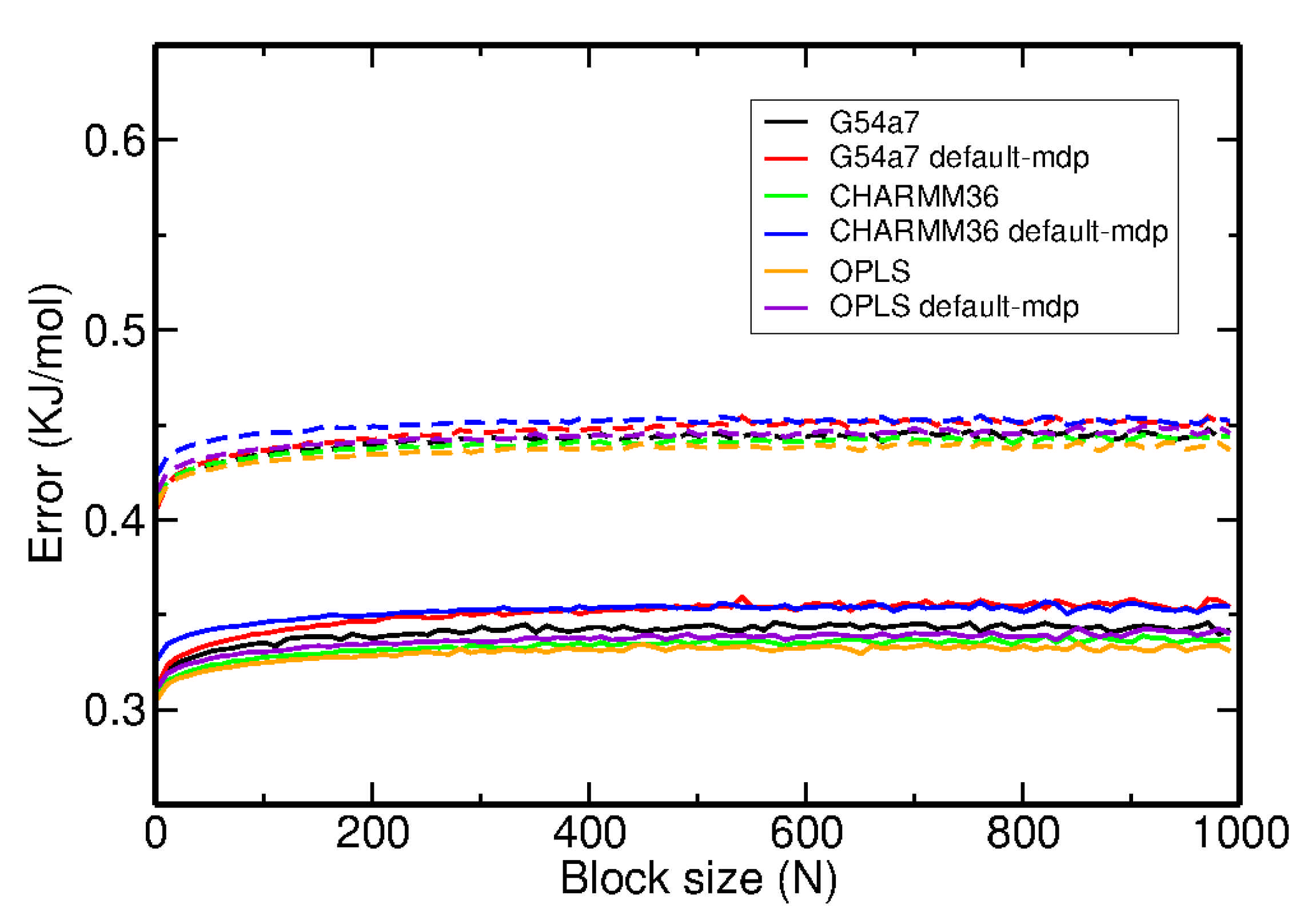
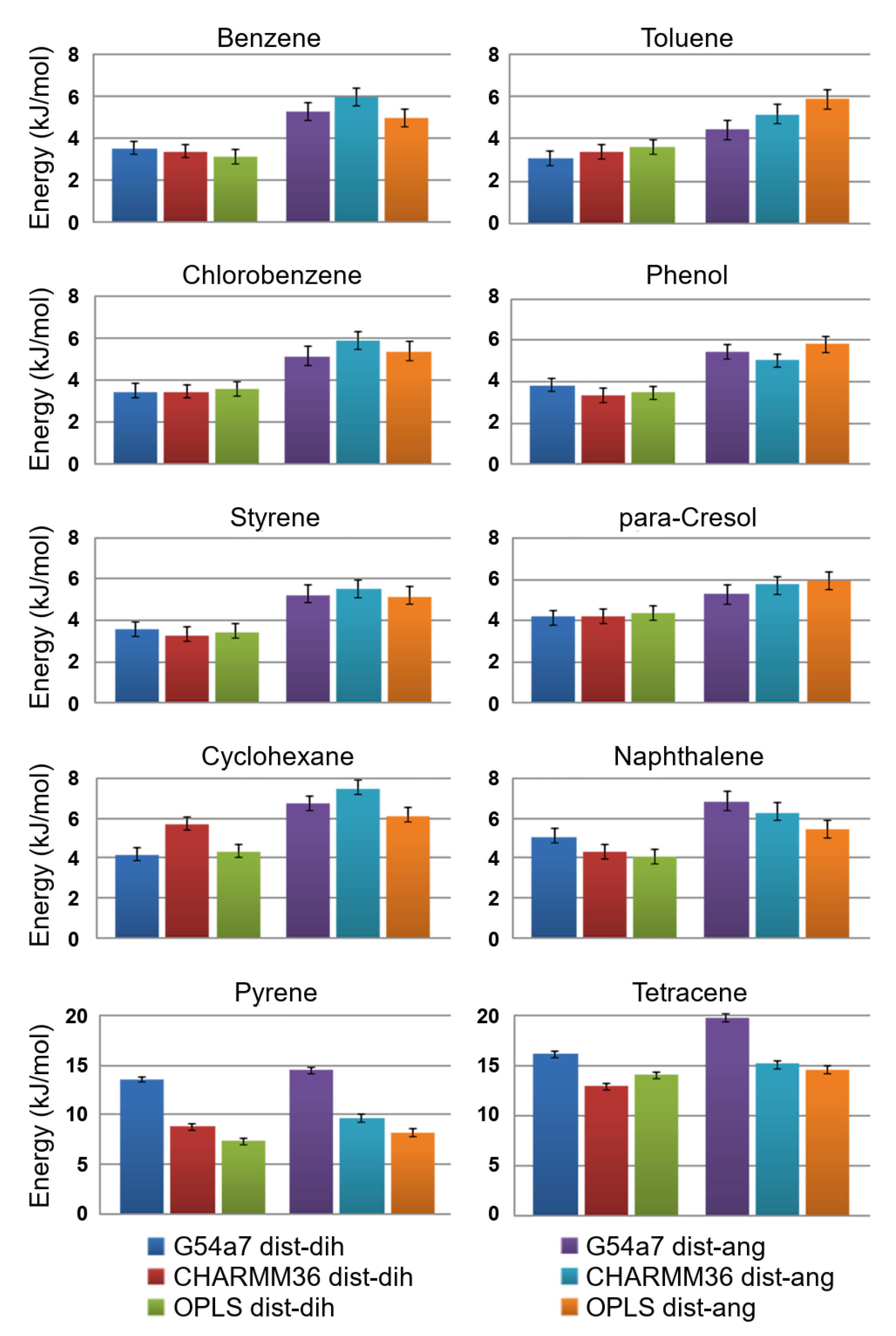
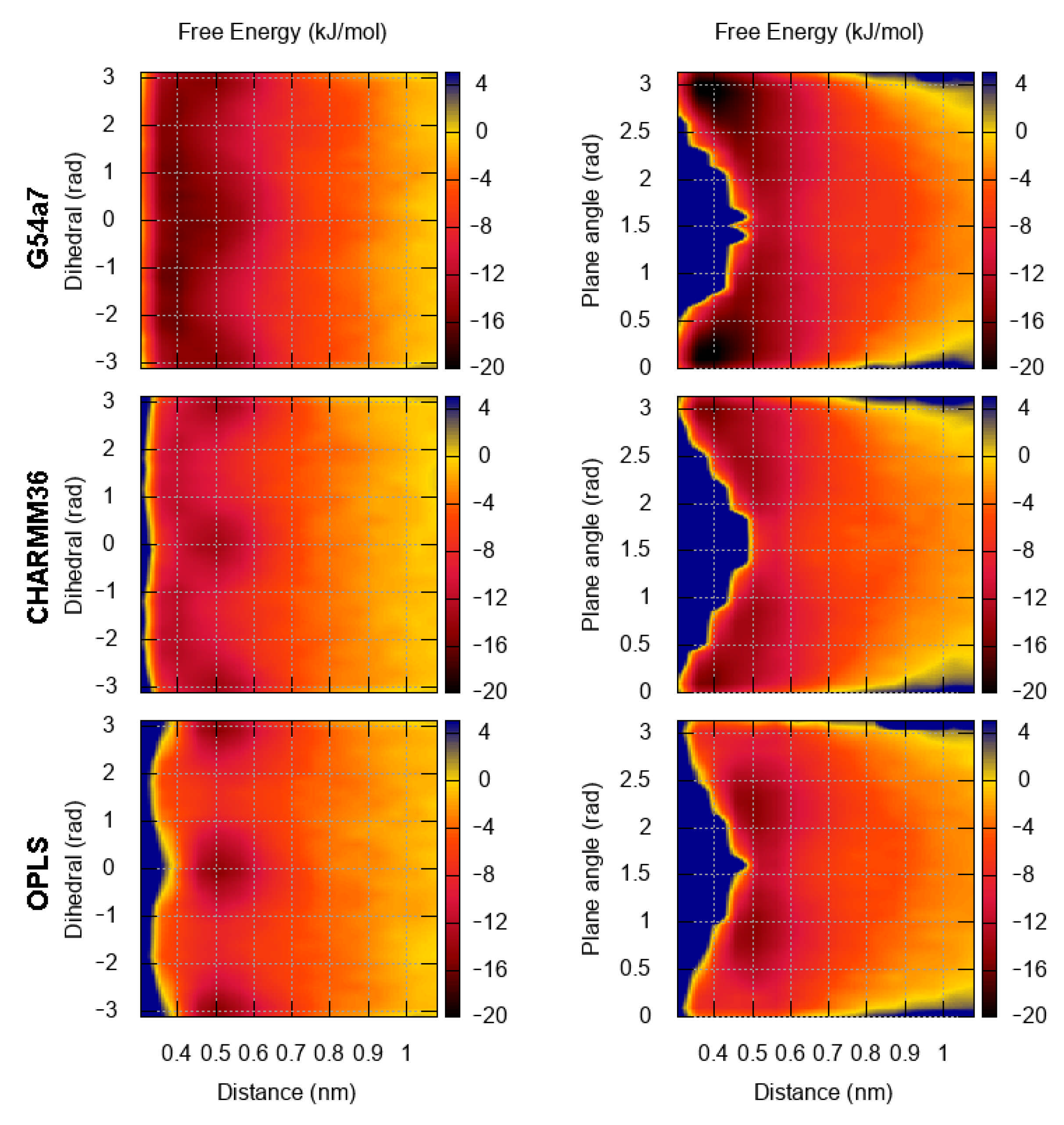
2.1. Comparison of Parameter Setups
2.2. Comparison of FES Profiles
3. Discussion
4. Methods
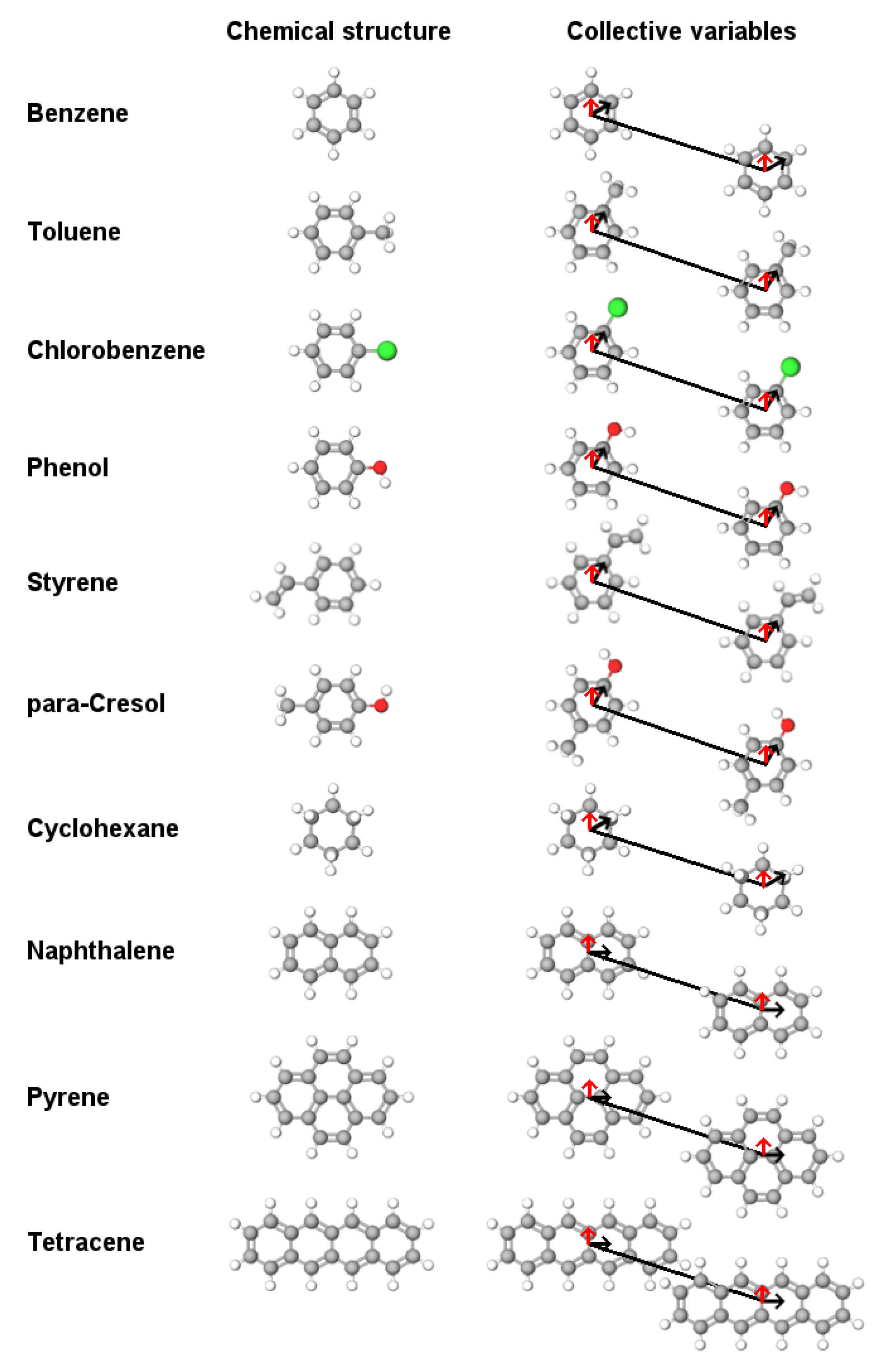
4.1. Molecule Parameters
4.2. MD Simulations
4.3. Metadynamics
4.4. Entropic Component of Free Energy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Polêto, M.D.; Rusu, V.H.; Grisci, B.I.; Dorn, M.; Lins, R.D.; Verli, H. Aromatic Rings Commonly Used in Medicinal Chemistry: Force Fields Comparison and Interactions With Water Toward the Design of New Chemical Entities. Front. Pharmacol. 2018, 9, 395. [Google Scholar] [CrossRef] [Green Version]
- Fleming, S.; Ulijn, R.V. Design of nanostructures based on aromatic peptide amphiphiles. Chem. Soc. Rev. 2014, 43, 8150–8177. [Google Scholar] [CrossRef] [PubMed]
- Diercks, T.; Coles, M.; Kessler, H. Applications of NMR in drug discovery. Curr. Opin. Chem. Biol. 2001, 5, 285–291. [Google Scholar] [CrossRef]
- Aguirre, C.; Cala, O.; Krimm, I. Overview of Probing Protein-Ligand Interactions Using NMR. Curr. Protoc. Protein Sci. 2015, 81, 17.18.1–17.18.24. [Google Scholar] [CrossRef] [PubMed]
- Everett, J.R. Drug Discovery and Development: The Role of NMR. In eMagRes; American Cancer Society: Atlanta, GA, USA, 2015; pp. 137–150. [Google Scholar] [CrossRef]
- Leavitt, S.; Freire, E. Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 2001, 11, 560–566. [Google Scholar] [CrossRef]
- Damian, L. Isothermal Titration Calorimetry for Studying Protein-Ligand Interactions. In Protein-Ligand Interactions: Methods and Applications; Williams, M.A., Daviter, T., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 103–118. [Google Scholar] [CrossRef]
- Cubberley, M.S.; Iverson, B.L. (1)H NMR investigation of solvent effects in aromatic stacking interactions. J. Am. Chem. Soc. 2001, 123, 7560–7563. [Google Scholar] [CrossRef] [PubMed]
- Frederix, P.W.J.M.; Patmanidis, I.; Marrink, S.J. Molecular simulations of self-assembling bio-inspired supramolecular systems and their connection to experiments. Chem. Soc. Rev. 2018, 47, 3470–3489. [Google Scholar] [CrossRef] [Green Version]
- Chipot, C.; Jaffe, R.; Maigret, B.; Pearlman, D.A.; Kollman, P.A. Benzene Dimer: A Good Model for π-π Interactions in Proteins? A Comparison between the Benzene and the Toluene Dimers in the Gas Phase and in an Aqueous Solution. J. Am. Chem. Soc. 1996, 118, 11217–11224. [Google Scholar] [CrossRef]
- Hobza, P.; Selzle, H.L.; Schlag, E.W. Potential Energy Surface for the Benzene Dimer. Results of ab Initio CCSD(T) Calculations Show Two Nearly Isoenergetic Structures: T-Shaped and Parallel-Displaced. J. Phys. Chem. 1996, 100, 18790–18794. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, V.; Bonomi, M.; Parrinello, M. Funnel metadynamics as accurate binding free-energy method. Proc. Natl. Acad. Sci. USA 2013, 110, 6358–6363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvalaglio, M.; Giberti, F.; Parrinello, M. 1,3,5-Tris(4-bromophenyl)benzene prenucleation clusters from metadynamics. Acta Crystallogr. Sect. C 2014, 70, 132–136. [Google Scholar] [CrossRef]
- Bochicchio, D.; Salvalaglio, M.; Pavan, G.M. Into the Dynamics of a Supramolecular Polymer at Submolecular Resolution. Nat. Commun. 2017, 8, 147. [Google Scholar] [CrossRef]
- Capelli, R.; Bochicchio, A.; Piccini, G.M.; Casasnovas, R.; Carloni, P.; Parrinello, M. Chasing the Full Free Energy Landscape of Neuroreceptor/Ligand Unbinding by Metadynamics Simulations. J. Chem. Theory Comput. 2019, 15, 3354–3361. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; van Gunsteren, W.F. The GROMOS Biomolecular Simulation Program Package. J. Phys. Chem. A 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Guvench, O.; MacKerell, A.D., Jr. Comparison of protein force fields for molecular dynamics simulations. Methods Mol. Biol. 2011, 443, 2240–2252. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, J.A.; Schall, J.D.; Maskey, S.; Mikulski, P.T.; Knippenberg, M.T.; Morrow, B.H. Review of force fields and intermolecular potentials used in atomistic computational materials research. Appl. Phys. Rev. 2018, 5, 031104. [Google Scholar] [CrossRef]
- Ponder, J.W.; Case, D.A. Force Fields for Protein Simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar] [CrossRef] [PubMed]
- González, M.A. Force fields and molecular dynamics simulations. JDN 2011, 12, 169–200. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [Green Version]
- Jing, Z.; Liu, C.; Cheng, S.Y.; Qi, R.; Walker, B.D.; Piquemal, J.; Ren, P. Polarizable Force Fields for Biomolecular Simulations: Recent Advances and Applications. Annu. Rev. Biophys. 2019, 48, 371–394. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, W. QM/MM Methods for Biomolecular Systems. Angew. Chem. Int. Ed. 2009, 48, 1198–1229. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Thallmair, S.; Conflitti, P.; Ramírez-Palacios, C.; Alessandri, R.; Raniolo, S.; Limongelli, V.; Marrink, S.J. Protein–ligand binding with the coarse-grained Martini model. Nat. Commun. 2020, 11, 3714. [Google Scholar] [CrossRef]
- Alessandri, R.; Grünewald, F.; Marrink, S.J. The Martini Model in Materials Science. Adv. Mater. 2021, 33, 2008635. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, E.M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843. [Google Scholar] [CrossRef]
- De Jong, D.H.; Periole, X.; Marrink, S.J. Dimerization of Amino Acid Side Chains: Lessons from the Comparison of Different Force Fields. J. Chem. Theory Comput. 2012, 8, 1003–1014. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Tian, S.X. A Comparative Study for Molecular Dynamics Simulations of Liquid Benzene. J. Chem. Theory Comput. 2008, 7, 63–88. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Severance, D.L. Aromatic-aromatic interactions: Free energy profiles for the benzene dimer in water, chloroform, and liquid benzene. J. Am. Chem. Soc. 1990, 112, 4768–4774. [Google Scholar] [CrossRef]
- Lee, E.C.; Kim, D.; Jurečka, P.; Tarakeshwar, P.; Hobza, P.; Kim, K.S. Understanding of Assembly Phenomena by Aromatic-Aromatic Interactions: Benzene Dimer and the Substituted Systems. J. Phys. Chem. A 2007, 111, 3446–3457. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Slipchenko, L.V.; Gordon, M.S. Modeling π–π Interactions with the Effective Fragment Potential Method: The Benzene Dimer and Substituents. J. Phys. Chem. A 2008, 112, 5286–5294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udier–Blagović, M.; Morales De Tirado, P.; Pearlman, S.A.; Jorgensen, W.L. Accuracy of free energies of hydration using CM1 and CM3 atomic charges. J. Comput. Chem. 2004, 25, 1322–1332. [Google Scholar] [CrossRef]
- Kashefolgheta, S.; Oliveira, M.P.; Rieder, S.R.; Horta, B.A.C.; Acree, W.E.; Hünenberger, P.H. Evaluating Classical Force Fields against Experimental Cross-Solvation Free Energies. J. Chem. Theory Comput. 2020, 16, 7556–7580. [Google Scholar] [CrossRef]
- Fröhlking, T.; Bernetti, M.; Calonaci, N.; Bussi, G. Toward empirical force fields that match experimental observables. J. Chem. Phys. 2020, 152, 230902. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [Green Version]
- Dodda, L.S.; Vilseck, J.Z.; Tirado-Rives, J.; Jorgensen, W.L. 1.14*CM1A-LBCC: Localized Bond-Charge Corrected CM1A Charges for Condensed-Phase Simulations. J. Phys. Chem. B 2017, 121, 3864–3870. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling Through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Di Nola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Tironi, I.G.; Sperb, R.; Smith, P.E.; van Gunsteren, W.F. A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys. 1995, 102, 5451–5459. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces, Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry, Jerusalem, Israel, 13–16 April 1981; Springer: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, I.S.; Lin, F.Y.; Vanommeslaeghe, K.; Lemkul, J.A.; Armacost, K.A.; Brooks, C.L.; MacKerell, A.D. Parametrization of halogen bonds in the CHARMM general force field: Improved treatment of ligand-protein interactions. Bioorg. Med. Chem. 2016, 24, 4812–4825. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Schyman, P. Treatment of Halogen Bonding in the OPLS-AA Force Field: Application to Potent Anti-HIV Agents. J. Chem. Theory Comput. 2012, 8, 3895–3901. [Google Scholar] [CrossRef] [Green Version]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [Green Version]
- Huber, T.; Torda, A.E.; van Gunsteren, W.F. Local elevation: A method for improving the searching properties of molecular dynamics simulation. J. Comput. Aided Mol. Des. 1994, 8, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Grubmüller, H. Predicting slow structural transitions in macromolecular systems: Conformational flooding. Phys. Rev. E 1995, 52, 2893–2906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branduardi, D.; Bussi, G.; Parrinello, M. Metadynamics with Adaptive Gaussians. J. Chem. Theory Comput. 2012, 8, 2247–2254. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patmanidis, I.; Alessandri, R.; de Vries, A.H.; Marrink, S.J. Comparing Dimerization Free Energies and Binding Modes of Small Aromatic Molecules with Different Force Fields. Molecules 2021, 26, 6069. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26196069
Patmanidis I, Alessandri R, de Vries AH, Marrink SJ. Comparing Dimerization Free Energies and Binding Modes of Small Aromatic Molecules with Different Force Fields. Molecules. 2021; 26(19):6069. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26196069
Chicago/Turabian StylePatmanidis, Ilias, Riccardo Alessandri, Alex H. de Vries, and Siewert J. Marrink. 2021. "Comparing Dimerization Free Energies and Binding Modes of Small Aromatic Molecules with Different Force Fields" Molecules 26, no. 19: 6069. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26196069