
Molecular Design in Practice: A Review of Selected Projects in a French Research Institute That Illustrates the Link between Chemical Biology and Medicinal Chemistry


 , , and
, , and
Abstract
:1. Introduction
2. Target Validation and Engagement
2.1. Chemical Biology Approaches to Reprogram the Transcriptome of Bacteria and Select Drug Candidates
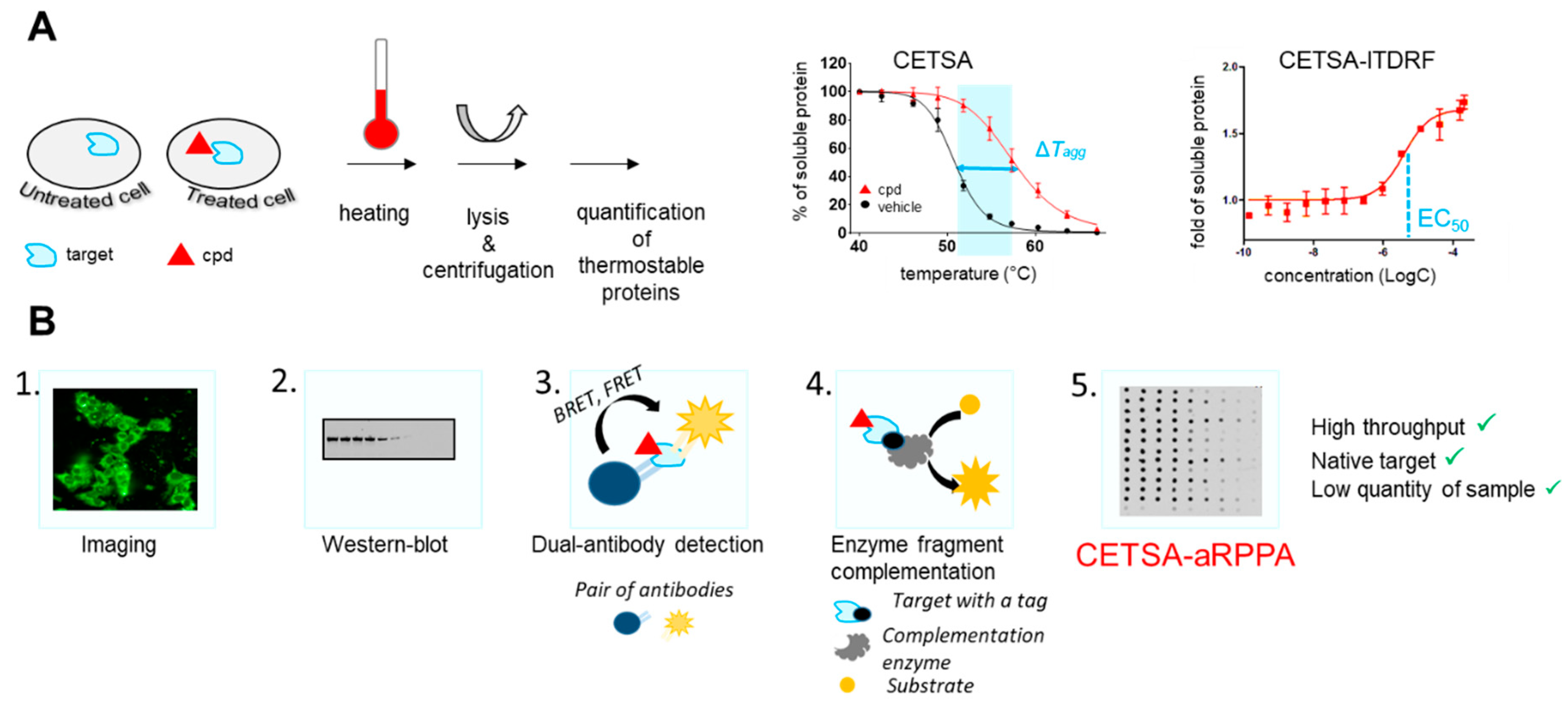
2.2. Chemical Biology Strategies to Quantify Target Engagement
3. Chemical Biology Strategies to Identify New Hits
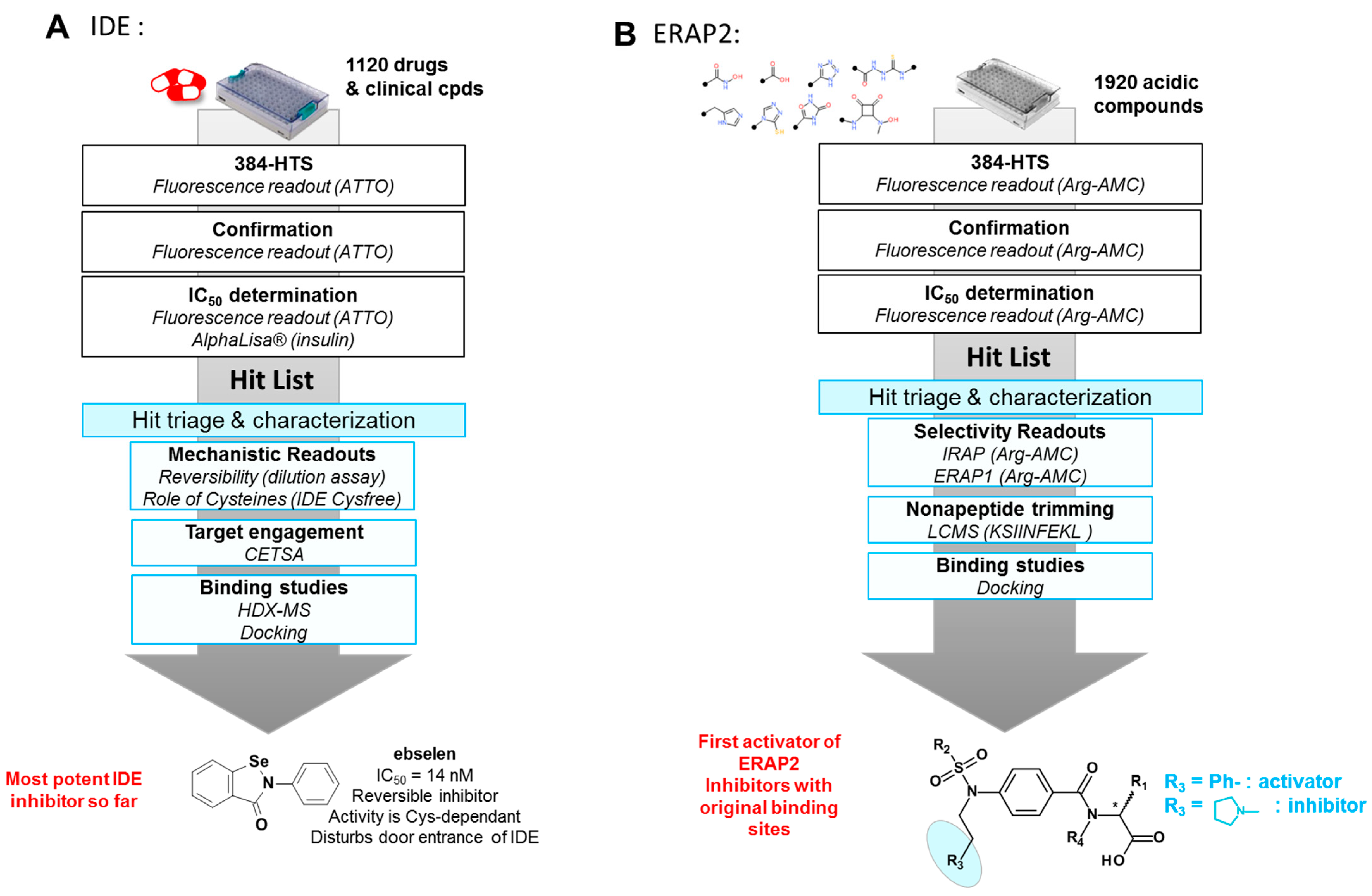
3.1. Screening of Focused Libraries or Clinical Compounds Libraries on Metalloproteases
3.2. Kinetic Target Guided Synthesis
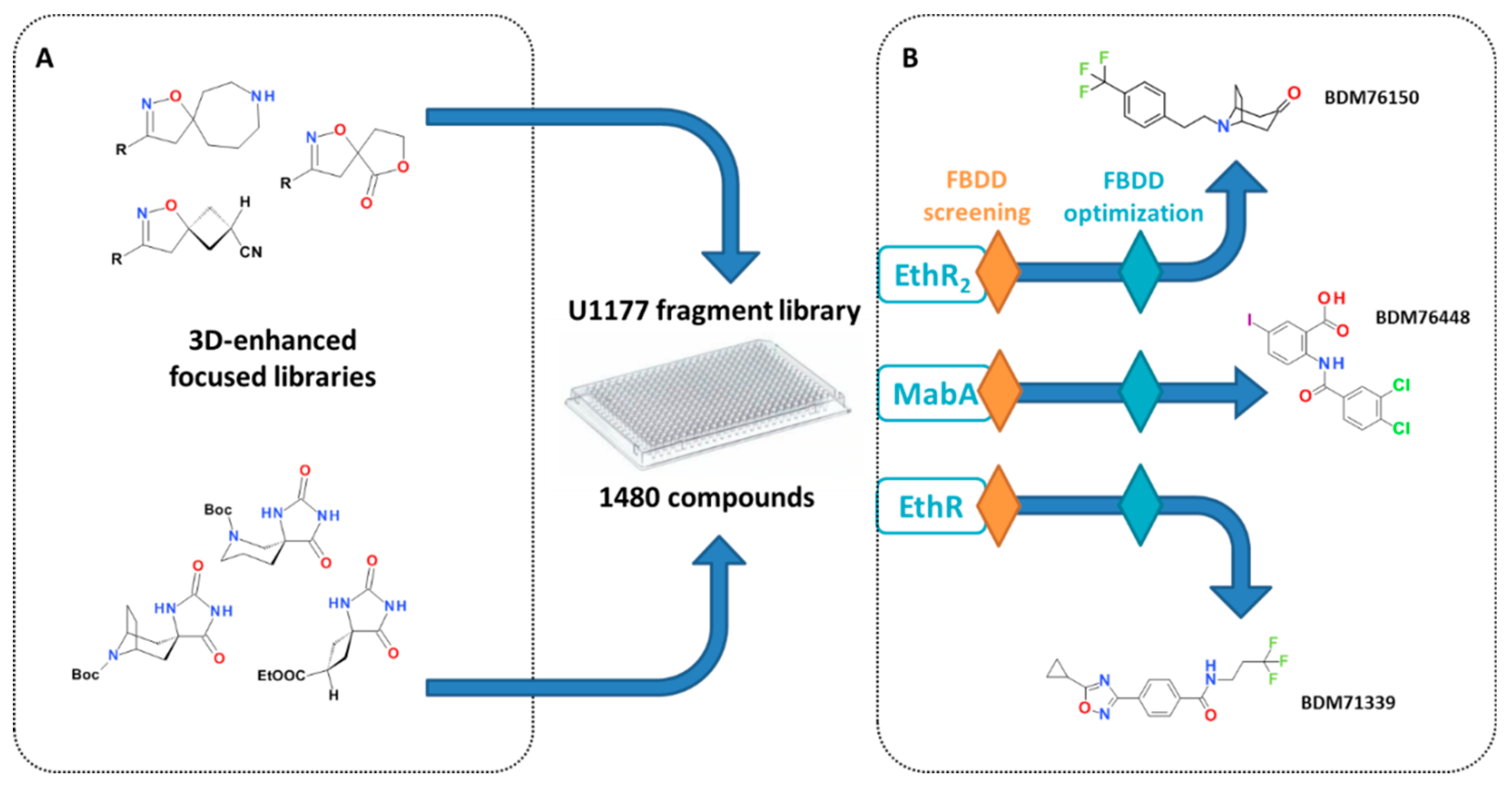
3.3. Fragment-Based Drug Discovery
3.3.1. Discovery of Fragments Targeting EthR and EthR2
3.3.2. Discovery of Fragments as Inhibitors of MabA
4. Chemical Biology Approaches to ADME Properties Modulation
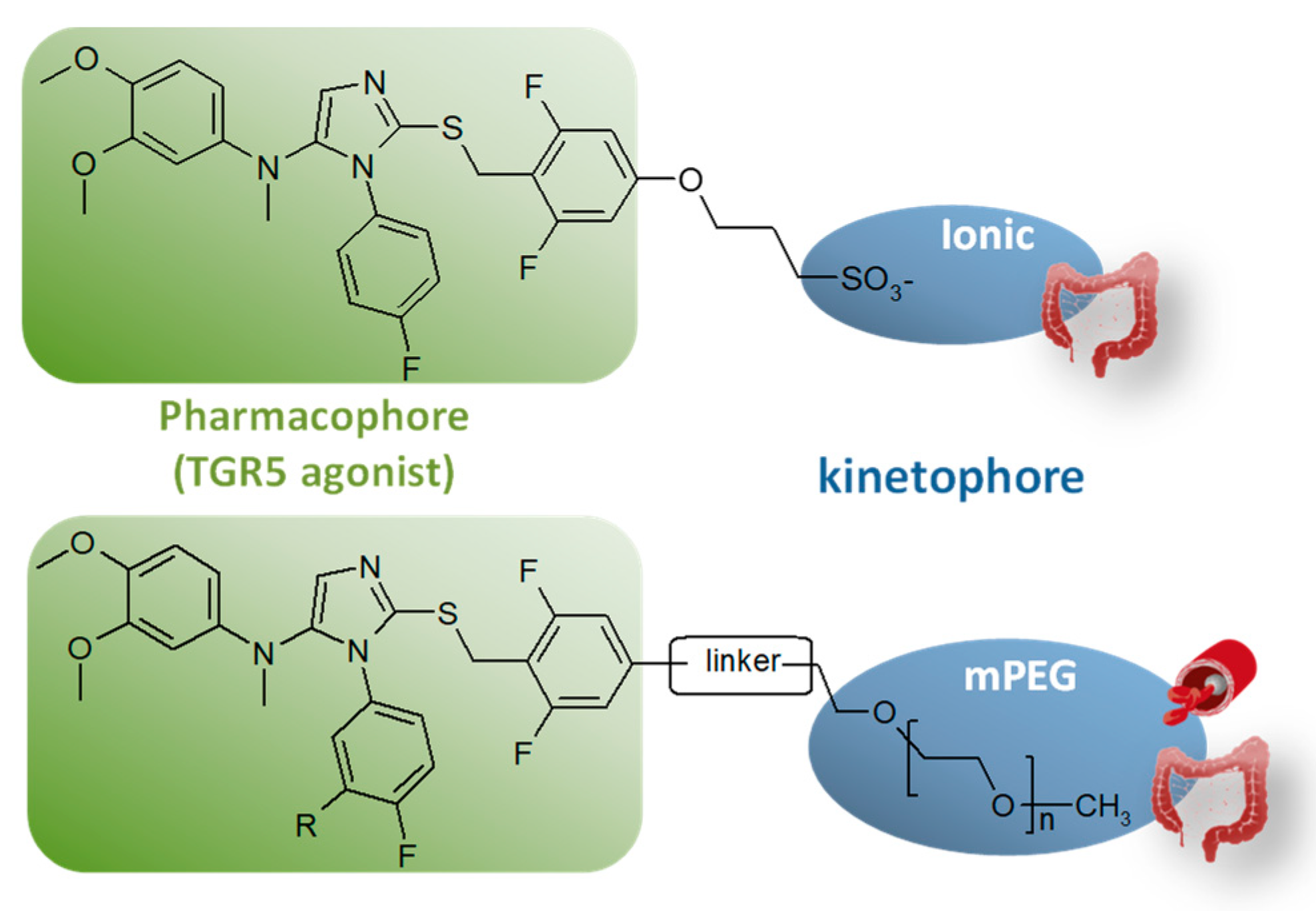
4.1. Controlling Target Engagement by Innovative Molecular Engineering
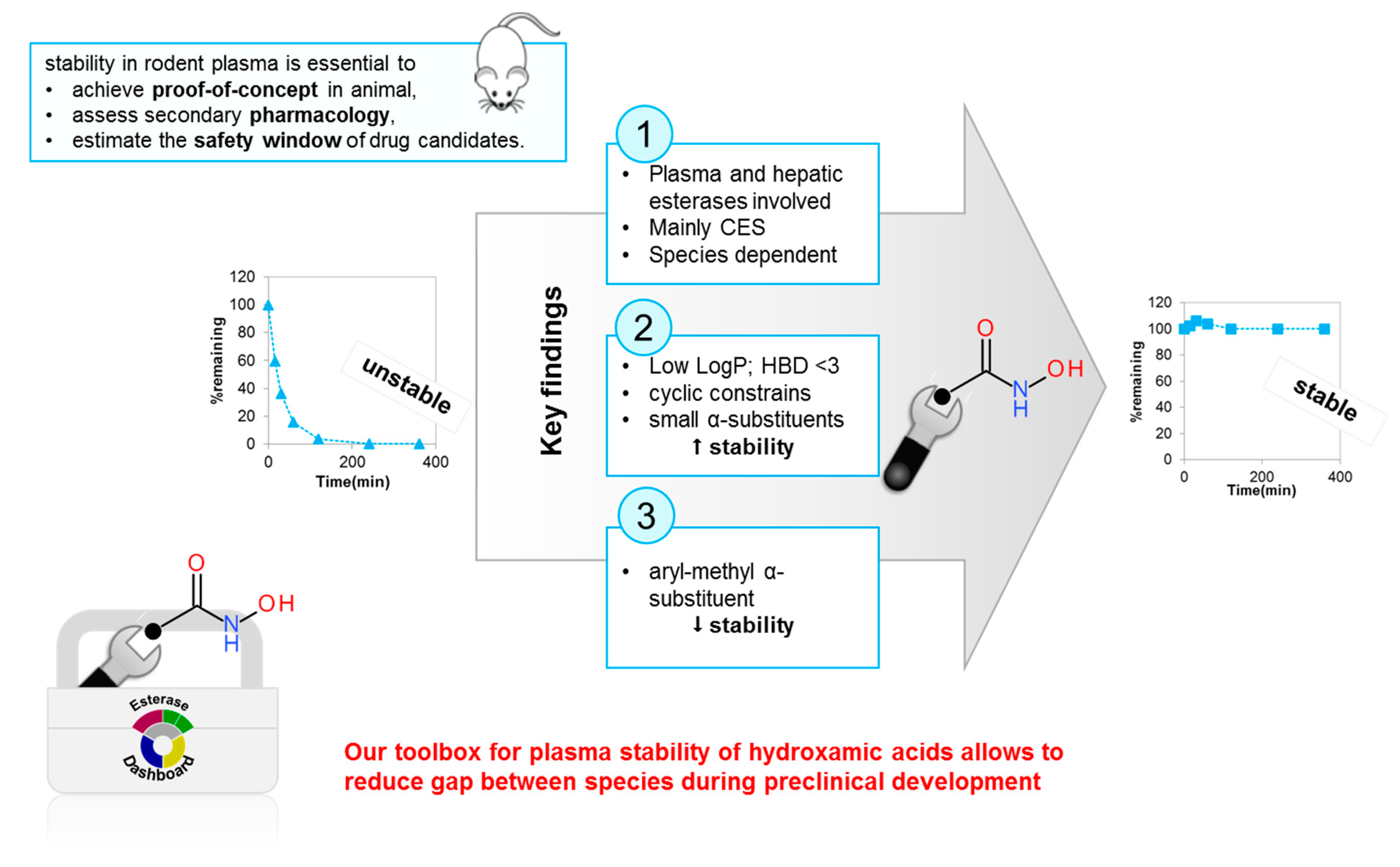
4.2. Rationalizing and Optimizing Plasma Stability
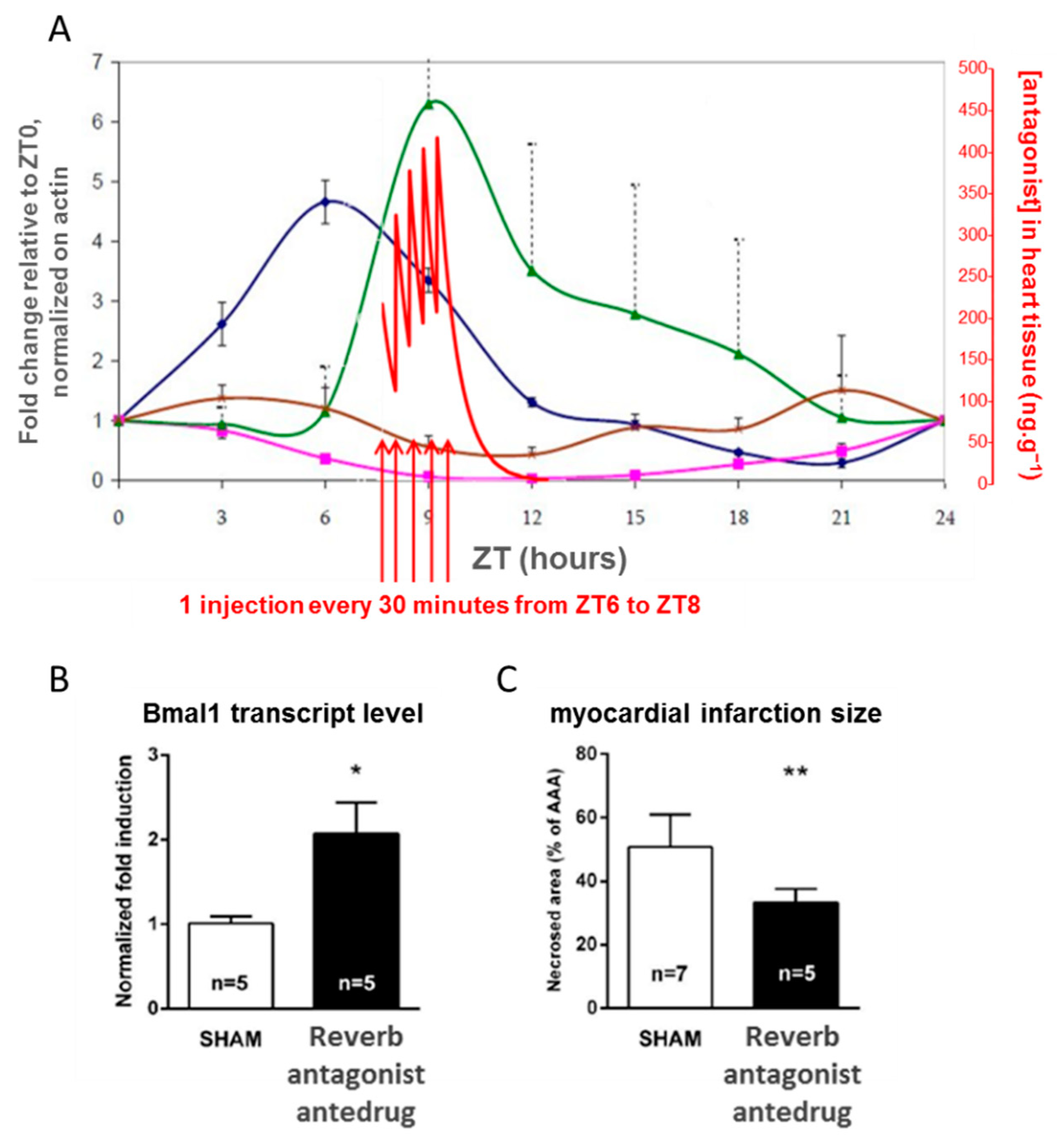
4.3. Controlling the Cell Clock In Vivo with an Antedrug to Understand a Clinical Observation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laborde, J.; Deraeve, C.; Bernardes-Génisson, V. Update of Antitubercular Prodrugs from a Molecular Perspective: Mechanisms of Action, Bioactivation Pathways, and Associated Resistance. ChemMedChem 2017, 12, 1657–1676. [Google Scholar] [CrossRef] [PubMed]
- Morlock, G.P.; Metchock, B.; Sikes, D.; Crawford, J.T.; Cooksey, R.C. ethA, inhA, and katG Loci of Ethionamide-Resistant Clinical Mycobacterium tuberculosis Isolates. Antimicrob. Agents Chemother. 2003, 47, 3799–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baulard, A.; Betts, J.; Engohang-Ndong, J.; Quan, S.; McAdam, R.A.; Brennan, P.J.; Locht, C.; Besra, G. Activation of the Pro-drug Ethionamide Is Regulated in Mycobacteria. J. Biol. Chem. 2000, 275, 28326–28331. [Google Scholar] [CrossRef] [Green Version]
- Willand, N.; Dirié, B.; Carette, X.; Bifani, P.; Singhal, A.; Desroses, M.; Leroux, F.; Willery, E.; Mathys, V.; Deprez-Poulain, R.; et al. Synthetic EthR inhibitors boost antituberculous activity of ethionamide. Nat. Med. 2009, 15, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, B.; Flipo, M.; Blondiaux, N.; Crauste, C.; Malaquin, S.; Leroux, F.; Piveteau, C.; Villeret, V.; Brodin, P.; Villoutreix, B.O.; et al. Ligand Efficiency Driven Design of New Inhibitors of Mycobacterium tuberculosis Transcriptional Repressor EthR Using Fragment Growing, Merging, and Linking Approaches. J. Med. Chem. 2014, 57, 4876–4888. [Google Scholar] [CrossRef] [Green Version]
- Flipo, M.; Desroses, M.; Lecat-Guillet, N.; Dirié, B.; Carette, X.; Leroux, F.; Piveteau, C.; Demirkaya, F.; Lens, Z.; Rucktooa, P.; et al. Ethionamide Boosters: Synthesis, Biological Activity, and Structure−Activity Relationships of a Series of 1,2,4-Oxadiazole EthR Inhibitors. J. Med. Chem. 2011, 54, 2994–3010. [Google Scholar] [CrossRef]
- Flipo, M.; Desroses, M.; Lecat-Guillet, N.; Villemagne, B.; Blondiaux, N.; Leroux, F.; Piveteau, C.; Mathys, V.; Flament, M.-P.; Siepmann, J.; et al. Ethionamide Boosters. 2. Combining Bioisosteric Replacement and Structure-Based Drug Design To Solve Pharmacokinetic Issues in a Series of Potent 1,2,4-Oxadiazole EthR Inhibitors. J. Med. Chem. 2011, 55, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Bernard, C.; Willand, N.; Déprez, B.; Jarlier, V.; Baulard, A.; Veziris, N. EthR inhibitor BDM41906 boosts the in vivo antituberculous activity of ethionamide in a murine model. In Proceedings of the 22th European Congress of Clinical Microbiology and Infectious Diseases, London, UK, 31 March – 3 April 2012. [Google Scholar]
- Villemagne, B.; Machelart, A.; Tran, N.C.; Flipo, M.; Moune, M.; Leroux, F.; Piveteau, C.; Wohlkönig, A.; Wintjens, R.; Li, X.; et al. Fragment-Based Optimized EthR Inhibitors with in Vivo Ethionamide Boosting Activity. ACS Infect. Dis. 2020, 6, 366–378. [Google Scholar] [CrossRef]
- Blondiaux, N.; Moune, M.; Desroses, M.; Frita, R.; Flipo, M.; Mathys, V.; Soetaert, K.; Kiass, M.; Delorme, V.; Djaout, K.; et al. Reversion of antibiotic resistance inMycobacterium tuberculosisby spiroisoxazoline SMARt-420. Science 2017, 355, 1206–1211. [Google Scholar] [CrossRef]
- Comess, K.M.; McLoughlin, S.M.; Oyer, J.A.; Richardson, P.L.; Stöckmann, H.; Vasudevan, A.; Warder, S.E. Emerging Approaches for the Identification of Protein Targets of Small Molecules—A Practitioners’ Perspective. J. Med. Chem. 2018, 61, 8504–8535. [Google Scholar] [CrossRef]
- Lundgren, S. Focusing on Relevance: CETSA-Guided Medicinal Chemistry and Lead Generation. ACS Med. Chem. Lett. 2019, 10, 690–693. [Google Scholar] [CrossRef] [Green Version]
- Henderson, M.J.; Holbert, M.A.; Simeonov, A.; Kallal, L.A. High-Throughput Cellular Thermal Shift Assays in Research and Drug Discovery. SLAS Discov. Adv. Life Sci. R D 2019, 25, 137–147. [Google Scholar] [CrossRef]
- Shaw, J.; Leveridge, M.; Norling, C.; Karén, J.; Molina, D.M.; O’Neill, D.; Dowling, J.E.; Davey, P.; Cowan, S.; Dabrowski, M.; et al. Determining direct binders of the Androgen Receptor using a high-throughput Cellular Thermal Shift Assay. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Martinez, N.J.; Asawa, R.; Cyr, M.G.; Zakharov, A.; Urban, D.J.; Roth, J.S.; Wallgren, E.; Klumpp-Thomas, C.; Coussens, N.P.; Rai, G.; et al. A widely-applicable high-throughput cellular thermal shift assay (CETSA) using split Nano Luciferase. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Herledan, A.; Andres, M.; Lejeune-Dodge, A.; Leroux, F.; Biela, A.; Piveteau, C.; Warenghem, S.; Couturier, C.; Deprez, B.; Deprez-Poulain, R. Drug Target Engagement Using Coupled Cellular Thermal Shift Assay—Acoustic Reverse-Phase Protein Array. SLAS Discov. Adv. Life Sci. R&D 2019, 25, 207–214. [Google Scholar] [CrossRef]
- Maingot, L.; Elbakali, J.; Dumont, J.; Bosc, D.; Cousaert, N.; Urban, A.; Deglane, G.; Villoutreix, B.; Nagase, H.; Sperandio, O.; et al. Aggrecanase-2 inhibitors based on the acylthiosemicarbazide zinc-binding group. Eur. J. Med. Chem. 2013, 69, 244–261. [Google Scholar] [CrossRef] [PubMed]
- Charton, J.; Gauriot, M.; Guo, Q.; Hennuyer, N.; Marechal, X.; Dumont, J.; Hamdane, M.; Pottiez, V.; Landry, V.; Sperandio, O.; et al. Imidazole-derived 2-[N-carbamoylmethyl-alkylamino]acetic acids, substrate-dependent modulators of insulin-degrading enzyme in amyloid-β hydrolysis. Eur. J. Med. Chem. 2014, 79, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Grasso, G.; Gioia, M.; Coletta, A.; Polticelli, F.; Di Pierro, D.; Milardi, D.; Van Endert, P.; et al. Multiple functions of insulin-degrading enzyme: A metabolic crosslight? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 554–582. [Google Scholar] [CrossRef] [PubMed]
- Leroux, F.; Bosc, D.; Beghyn, T.; Hermant, P.; Warenghem, S.; Landry, V.; Pottiez, V.; Guillaume, V.; Charton, J.; Herledan, A.; et al. Identification of ebselen as a potent inhibitor of insulin degrading enzyme by a drug repurposing screening. Eur. J. Med. Chem. 2019, 179, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Saulle, I.; Vicentini, C.; Clerici, M.; Biasin, M. An Overview on ERAP Roles in Infectious Diseases. Cells 2020, 9, 720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babaie, F.; Hosseinzadeh, R.; Ebrazeh, M.; Seyfizadeh, N.; Aslani, S.; Salimi, S.; Hemmatzadeh, M.; Azizi, G.; Jadidi-Niaragh, F.; Mohammadi, H. The roles of ERAP1 and ERAP2 in autoimmunity and cancer immunity: New insights and perspective. Mol. Immunol. 2020, 121, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Medve, L.; Gealageas, R.; Lam, B.V.; Guillaume, V.; Castillo-Aguilera, O.; Camberlein, V.; Piveteau, C.; Rosell, M.; Fleau, C.; Warenghem, S.; et al. Modulators of hERAP2 discovered by high-throughput screening. Eur. J. Med. Chem. 2021, 211, 113053. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.; Gierse, R.M.; Hirsch, A.K.H. Protein-Templated Dynamic Combinatorial Chemistry: Brief Overview and Experimental Protocol. Eur. J. Org. Chem. 2019, 2019, 3581–3590. [Google Scholar] [CrossRef] [PubMed]
- Bosc, D.; Camberlein, V.; Gealageas, R.; Castillo-Aguilera, O.; Deprez, B.; Deprez-Poulain, R. Kinetic Target-Guided Synthesis: Reaching the Age of Maturity. J. Med. Chem. 2019, 63, 3817–3833. [Google Scholar] [CrossRef]
- Bosc, D.; Jakhlal, J.; Deprez, B.; Deprez-Poulain, R. Kinetic target-guided synthesis in drug discovery and chemical biology: A comprehensive facts and figures survey. Futur. Med. Chem. 2016, 8, 381–404. [Google Scholar] [CrossRef] [Green Version]
- Oueis, E.; Sabot, C.; Renard, P.-Y. New insights into the kinetic target-guided synthesis of protein ligands. Chem. Commun. 2015, 51, 12158–12169. [Google Scholar] [CrossRef]
- Rani, A.; Singh, G.; Singh, A.; Maqbool, U.; Kaur, G.; Singh, J. CuAAC-ensembled 1,2,3-triazole-linked isosteres as pharmacophores in drug discovery: Review. RSC Adv. 2020, 10, 5610–5635. [Google Scholar] [CrossRef]
- Agalave, S.; Maujan, S.R.; Pore, V.S. Click Chemistry: 1,2,3-Triazoles as Pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef]
- Willand, N.; Desroses, M.; Toto, P.P.; Dirié, B.; Lens, Z.; Villeret, V.; Rucktooa, P.; Locht, C.; Baulard, A.; Deprez, B. Exploring Drug Target Flexibility Using in Situ Click Chemistry: Application to a Mycobacterial Transcriptional Regulator. ACS Chem. Biol. 2010, 5, 1007–1013. [Google Scholar] [CrossRef]
- Deprez-Poulain, R.; Hennuyer, N.; Bosc, D.; Liang, W.G.; Enée, E.; Marechal, X.; Charton, J.; Totobenazara, J.; Berte, G.; Jahklal, J.; et al. Catalytic site inhibition of insulin-degrading enzyme by a small molecule induces glucose intolerance in mice. Nat. Commun. 2015, 6, 8250. [Google Scholar] [CrossRef] [Green Version]
- Li, Q. Application of Fragment-Based Drug Discovery to Versatile Targets. Front. Mol. Biosci. 2020, 7, 180. [Google Scholar] [CrossRef]
- Hall, R.J.; Mortenson, P.; Murray, C.W. Efficient exploration of chemical space by fragment-based screening. Prog. Biophys. Mol. Biol. 2014, 116, 82–91. [Google Scholar] [CrossRef]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular Complexity and Its Impact on the Probability of Finding Leads for Drug Discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.; Macdonald, S.J. The impact of aromatic ring count on compound developability—Are too many aromatic rings a liability in drug design? Drug Discov. Today 2009, 14, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.C.; Dhondt, H.; Flipo, M.; Deprez, B.; Willand, N. Synthesis of functionalized 2-isoxazolines as three-dimensional fragments for fragment-based drug discovery. Tetrahedron Lett. 2015, 56, 4119–4123. [Google Scholar] [CrossRef]
- Prevet, H.; Flipo, M.; Roussel, P.; Deprez, B.; Willand, N. Microwave-assisted synthesis of functionalized spirohydantoins as 3-D privileged fragments for scouting the chemical space. Tetrahedron Lett. 2016, 57, 2888–2894. [Google Scholar] [CrossRef]
- Moreira, W.; Lim, J.J.; Yeo, S.Y.; Ramanujulu, P.M.; Dymock, B.W.; Dick, T. Fragment-Based Whole Cell Screen Delivers Hits against M. tuberculosis and Non-tuberculous Mycobacteria. Front. Microbiol. 2016, 7, 1392. [Google Scholar] [CrossRef] [Green Version]
- Prevet, H.; Moune, M.; Tanina, A.; Kemmer, C.; Herledan, A.; Frita, R.; Wohlkönig, A.; Bourotte, M.; Villemagne, B.; Leroux, F.; et al. A fragment-based approach towards the discovery of N-substituted tropinones as inhibitors of Mycobacterium tuberculosis transcriptional regulator EthR2. Eur. J. Med. Chem. 2019, 167, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Faïon, L.; Djaout, K.; Frita, R.; Pintiala, C.; Cantrelle, F.-X.; Moune, M.; Vandeputte, A.; Bourbiaux, K.; Piveteau, C.; Herledan, A.; et al. Discovery of the first Mycobacterium tuberculosis MabA (FabG1) inhibitors through a fragment-based screening. Eur. J. Med. Chem. 2020, 200, 112440. [Google Scholar] [CrossRef]
- Kramer, W. Bile Acid Reabsorption Inhibitors (BARI): Novel Hypolipidemic Drugs. Curr. Med. Chem. 2006, 13, 997–1016. [Google Scholar] [CrossRef] [PubMed]
- Charmot, D. Non-Systemic Drugs: A Critical Review. Curr. Pharm. Des. 2012, 18, 1434–1445. [Google Scholar] [CrossRef] [Green Version]
- Lasalle, M.; Hoguet, V.; Hennuyer, N.; Leroux, F.; Piveteau, C.; Belloy, L.; Lestavel, S.; Vallez, E.; Dorchies, E.; Duplan, I.; et al. Topical Intestinal Aminoimidazole Agonists of G-Protein-Coupled Bile Acid Receptor 1 Promote Glucagon Like Peptide-1 Secretion and Improve Glucose Tolerance. J. Med. Chem. 2017, 60, 4185–4211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoguet, V.; Lasalle, M.; Maingot, M.; Dequirez, G.; Boulahjar, R.; Leroux, F.; Piveteau, C.; Herledan, A.; Biela, A.; Dumont, J.; et al. Beyond the Rule of 5: Impact of PEGylation with Various Polymer Sizes on Pharmacokinetic Properties, Structure–Properties Relationships of mPEGylated Small Agonists of TGR5 Receptor. J. Med. Chem. 2021, 64, 1593–1610. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Musson, D.G.; Wang, A.Q. Stability studies of vorinostat and its two metabolites in human plasma, serum and urine. J. Pharm. Biomed. Anal. 2006, 42, 556–564. [Google Scholar] [CrossRef]
- Hermant, P.; Bosc, D.; Piveteau, C.; Gealageas, R.; Lam, B.; Ronco, C.; Roignant, M.; Tolojanahary, H.; Jean, L.; Renard, P.-Y.; et al. Controlling Plasma Stability of Hydroxamic Acids: A MedChem Toolbox. J. Med. Chem. 2017, 60, 9067–9089. [Google Scholar] [CrossRef]
- Montaigne, D.; Marechal, X.; Modine, T.; Coisne, A.; Mouton, S.; Fayad, G.; Ninni, S.; Klein, C.; Ortmans, S.; Seunes, C.; et al. Daytime variation of perioperative myocardial injury in cardiac surgery and its prevention by Rev-Erbα antagonism: A single-centre propensity-matched cohort study and a randomised study. Lancet 2018, 391, 59–69. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | R | H |  |
| Cpd | 3 | 24 | |
| In vitro activity | |||
| hTGR5 EC50 (nM) | 35 | 24 | |
| mTGR5 EC50 (nM) | 0.8 | 0.4 | |
| In vitro ADME properties | |||
| Solubility (µM) a | 6.3 | >200 | |
| LogD (7.4) b | 3.5 | 0.8 | |
| Papp A-B c | 5.32 | 0.031 | |
| Papp B-A c | 5.47 | 19 | |
| Efflux ratio c | 1 | 636 | |
| Clint d | 1287 | 1211 | |
| In vivo PK properties e | |||
| [C]plasmamax (nM) | NT | 102 | |
| Fecal recovery (%) f | NT | 100 | |
 | R | H |  | |||
| Cpd | 2 | P7 | P9 | P11 | P12 | |
| n (PEG unit) | - | 4–13 | 11–25 | 31–57 | 100–138 | |
| MW (g/mol) | 516 | 824–1220 | 1132–1748 | 2012–3156 | 5048–6720 | |
| In vitro activity | ||||||
| hTGR5 EC50 (nM) | 20 | 60 | 145 | 515 | 1102 | |
| mTGR5 EC50 (nM) | 0.8 | 5 | 13 | 25 | 63 | |
| In vitro ADME properties | ||||||
| Solubility (µM) a | 8.8 | 151 | >200 | >200 | >2 00 | |
| LogD7.4 | 3.97 | 3.42 | 1.58 | −1.53 | −1.58 | |
| Clint b | 1254 | 524 | 17 | 8 | 11 | |
| Papp A-B c | 9.3 | 4.9 | <0.02 | <0.12 | NT | |
| Papp B-A c | 8.5 | 19.5 | 9.1 | <0.03 | NT | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deprez, B.; Bosc, D.; Charton, J.; Couturier, C.; Deprez-Poulain, R.; Flipo, M.; Leroux, F.; Villemagne, B.; Willand, N. Molecular Design in Practice: A Review of Selected Projects in a French Research Institute That Illustrates the Link between Chemical Biology and Medicinal Chemistry. Molecules 2021, 26, 6083. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26196083
Deprez B, Bosc D, Charton J, Couturier C, Deprez-Poulain R, Flipo M, Leroux F, Villemagne B, Willand N. Molecular Design in Practice: A Review of Selected Projects in a French Research Institute That Illustrates the Link between Chemical Biology and Medicinal Chemistry. Molecules. 2021; 26(19):6083. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26196083
Chicago/Turabian StyleDeprez, Benoit, Damien Bosc, Julie Charton, Cyril Couturier, Rebecca Deprez-Poulain, Marion Flipo, Florence Leroux, Baptiste Villemagne, and Nicolas Willand. 2021. "Molecular Design in Practice: A Review of Selected Projects in a French Research Institute That Illustrates the Link between Chemical Biology and Medicinal Chemistry" Molecules 26, no. 19: 6083. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26196083