
Design, Synthesis and Biological Evaluation of Arylpyridin-2-yl Guanidine Derivatives and Cyclic Mimetics as Novel MSK1 Inhibitors. An Application in an Asthma Model

 , , , and
, , , and
Abstract
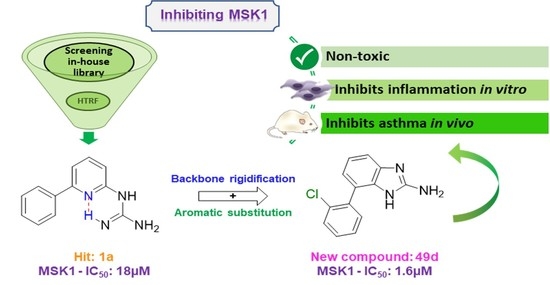
:
1. Introduction
2. Results
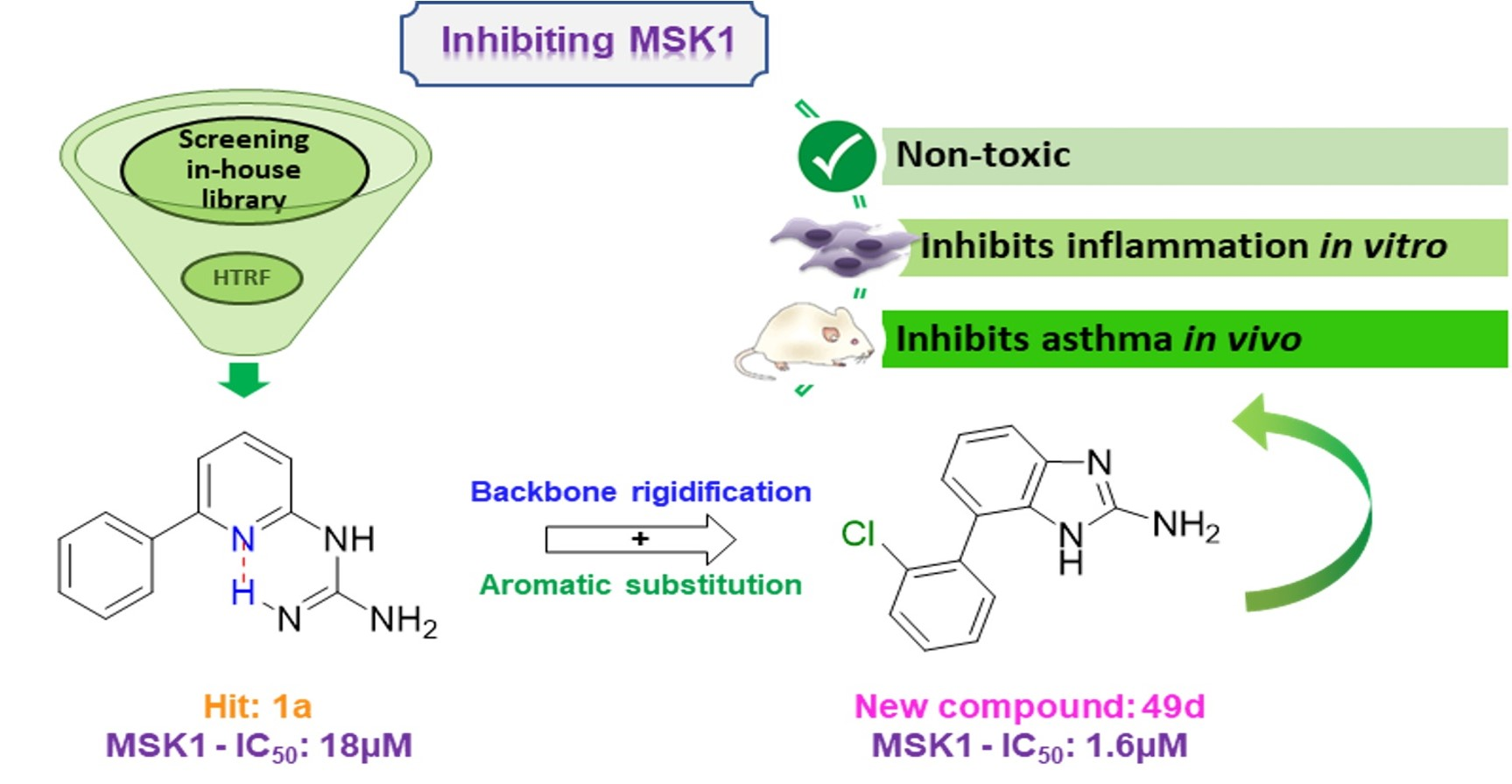
2.1. Screening and Hit Selection
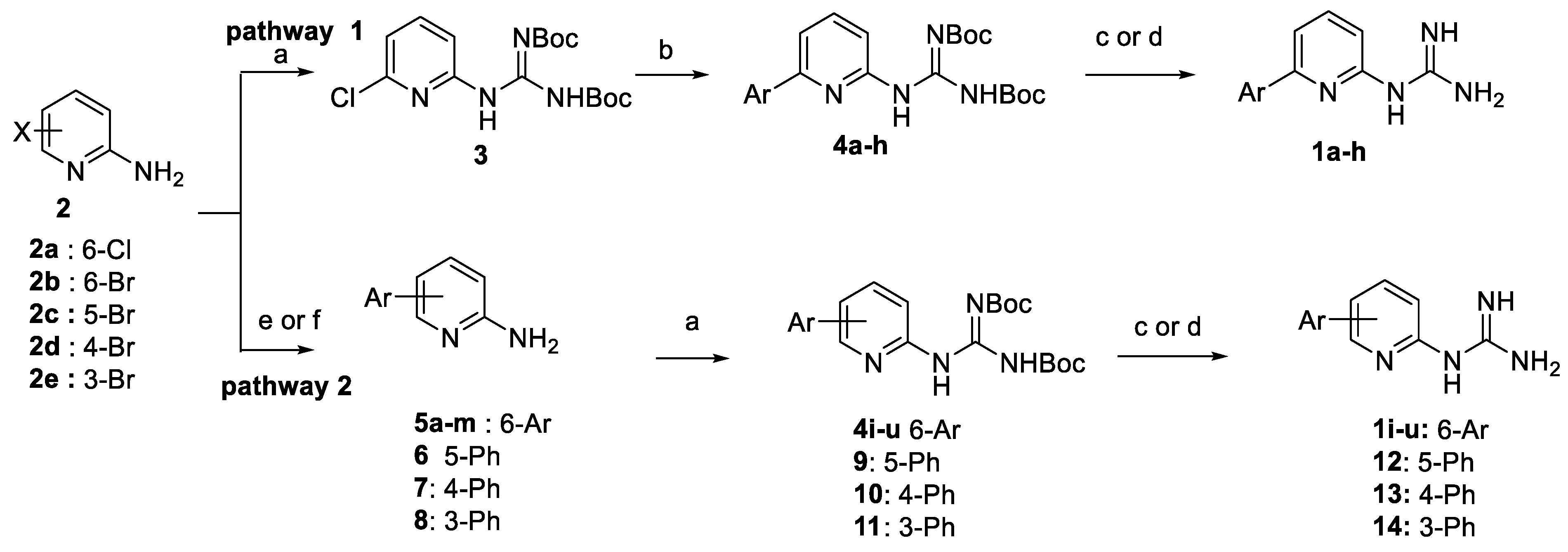
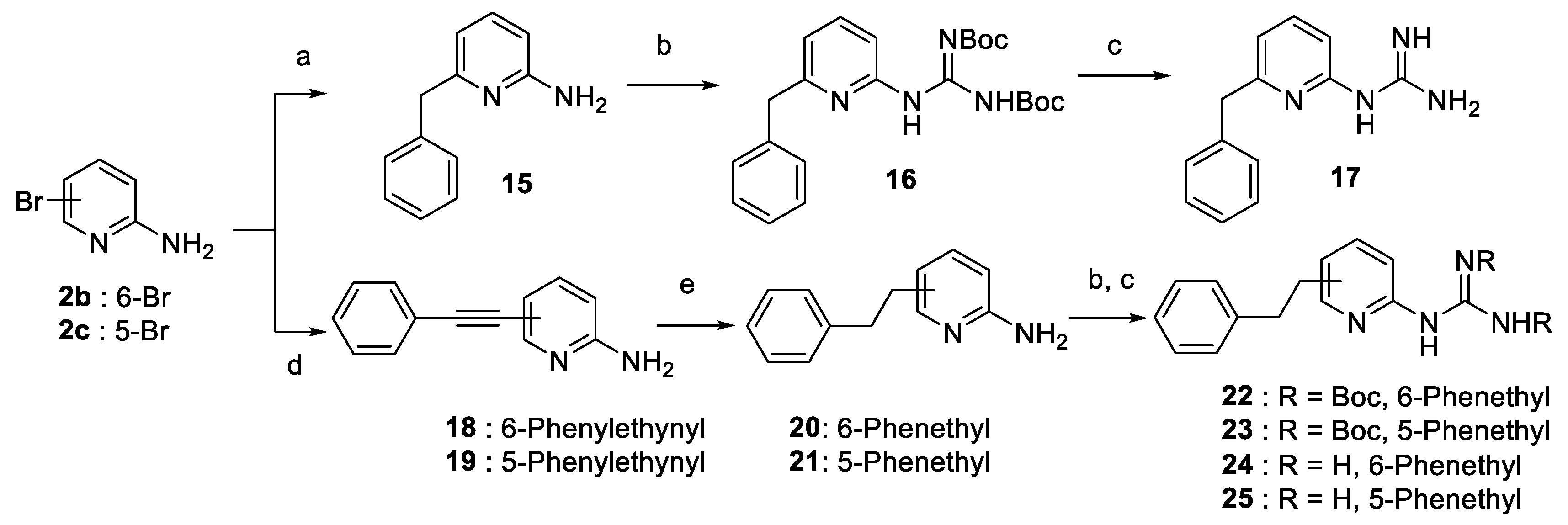
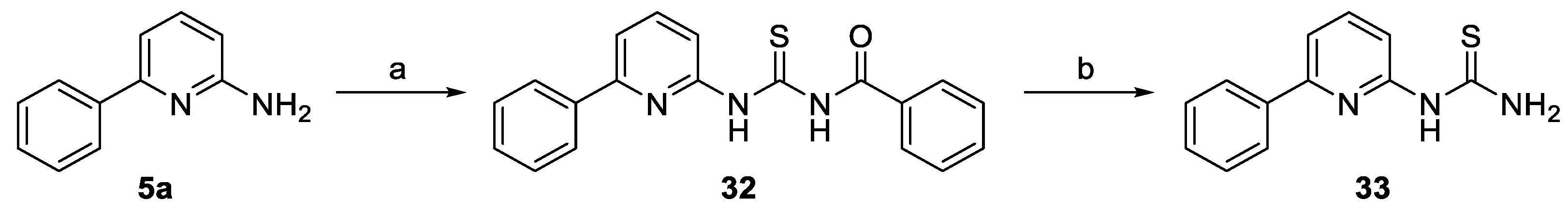
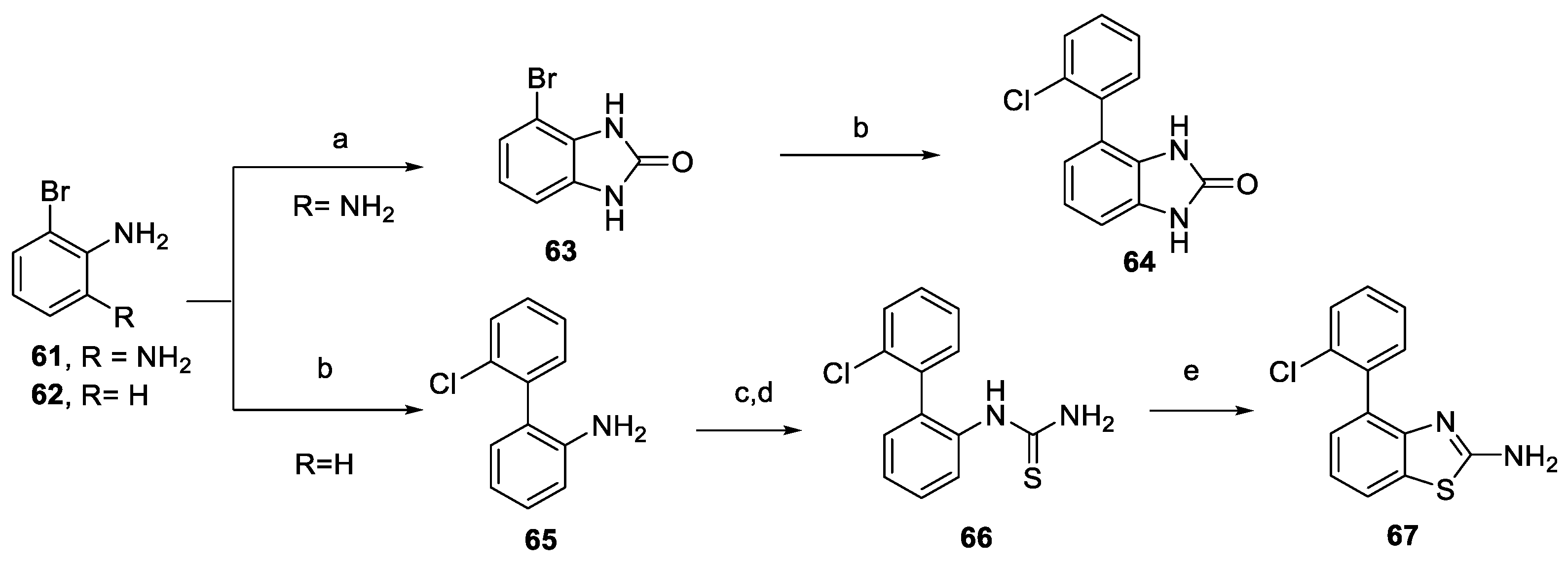
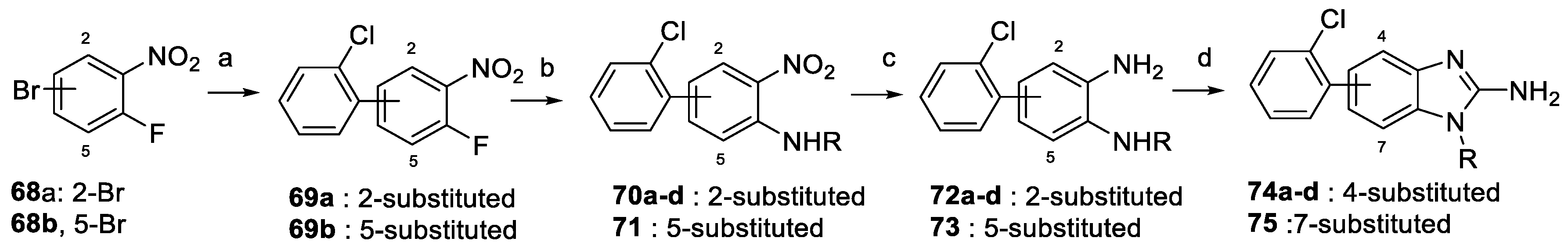
2.2. Synthesis of the Compounds
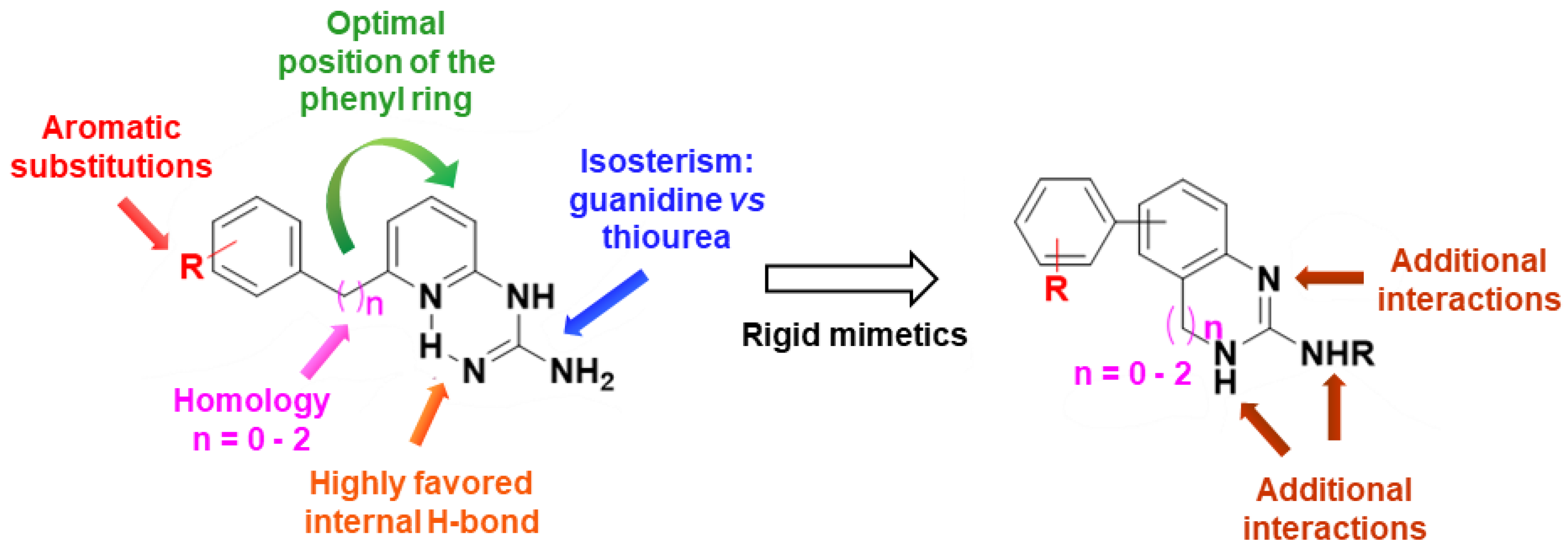
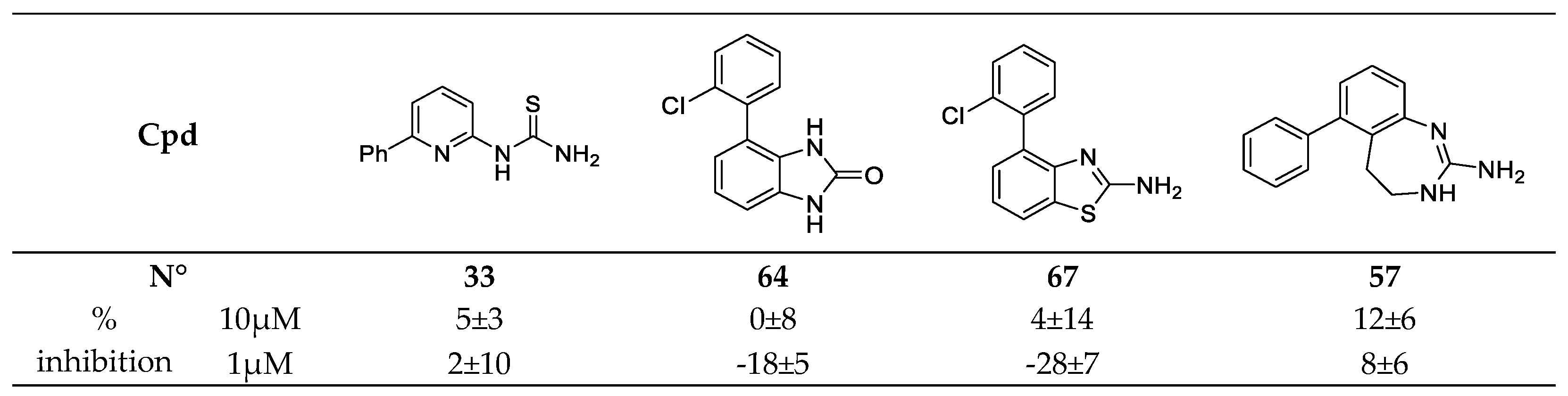
2.3. Evaluation of the New Compounds in an Enzymatic Assay for MSK1 Inhibition
2.4. In Vitro Evaluation of Compounds
2.5. In Vivo Evaluation of Compounds
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis
4.1.1. General Information
4.1.2. General Method A: Preparation of Boc-Protected Guanidines Derivatives 3, 4j, 4l, 4m, 4o–u, 16
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-chloropyridine (3). Following general method A and starting from 2-amino-6-chloropyridine (2a, 1.00 g, 7.78 mmol), 3 was obtained as a white solid (2.36 g, 6.37 mmol, 86%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.53 (s, 18H), 7.03 (d, 1H, J = 7.9 Hz), 7.65 (t, 1H, J = 7.9 Hz), 8.36 (d, 1H, J = 7.9 Hz), 10.85 (s, 1H), 11.50 (s, 1H); 13C-NMR (101 MHz, CDCl3) δ ppm 28.0, 28.1, 80.2, 84.2, 114.2, 119.8, 140.6, 153.0.
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(2-fluorophenyl)pyridine (4j). Following general method A and starting from 5b (258 mg, 1.37 mmol), compound 4j was obtained as a white solid (495 mg, 1.15 mmol, 88%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.51 (s, 9H), 1.54 (s, 9H), 7.16 (d, 1H, J = 7.4 Hz), 7.47–7.52 (m, 2H), 7.59 (t, 1H, J = 7.4 Hz), 7.73–7.79 (m, 2H), 8.41 (br s, 1H), 10.83 (s, 1H), 11.56 (s, 1H); 13C-NMR (101 MHz, CDCl3) δ ppm 28.0, 28.2, 79.7, 79.9, 115.1, 120.0, 122.7, 125.4, 126.3 (q, J = 5.9 Hz), 128.3, 128.2, 131.5, 131.6, 138.2, 139.5. 150.0, 153.3
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(2-methylphenyl)pyridine (4l). Following general method A and starting from 5d (100 mg, 0.54 mmol), compound 4l was obtained as a colorless oil (216 mg, 0.51 mmol, 98%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.44 (s, 9H), 1.47 (s, 9H), 2.30 (s, 3H), 7.06 (d, 1H, J = 7.5 Hz), 7.17–7.22 (m, 3H), 7.31 (d, 1H, J = 7.5 Hz), 7.69 (t, 1H, J = 7.5 Hz), 8.25 (br s, 1H), 10.73 (s, 1H), 11.49 (s, 1H); 13C-NMR DEPT-135 (101 MHz, CDCl3) δ ppm 20.5, 28.0, 28.2, 120.1, 125.8, 128.3, 129.6, 130.7, 138.3.
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(2-trifluoromethylphenyl)pyridine (4m). Following general method A and starting from 5e (304 mg, 1.28 mmol), 4m was obtained as a white solid (411 mg, 0.86 mmol, 71%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.54 (s, 18H), 7.13 (dd, 1H, J = 8.7 Hz, J = 11.2 Hz), 7.24 (t, 1H, J = 7.4 Hz), 7.33–7.39 (m, 1H), 7.57 (d, 1H, J = 6.4 Hz), 7.78 (t, 1H, J = 7.9 Hz), 8.04 (br s, 1H), 8.37 (br s, 1H), 10.86 (s, 1H), 11.58 (s, 1H); 13C DEPT-135 NMR (101 MHz, CDCl3) δ ppm 28.1, 28.2, 116.1, 116.3, 120.6, 124.4 (q, J = 3.7 Hz), 130.4 (q, J = 8.1 Hz), 131.1, 138.6.
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(2,3-dichlorophenyl)pyridine (4o). Following general method A and starting from 5g (158 mg, 0.66 mmol), 4o was obtained as a white solid (269 mg, 0.56 mmol, 89%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.52 (s, 18H), 7.24–7.29 (m, 2H), 7.42 (dd, 1H, J = 1.6 Hz, J = 7.9 Hz), 7.49 (dd, 1H, J = 1.6 Hz, J = 7.9 Hz), 7.78 (t, 1H, J = 7.9 Hz), 8.39 (br s, 1H), 10.81 (s, 1H), 11.57 (s, 1H); 13C-NMR (101 MHz, CDCl3) δ ppm 28.1, 79.9, 80.1, 120.8, 127.2, 129.7, 130.3, 130.9, 133.6, 138.2, 140.9, 142.2, 146.5, 146.7, 153.5, 155.2.
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(2,4-dichlorophenyl)pyridine (4p). Following general method A and starting from 5h (182 mg, 0.76 mmol), 4p was obtained as a colorless oil (330 mg, 0.69 mmol, 95%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.53 (s, 18H), 7.32 (dd, 1H, J = 2.0 Hz, J = 8.4 Hz), 7.35–7.37 (m, 1H), 7.47 (d, 1H, J = 2.0 Hz), 7.54 (d, 1H, J = 8.4 Hz), 7.77 (t, 1H, J = 7.9 Hz), 8.38 (br s, 1H), 10.81 (s, 1H), 11.57 (s, 1H); 13C-NMR (101 MHz, CDCl3) δ ppm 28.2, 80.0, 80.1, 115.3, 120.9, 127.2, 128.6, 129.8, 132.6, 132.9, 134.8, 139.9, 143.4, 145.0, 149.6, 150.5, 153.2, 155.5.
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(2,5-dichlorophenyl)pyridine (4q). Following general method A and starting from 5i (143 mg, 0.60 mmol), 4q was obtained as a yellow solid (264 mg, 0.55 mmol, 97%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.54 (s, 18H), 7.28 (dd, 1H, J = 2.6 Hz, J = 8.5 Hz), 7.36–7.39 (m, 2H), 7.57 (s, 1H), 7.79 (t, 1H, J = 7.9 Hz), 8.39 (br s, 1H), 10.84 (s, 1H), 11.57 (s, 1H); 13C-NMR DEPT-135 (101 MHz, CDCl3) δ ppm 28.0, 28.2, 115.3, 120.9, 129.5, 130.5, 131.2, 131.5, 132.8, 138.2, 139.9.
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(4-pyridyl)pyridine (4r). Following general method A and starting from 5j (99 mg, 0.58 mmol), 4r was obtained as a white solid (156 mg, 0.38 mmol, 68%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.55 (s, 18H), 7.54 (d, 1H, J = 7.8 Hz), 7.83 (t, 1H, J = 7.8 Hz), 7.89 (br s, 2H), 8.48 (br s, 1H), 8.70 (d, 2H, J = 6.3 Hz), 10.94 (s, 1H), 11.59 (s, 1H); 13C DEPT-135 NMR (101 MHz, CDCl3) δ ppm 28.1, 116.5, 120.9, 139.3, 150.3.
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(3-pyridyl)pyridine (4s). Following general method A and starting from 6-(pyridin-3-yl)pyridin-2-amine 5k (15 mg, 0.07 mmol), 4s was obtained as a white solid (20 mg, 62%) and immediately used in the BOC deprotection step without analytical characterization.
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(2-chloropyridin-3-yl)pyridine (4t). Following general method A and starting from 5l (75 mg, 0.36 mmol), 4t was obtained as a white solid (121 mg, 0.27 mmol, 74%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.52 (s, 18H), 7.33 (dd, J = 7.6, 4.8 Hz, 1H), 7.48 (d, J = 7.1 Hz, 1H), 7.80 (t, J = 7.9 Hz, 1H), 7.96 (d, J = 7.0 Hz, 1H), 8.41 (dd, J = 4.7, 1.9 Hz, 2H), 10.85 (s, 1H), 11.57 (s, 1H).
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(2-methoxypyridin-3-yl)pyridine (4u). Following general method A and starting from 5m (145 mg, 0.72 mmol), 4u was obtained as a white solid (241 mg, 0.54 mmol, 76%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.54 (s, 18H), 4.03 (s, 3H), 7.02 (dd, J = 7.2, 5.1 Hz, 1H), 7.78 (dt, J = 15.6, 7.6 Hz, 2H), 8.19 (dd, J = 4.9, 1.9 Hz, 1H), 8.33 (d, J = 5.6 Hz, 1H), 10.82 (s, 1H), 11.59 (s, 1H).
- ∗
- 6-Benzyl-1-[2,3-di(tert-butoxycarbonyl)guanidino]pyridine (16). Following general method A and starting from 15 (111 mg, 0.60 mmol), 16 was obtained as a white solid (73 mg, 0.17 mmol, 29%). 1H-NMR (300 MHz, CDCl3) δ ppm 1.52 (s, 9H), 1.55 (s, 9H), 4.05 (s, 2H), 6.80 (d, 1H, J = 7.6 Hz), 7.20–7.32 (m, 6H), 7.58 (t, 1H, J = 7.6 Hz).
4.1.3. General Method B: Pd-Catalyzed Suzuki-Miyaura Cross-Coupling Using Pd(PPh3)4. Preparation of 5a, 5b, 35b–g, 45a, 45b
- ∗
- 6-Phenylpyridin-2-amine (5a) [21]. Following general method B and starting from 2-amino-6-chloropyridine (2a, 200 mg, 1.56 mmol) and phenylboronic acid (228 mg, 1.87 mmol), 5a was obtained as a yellow oil (242 mg, 1.42 mmol, 92%). 1H-NMR (400 MHz, CDCl3) δ ppm 4.54 (s, 2H), 6.45 (d, 1H, J = 7.8 Hz), 7.10 (d, 1H, J = 7.8 Hz), 7.38 (t, 1H, J = 7.7 Hz), 7.44 (t, 2H, J = 7.7 Hz), 7.50 (t, 1H, J = 7.8 Hz), 7.94 (d, 2H, J = 7.7 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 107.1, 111.0, 126.8, 128.5, 128.6, 138.4, 139.7, 156.2, 158.3.
- ∗
- 6-(2-Fluorophenyl)pyridin-2-amine (5b). Following general method B and starting from 2-amino-6-chloropyridine (2a, 200 mg, 1.56 mmol) and 2-fluorophenylboronic acid (261 mg, 1.87 mmol), 5b was obtained as a white solid (274 mg, 1.45 mmol, 93%). 1H-NMR (400 MHz, CDCl3) δ ppm 4.52 (s, 2H), 6.48 (d, 1H, J = 8.2 Hz), 7.10–7.16 (m, 2H), 7.22 (td, 1H, J = 1.3 Hz, J = 7.8 Hz), 7.30–7.36 (m, 1H), 7.50 (t, 1H, J = 7.8 Hz), 7.90 (td, 1H, J = 1.9 Hz, J = 7.8 Hz); 19F-NMR (376 MHz, CDCl3) δ ppm −116.6; 13C-NMR (101 MHz, CDCl3) δ ppm 107.5, 115.0 (d, J = 8.8 Hz), 116.1 (d, J = 23.5 Hz), 124.3 (d, J = 3.7 Hz), 127.7 (d, J = 11.7 Hz), 129.9 (d, J = 8.8 Hz), 130.8 (d, J = 2.9 Hz), 138.1, 151.7, 158.3, 160.4 (d, J = 248.7 Hz).
- ∗
- 6-(2-Methylphenyl)pyridin-2-amine (5d). Following general method B and starting from 2-amino-6-chloropyridine (2a, 150 mg, 1.17 mmol) and 2-tolylboronic acid (190 mg, 1.40 mmol), 5d was obtained as a yellow solid (195 mg, 1.06 mmol, 90%). 1H-NMR (400 MHz, CDCl3) δ ppm 2.27 (s, 3H), 4.43 (s, 2H), 6.37 (d, 1H, J = 7.9 Hz), 6.65 (d, 1H, J = 7.3 Hz), 7.15–7.19 (m, 3H), 7.26–7.29 (m, 1H), 7.41 (t, 1H, J = 7.9 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 20.3, 106.5, 114.3, 125.7, 127.9, 129.3, 130.6, 135.6, 137.8, 140.8, 157.8, 158.6.
- ∗
- 6-[2-(Trifluoromethyl)phenyl]pyridin-2-amine (5e). Following general method B and starting from 2-amino-6-chloropyridine (2a, 200 mg, 1.56 mmol) and 2-trifluoromethylphenylboronic acid (355 mg, 1.87 mmol), 5e was obtained as a white solid (325 mg, 1.36 mmol, 88%). 1H-NMR (400 MHz, CDCl3) δ ppm 4.52 (s, 2H), 6.50 (dd, 1H, J = 0.6 Hz, J = 8.3 Hz), 6.75 (d, 1H, J = 7.4 Hz), 7.46–7.50 (m, 3H), 7.58 (t, 1H, J = 7.4 Hz), 7.73 (d, 1H, J = 7.4 Hz); 19F-NMR (376 MHz, CDCl3) δ ppm -56.9; 13C-NMR (101 MHz, CDCl3) δ ppm 107.5, 114.2 (q, J = 2.2 Hz), 124.1 (q, J = 274.4 Hz), 126.3 (q, J = 5.1 Hz), 128.0, 131.3, 131.5, 137.7, 140.3, 156.2, 157.7.
- ∗
- 6-(2,3-Dichlorophenyl)pyridin-2-amine (5g). Following general method B and starting from 2-amino-6-bromopyridine (2b, 200 mg, 1.16 mmol) and 2,3-dichlorophenylboronic acid (265 mg, 1.39 mmol), 5g was obtained as a yellow solid (178 mg, 0.74 mmol, 64%). 1H-NMR (400 MHz, CDCl3) δ ppm 4.55 (s, 2H), 6.51 (d, 1H, J = 8.0 Hz), 6.87 (d, 1H, J = 8.0 Hz), 7.25 (t, 1H, J = 7.8 Hz), 7.38 (dd, 1H, J = 1.6 Hz, J = 7.8 Hz), 7.47 (dd, 1H, J = 1.6 Hz, J = 7.8 Hz), 7.51 (t, 1H, J = 8.0 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 107.7, 114.8, 127.2, 129.3, 130.0, 130.8, 133.6, 137.8, 141.9, 155.3, 158.1.
- ∗
- 6-(2,4-Dichlorophenyl)pyridin-2-amine (5h). Following general method B and starting from 2-amino-6-bromopyridine (2b, 200 mg, 1.16 mmol) and 2,4-dichlorophenylboronic acid (265 mg, 1.39 mmol), 5h was obtained as a white solid (207 mg, 0.86 mmol, 75%). 1H-NMR (400 MHz, CDCl3) δ ppm 4.56 (s, 2H), 6.50 (dd, 1H, J = 0.5 Hz, J = 8.2 Hz), 6.92 (dd, 1H, J = 0.5 Hz, J = 8.2 Hz), 7.31 (dd, 1H, J = 2.0 Hz, J = 8.4 Hz), 7.46 (d, 1H, J = 2.0 Hz), 7.47–7.52 (m, 2H); 13C-NMR (101 MHz, CDCl3) δ ppm 107.7, 114.9, 127.2, 129.8, 132.2, 132.9, 134.4, 137.7, 138.0, 154.2, 158.2.
- ∗
- 6-(2,5-Dichlorophenyl)pyridin-2-amine (5i). Following general method B and starting from 2-amino-6-bromopyridine (2b, 50 mg, 0.29 mmol) and 2,5-dichlorophenylboronic acid (66 mg, 0.35 mmol), 5i was obtained as a white solid (56 mg, 0.23 mmol, 81%). 1H-NMR (400 MHz, CDCl3) δ ppm 4.54 (s, 2H), 6.51 (dd, 1H, J = 0.4 Hz, J = 7.9 Hz), 6.95 (dd, 1H, J = 0.4 Hz, J = 7.9 Hz), 7.26 (dd, 1H, J = 2.5 Hz, J = 8.5 Hz), 7.37 (d, 1H, J = 8.5 Hz), 7.51 (t, 1H, J = 7.9 Hz), 7.55 (d, 1H, J = 2.6 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 107.8, 114.9, 129.1, 130.5, 131.1, 131.2, 132.7, 137.8, 140.8, 154.0, 158.2.
- ∗
- 6-(Pyridin-4-yl)pyridin-2-amine (5j). Following general method B and starting from 2-amino-6-chloropyridine (2a, 100 mg, 0.78 mmol) and 4-pyridineboronic acid (115 mg, 0.93 mmol), 5j was obtained as a yellow solid (114 mg, 0.66 mmol, 85%). 1H-NMR (400 MHz, DMSO-d6) δ ppm 6.14 (s, 2H), 6.53 (d, 1H, J = 8.0 Hz), 7.20 (d, 1H, J = 8.0 Hz), 7.52 (t, 1H, J = 8.0 Hz), 7.92 (d, 2H, J = 6.1 Hz), 8.62 (d, 2H, J = 6.1 Hz); 13C-NMR (101 MHz, DMSO-d6) δ ppm 109.5, 121.0, 138.6, 146.7, 150.5, 152.0, 160.2.
- ∗
- 6-(Pyridin-3-yl)pyridin-2-amine (5k). Following general method B and starting from 2-amino-6-chloropyridine (2a, 20.5 mg, 0.16 mmol) and 3-pyridineboronic acid (23.1 mg, 0.19 mmol), 5k was obtained as a white powder (17 mg, 0.1 mmol, 62%). 1H-NMR (500 MHz, CDCl3) δ ppm 4.63 (bs, 2H), 6.49 (d, J = 5 Hz, 1H), 7.09 (d, J = 5 Hz, 1H), 7.35 (m, 1H), 7.52 (t, J = 5 Hz, 1H), 8.23 (dt, J = 5 Hz, 1H), 8.60 (d, J = 5 Hz, 1H), 9.14 (s, 1H); 13C-NMR (125 MHz, CDCl3) δ ppm 107.9, 110.9, 123.4, 134.2, 138.5, 148.3, 149.5, 153.3, 158.5. Analytical data are consistent with previously reported characterization [41].
- ∗
- 6-(2-chloropyridin-3-yl)pyridin-2-amine (5l). Following general method B and starting from 2-amino-6-bromopyridine (2b, 173 mg, 0.58 mmol) and 2-chloropyridin-3-ylboronic acid (109 mg, 0.69 mmol), 5k was obtained as a beige solid (75 mg, 0.66 mmol, 63%). Crude solid was triturated in Et2O and used in the next step without further purification. 1H-NMR (400 MHz, CDCl3) δ ppm 4.55 (bs, 2H), 6.55 (dd, J = 8.2, 0.8 Hz, 1H), 7.05 (dd, J = 7.4, 0.7 Hz, 1H), 7.34 (dd, J = 7.6, 4.8 Hz, 1H), 7.55 (dd, J = 8.2, 7.5 Hz, 1H), 7.92 (dd, J = 7.6, 2.0 Hz, 1H), 8.42 (dd, J = 4.8, 2.0 Hz, 1H).
- ∗
- 6-(2-methoxypyridin-3-yl)pyridin-2-amine (5m). Following general method B and starting from 2-amino-6-chloropyridine 2a (100 mg, 0.78 mmol) and 2-methoxypyridin-3-ylboronic acid (143 mg, 0.93 mmol), 5l as a yellow solid (143 mg, 0.71 mmol, 91%). Crude solid was triturated in Et2O and used without further purification. 1H-NMR (400 MHz, CDCl3) δ 3.74 (s, 3H), 4.21 (bs, 2H), 6.48 (dd, J = 8.1, 0.6 Hz, 1H), 7.00 (dd, J = 7.4, 5.0 Hz, 1H), 7.33 (dd, J = 7.6, 0.7 Hz, 1H), 7.50 (t, J = 7.8 Hz, 1H), 8.24–8.08 (m, 2H).
- ∗
- 2-Amino-6-(4-chlorophenyl)benzonitrile (35b). Following general method B and starting from 2-amino-6-iodobenzonitrile (34, 200 mg, 0.82 mmol) and 4-chlorophenylboronic acid (154 mg, 0.98 mmol), 35b was obtained as a white solid (165 mg, 0.72 mmol, 88%). 1H-NMR (400 MHz, CDCl3) δ ppm 4.54 (s 2H), 6.73–6.75 (m, 2H), 7.35 (t, 1H, J = 8.0 Hz), 7.43 (d, 2H, J = 8.8 Hz), 7.47 (d, 2H, J = 8.8 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 113.9, 117.1, 118.8, 128.8, 129.9, 133.6, 134.7, 137.3, 144.6, 150.7. Analytical data are consistent with the previously reported characterization [42].
- ∗
- 2-Amino-6-(4-methoxyphenyl)benzonitrile (35c). Following general method B and starting from 2-amino-6-iodobenzonitrile (34, 200 mg, 0.82 mmol) and 4-methoxyphenylboronic acid (149 mg, 0.98 mmol), 35c was obtained as a yellow solid (169 mg, 0.76 mmol, 92%). 1H-NMR (400 MHz, CDCl3) δ ppm 3.85 (s, 3H), 4.51 (s, 2H), 6.69 (dd, 1H, J = 0.9 Hz, J = 8.0 Hz), 6.76 (dd, 1H, J = 0.9 Hz, J = 8.0 Hz), 6.99 (d, 2H, J = 8.8 Hz), 7.32 (t, 1H, J = 8.0 Hz), 7.49 (d, 2H, J = 8.8 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 55.3, 113.1, 114.0, 117.6, 118.8, 129.8, 131.3, 133.4, 145.6, 150.6, 159.9. Analytical data are consistent with the previously reported characterization [42].
- ∗
- 2-Amino-6-(3-chlorophenyl)benzonitrile (35d). Following general method B and starting from 2-amino-6-iodobenzonitrile (34, 200 mg, 0.82 mmol) and 3-chlorophenylboronic acid (154 mg, 0.98 mmol), 35d was obtained as a yellow solid (176 mg, 0.77 mmol, 94%). 1H-NMR (400 MHz, CDCl3) δ ppm 4.56 (s, 2H), 6.75 (d, 2H, J = 7.9 Hz), 7.35 (t, 1H, J = 7.9 Hz), 7.38–7.45 (m, 3H), 7.49–7.50 (m, 1H); 13C-NMR (101 MHz, CDCl3) δ ppm 114.1, 117.0, 118.8, 126.8, 128.6, 128.7, 129.8, 133.6, 134.5, 140.6, 144.3, 150.7.
- ∗
- 2-Amino-6-(3-methoxyphenyl)benzonitrile (35e). Following general method B and starting from 2-amino-6-iodobenzonitrile (34, 200 mg, 0.82 mmol) and 3-methoxyphenylboronic acid (149 mg, 0.98 mmol), 35e was obtained as a yellow solid (161 mg, 0.72 mmol, 87%). 1H-NMR (400 MHz, CDCl3) δ ppm 3.86 (s, 3H), 4.53 (s, 2H), 6.7 3 (dd, 1H, J = 0.9 Hz, J = 8.3 Hz), 6.79 (dd, 1H, J = 0.9 Hz, J = 7.7 Hz), 6.96 (ddd, 1H, J = 0.9 Hz, J = 2.6 Hz, J = 8.3 Hz), 7.07 (dd, 1H, J = 1.6 Hz, J = 2.6 Hz), 7.12 (ddd, 1H, J = 0.9 Hz, J = 1.6 Hz, J = 7.7 Hz), 7.32–7.39 (m, 2H); 13C-NMR (101 MHz, CDCl3) δ ppm 55.4, 113.6, 114.1, 114.3, 117.3, 118.9, 121.0, 129.6, 133.4, 140.2, 145.7, 150.6, 159.6.
- ∗
- 2-Amino-6-(2-chlorophenyl)benzonitrile (35f). Following general method B and starting from 2-amino-6-iodobenzonitrile (34, 393 mg, 1.61 mmol) and 2-chlorophenylboronic acid (302 mg, 1.93 mmol), 35f was obtained as an orange solid (319 mg, 1.40 mmol, 87%). 1H-NMR (400 MHz, CDCl3) δ ppm 4.51 (s, 2H), 6.71 (dd, 1H, J = 0.9 Hz, J = 7.4 Hz), 6.77 (dd, 1H, J = 0.9 Hz, J = 8.3 Hz), 7.32–7.38 (m, 4H), 7.47–7.52 (m, 1H); 13C-NMR (101 MHz, CDCl3) δ ppm 114.2, 116.5, 119.6, 126.8, 129.8, 129.9, 131.0, 132.9, 133.2, 137.7, 143.3, 150.0.
- ∗
- 2-Amino-6-(2-methoxyphenyl)benzonitrile (35g). Following general method B and starting from 2-amino-6-iodobenzonitrile (34, 300 mg, 1.23 mmol) and 2-methoxyphenylboronic acid (224 mg, 1.48 mmol), 35g was obtained as a white solid (221 mg, 0.99 mmol, 80%). 1H-NMR (400 MHz, CDCl3) δ ppm 3.85 (s, 3H), 4.45 (s, 2H), 6.73 (dd, 2H, J = 1.0 Hz, J = 8.3 Hz), 7.01 (d, 1H, J = 8.3 Hz), 7.03 (td, 1H, J = 1.0 Hz, J = 7.5 Hz), 7.24 (dd, 1H, J = 1.8 Hz, J = 7.4 Hz), 7.34 (t, 1H, J = 7.9 Hz), 7.39 (ddd, 1H, J = 1.8 Hz, J = 7.5 Hz, J = 8.2 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 55.5, 111.3, 113.6, 117.2, 119.8, 120.7, 127.9, 130.0, 130.8, 133.2, 142.9, 149.8, 156.5.
- ∗
- 2-nitro-3-phenylaniline (45a). Following general method B and starting from 3-bromo-2-nitroaniline (44a, 600 mg, 2.77 mmol) and phenylboronic acid (405 mg, 3.12 mmol) 45a was obtained as an orange solid (482 mg, 2.25 mmol, 81%).1H-NMR (400 MHz, CDCl3) δ ppm 4.86 (s, 2H), 6.57 (dd, 1H, J = 1.1 Hz, J = 7.4 Hz), 6.67 (dd, 1H, J = 1.0 Hz, J = 8.3 Hz), 7.15 (d, 1H, J = 8.3 Hz), 7.18 (dd, 2H, J = 1.9 Hz, J = 7.4 Hz), 7.23–7.30 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ ppm 117.1, 120.6, 127.4, 127.8, 128.6, 132.4, 138.5, 138.7, 141.8. Analytical data are consistent with the previously reported 1H-NMR characterization [43].
- ∗
- 3-(4-Chlorophenyl)-2-nitroaniline (45b). Following general method B and starting from 3-bromo-2-nitroaniline (44a, 100 mg, 0.46 mmol) and 4-chlorophenylboronic acid (86 mg, 0.55 mmol), 45b was obtained as an orange solid (89 mg, 0.36 mmol, 78%). 1H-NMR (400 MHz, CDCl3) δ ppm 5.09 (s, 2H), 6.66 (d, 1H, J = 7.3 Hz), 6.83 (d, 1H, J = 8.3 Hz), 7.24–7.32 (m, 3H), 7.39 (d, 2H, J = 8.3 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 117.5, 120.5, 128.7, 128.8, 132.6, 133.9, 137.3, 137.4, 142.1.
4.1.4. General Method C: Pd-Catalyzed Suzuki-Miyaura Cross-Coupling Using Pd(OAc)2 and S-Phos for the Preparation of 45c–g
- ∗
- 2-Nitro-3-phenylaniline (45a). Following general method C and starting from 3-bromo-2-nitroaniline (44a, 100 mg, 0.92 mmol) and phenylboronic acid (135 mg, 1.11 mmol) 45a was obtained as an orange solid (95 mg, 0.44 mmol, 48%). Analytical data are consistent with previously reported characterization (see method B). Besides 45a, 2-nitro-N1-(2-nitro-[1,1′-biphenyl]-3-yl)benzene-1,3-diamine was also isolated as a red solid (50 mg, 0.14 mmol, 30%).1H-NMR (400 MHz, CDCl3) δ ppm 6.03 (s, 2H), 6.26 (d, 1H, J = 8.2 Hz), 6.50 (d, 1H, J = 8.2 Hz), 7.08 (t, 1H, J = 8.2 Hz), 7.14 (d, 1H, J = 7.5 Hz), 7.35–7.48 (m, 6H), 7.52 (d, 1H, J = 7.5 Hz), 9.92 (s, 1H). 13C-NMR (101 MHz, CDCl3) δ ppm δ 104.5, 108.9, 122.9, 126.1, 127.7, 128.6, 128.9, 131.2, 134.1, 135.0, 136.8, 137.1, 141.8, 146.6.
- ∗
- 3-(4-Chlorophenyl)-2-nitroaniline (45b). Following general method C and starting from 3-bromo-2-nitroaniline (44a, 150 mg, 0.69 mmol) and 2-chlorophenylboronic acid (130 mg, 0.83 mmol), 45b was obtained as an orange solid (29 mg, 0.12 mmol, 17%). Analytical data are consistent with previously reported characterization (see method B). Besides 45b, 1-N-[3-(4-chlorophenyl)-2-nitrophenyl]-2-nitrobenzene-1,3-diamine was also isolated as a red solid (70 mg, 0.18 mmol, 52%). 1H-NMR (400 MHz, CDCl3) δ ppm 5.97 (br s, 2H), 6.23 (d, 1H, J = 8.4 Hz), 6.46 (d, 1H, J = 8.4 Hz), 7.02–7.06 (m, 2H), 7.21–7.26 (m, 2H), 7.35–7.37 (m, 2H), 7.41 (t, 1H, J = 8.0 Hz), 7.49 (d, 1H, J = 8.0 Hz), 9.88 (s, 1H). 13C-NMR (101 MHz, CDCl3) δ ppm 104.6, 109.2, 123.0, 125.8, 129.0, 129.2, 131.3, 134.4, 134.8, 134.9, 135.3, 136.0, 139.4, 141.5, 146.6.
- ∗
- 3-(4-Methoxyphenyl)-2-nitroaniline (45c). Following general method C and starting from 3-bromo-2-nitroaniline (44a, 150 mg, 0.69 mmol) and 4-methoxyphenylboronic acid (126 mg, 0.83 mmol), 45c was obtained as a yellow solid (112 mg, 0.46 mmol, 66%). 1H-NMR (400 MHz, CDCl3) δ ppm 3.87 (s, 3H), 4.93 (s, 2H), 6.72 (dd, 1H, J = 1.2 Hz, J = 7.5 Hz), 6.79 (dd, 1H, J = 1.2 Hz, J = 8.3 Hz), 6.96 (d, 2H, J = 8.8 Hz), 7.27 (d, 2H, J = 8.8 Hz), 7.27–7.31 (m, 1H); 13C-NMR (101 MHz, CDCl3) δ ppm 55.0, 113.8, 116.3, 120.2, 128.3, 130.6, 131.9, 137.6, 141.2, 159.1.
- ∗
- 3-(2-Chlorophenyl)-2-nitroaniline (45d). Following general method C and starting from 3-bromo-2-nitroaniline (44a, 150 mg, 0.69 mmol) and 2-chlorophenylboronic acid (130 mg, 0.83 mmol), 45d was obtained as a yellow solid (149 mg, 0.60 mmol, 87%). 1H-NMR (400 MHz, CDCl3) δ ppm 5.40 (s, 2H), 6.54 (dd, 1H, J = 1.3 Hz, J = 7.4 Hz), 6.82 (dd, 1H, J = 1.3 Hz, J = 8.3 Hz), 7.20–7.30 (m, 4H), 7.35–7.39 (m, 1H); 13C-NMR (101 MHz, CDCl3) δ ppm 118.4, 120.8, 126.8, 128.8, 129.3, 129.7, 132.4, 133.0, 136.5, 138.5, 143.0.
- ∗
- 3-(2,3-Dichlorophenyl)-2-nitroaniline (45e). Following general method C and starting from 3-bromo-2-nitroaniline (44a, 200 mg, 0.92 mmol) and 2,3-dichlorophenylboronic acid (211 mg, 1.11 mmol), 45e was obtained as a yellow solid (166 mg, 0.59 mmol, 64%). 1H-NMR (400 MHz, CDCl3) δ ppm 5.55 (s, 2H), 6.54 (dd, 1H, J = 1.3 Hz, J = 7.9 Hz), 6.87 (dd, 1H, J = 1.3 Hz, J = 7.9 Hz), 7.15 (dd, 1H, J = 1.6 Hz, J = 8.0 Hz), 7.24 (t, 1H, J = 7.9 Hz), 7.33 (t, 1H, J = 8.0 Hz), 7.45 (dd, 1H, J = 1.6 Hz, J = 8.0 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 118.7, 120.5, 127.3, 127.8, 129.6, 131.0, 133.1, 133.2, 136.3, 140.9, 143.4.
- ∗
- 3-(2,5-Dichlorophenyl)-2-nitroaniline (45f). Following general method C and starting from 3-bromo-2-nitroaniline (44a, 200 mg, 0.92 mmol) and 2,5-dichlorophenylboronic acid (211 mg, 1.11 mmol), 45f was obtained as a yellow solid (120 mg, 0.42 mmol, 46%). 1H-NMR (400 MHz, CDCl3) δ ppm 5.58 (s, 2H), 6.54 (dd, 1H, J = 1.3 Hz, J = 7.3 Hz), 6.88 (dd, 1H, J = 1.3 Hz, J = 8.4 Hz), 7.24–7.28 (m, 2H), 7.31–7.35 (m, 2H); 13C-NMR (101 MHz, CDCl3) δ ppm 119.0, 120.6, 128.8, 129.5, 130.3, 130.8, 132.6, 133.3, 135.4, 140.3, 143.4.
- ∗
- 3-(2-Methoxyphenyl)-2-nitroaniline (45g). Following general method C and starting from 3-bromo-2-nitroaniline (44a, 150 mg, 0.69 mmol) and 2-methoxyphenylboronic acid (126 mg, 0.83 mmol), 45g was obtained as a yellow solid (142 mg, 0.58 mmol, 84%). 1H-NMR (400 MHz, CDCl3) δ ppm 3.68 (s, 3H), 5.13 (s, 2H), 6.62 (dd, 1H, J = 1.3 Hz, J = 7.4 Hz), 6.75 (dd, 1H, J = 1.3 Hz, J = 8.3 Hz), 6.85 (d, 1H, J = 8.3 Hz), 7.02 (t, 1H, J = 7.4 Hz), 7.22–7.33 (m, 3H); 13C-NMR (101 MHz, CDCl3) δ ppm 55.3, 110.5, 117.6, 121.1, 121.2, 128.4, 129.3, 129.6, 132.8, 135.3, 141.9, 155.7.
Miscellaneous: Pd-Catalyzed Suzuki-Miyaura Cross-Coupling Using Pd(OAc)2 and S-Phos and K3PO4 for the Preparation of 15
- ∗
- 6-Benzylpyridin-2-amine (15). This preparation was adapted from the method described by Cee et al. [44]. A microwave vial (oven-dried and under argon) was charged with 2-amino-6-bromopyridine (2b, 100 mg, 0.58 mmol, 1.0 equiv.), B-Bn-9-BBN 0.5 M in THF (2.31 mL, 1.16 mmol, 2 equiv.), Pd(OAc)2 (6.5 mg, 0.03 mmol, 5 mol%), S-Phos (24 mg, 0.06 mmol, 10 mol%), K3PO4 (368 mg, 1.73 mmol, 3.0 equiv.) and anhydrous THF (3 mL). The vial was capped properly, flushed with argon and heated to 100 °C for 1.5 h. After it was cooled, the reaction mixture was concentrated under vacuum. The crude residue was diluted in water. The aqueous phase was extracted 3 times with EtOAc. The organic layers were combined, washed with brine, dried over Na2SO4, filtered, concentrated and purified by silica gel column chromatography (hexane:EtOAc 1:1 to 0:1), yielding 15 as a yellow solid (105 mg, 0.57 mmol, 99%). 1H-NMR (300 MHz, CDCl3) δ ppm 3.97 (s, 2H), 4.46 (br s, 2H), 6.32 (d, 1H, J = 8.1 Hz), 6.41 (d, 1H, J = 7.3 Hz), 7.20–7.35 (m, 6H); 13C-NMR (101 MHz, CDCl3) δ ppm 44.2, 106.1, 113.1, 126.2, 128.4, 129.2, 138.3.
4.1.5. General Method D: Pd-Catalyzed Suzuki-Miyaura Cross-Coupling Using Pd(OAc)2 and X-Phos Preparation of Compounds 4c, 6
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(4-trifluoromethylphenyl)pyridine (4c). Following general method D and starting from 3 (200 mg, 0.54 mmol) and 4-trifluoromethylphenylboronic acid (123 mg, 0.65 mmol), 4c was obtained as a white solid (220 mg, 0.46 mmol, 85%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.56 (s, 18H), 7.51 (d, 1H, J = 7.8 Hz), 7.70 (d, 2H, J = 8.3 Hz), 7.81 (t, 1H, J = 7.8 Hz), 8.12 (d, 2H, J = 8.3 Hz), 8.42 (br s, 1H), 10.91 (s, 1H), 11.59 (s, 1H); 19F-NMR (376 MHz, CDCl3) δ ppm −62.6; 13C-NMR DEPT-135 (101 MHz, CDCl3) δ ppm 28.1, 116.5, 125.5 (q, J = 4.0 Hz), 127.1, 139.2.
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(furan-3-yl)pyridine (4g). Following general method D and starting from 3 (150 mg, 0.40 mmol) and 3-furanboronic acid (54 mg, 0.48 mmol), 4g was obtained as a yellow solid (71 mg, 0.18 mmol, 44%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.54 (s, 18H), 6.89 (s, 1H), 7.17 (d, 1H, J = 7.7 Hz), 7.46 (t, 1H, J = 1.8 Hz), 7.68 (t, 1H, J = 7.7 Hz), 8.04 (br s, 1H), 8.27 (br s, 1H), 10.78 (s, 1H), 11.56 (s, 1H); 13C DEPT-135 NMR (101 MHz, CDCl3) δ ppm 28.1, 28.2, 108.6 (2C), 116.1, 138.8, 141.5, 143.8.
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(1-benzofuran-2-yl)pyridine (4h). Following general method D and starting from 3 (150 mg, 0.40 mmol) and benzofuran-2-boronic acid (79 mg, 0.48 mmol), 4h was obtained as a white solid (87 mg, 0.19 mmol, 47%). 1H-NMR (400 MHz, CDCl3) δ ppm 1.55 (s, 18H), 7.25 (t, 1H, J = 7.7 Hz), 7.33 (t, 1H, J = 8.2 Hz), 7.47 (s, 1H), 7.54 (d, 1H, J = 8.0 Hz), 7.61 (br s, 1H), 7.64 (d, 1H, J = 7.7 Hz), 7.80 (t, 1H, J = 7.7 Hz), 8.38 (br s, 1H), 10.88 (s, 1H), 11.59 (s, 1H); 13C-NMR (101 MHz, CDCl3) δ ppm 28.2, 78.6, 79.8, 105.1, 111.5, 114.2, 115.2, 121.6, 123.2, 125.2, 128.8, 139.0, 148.8, 152.7, 153.2, 153.8, 154.3, 154,7, 155.3.
- ∗
- 2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-(2-chloro-3-(trifluoromethyl)phenyl)pyridine (4i). Following general method D and starting from 3 (60 mg, 0.14 mmol) and 2-Chloro-3-(trifluoromethyl)-phenylboronic acid (42 mg, 0.19 mmol), 4i was obtained as an oil and the crude was immediately used in the deprotection step with TFA.
4.1.6. General Method E. Formation of Guanidinium Trifluoroacetate Salts. Preparation of cpds. 1c, 1g–j, 1l–m, 1o–u and 17
- ∗
- 1-[6-(4-Trifluoromethylphenyl)pyridin-2-yl] guanidinium trifluoroacetate (1c). Following general method E and starting from 4c (100 mg, 0.21 mmol), 1c was obtained as a white solid (81 mg, 0.20 mmol, 98%). Purity ≥ 98%; mp = 248–249 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.13 (d, 1H, J = 8.0 Hz), 7.81 (d, 1H, J = 8.0 Hz), 7.88 (d, 2H, J = 8.4 Hz), 8.03 (t, 1H, J = 8.0 Hz), 8.16 (d, 2H, J = 8.4 Hz), 8.53 (br s, 4H), 11.46 (s, 1H); 19F-NMR (376 MHz, CDCl3) δ ppm −74.0, −61.2; 13C-NMR (101 MHz, DMSO-d6) δ ppm 113.7, 117.5, 126.3 (q, J = 3.7 Hz), 128.0, 130.2 (q, J = 32.3 Hz), 141.2, 141.9, 152.6, 153.1, 155.8, 159.9 (q, J = 31.5 Hz); HRMS (M + H)+ 281.1003 (calcd for C13H11F3N4H+ 281.1009).
- ∗
- 1-[6-(Furan-3-yl)pyridin-2-yl] guanidinium trifluoroacetate (1g). Following general method E and starting from 4g (58 mg, 0.14 mmol), 1g was obtained as a white solid (36 mg, 0.11 mmol, 80%). Purity ≥ 98%; mp = 219–221 °C; 1H-NMR (400 MHz, DMSO-d6+D2O) δ ppm 6.90 (d, 1H, J = 7.9 Hz), 7.04 (d, 1H, J = 1.5 Hz), 7.48 (d, 1H, J = 7.9 Hz), 7.78 (t, 1H, J = 1.5 Hz), 7.87 (t, 1H, J = 7.9 Hz), 8.42 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 109.1, 111.9, 116.0, 126.1, 140.8, 143.0, 145.3, 149.1, 152.4, 155.8, 159.9 (q, J = 32.3 Hz); HRMS (M + H)+ 203.0914 (calcd for C10H10N4OH+ 203.0927).
- ∗
- 1-[6-(1-Benzofuran-2-yl)pyridin-2-yl] guanidinium trifluoroacetate (1h). Following general method E and starting from 4h (54 mg, 0.12 mmol), 1h was obtained as a white solid (31 mg, 0.08 mmol, 71%). Purity ≥ 98%; mp = 269–270 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.07 (d, 1H, J = 8.2 Hz), 7.33 (td, 1H, J = 1.3 Hz, J = 7.8 Hz), 7.42 (td, 1H, J = 1.3 Hz, J = 7.8 Hz), 7.68 (d, 1H, J = 8.2 Hz), 7.73–7.76 (m, 3H), 8.02 (t, 1H, J = 7.8 Hz), 8.53 (br s, 4H), 11.44 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 106.4, 112.0, 113.6, 115.6, 122.4, 124.1, 126.4, 128.7, 141.1, 145.8, 152.6, 153.7, 155.2, 155.8, 159.8 (q, J = 31.5 Hz); HRMS (M + H)+ 253.1076 (calcd for C14H12N4OH+ 253.1084).
- ∗
- 1-[6-(2-chloro-3-trifluoromethylphenyl)pyridin-2-yl] guanidinium trifluoroacetate (1i). Following general method D and starting from 3 (60 mg, 0.14 mmol) and 2-chloro-3-(trifluoromethyl)phenylboronic acid (42 mg, 0.19 mmol), 4i was obtained as an oil and without purification dissolved in a mixture of TFA:DCM (1:1, 1 mL) according to method E. The solution was stirred at room temperature for 2 h. The solution was concentrated under vacuum and purified by reverse chromatography (MeOH/H2O + 0.05%TFA) to yield 1i as a white solid (27 mg, 22%). 1H-NMR (400 MHz, DMSO-d6) δ 7.17 (d, J = 8.7 Hz, 1H), 7.52–7.42 (m, 1H), 7.71 (t, J = 7.8, 7.8 Hz, 1H), 7.90 (dd, J = 7.7, 1.3 Hz, 1H), 8.00 (dd, J = 7.9, 1.4 Hz, 1H), 8.05 (t, J = 7.9Hz, 1H), 8.35 (bs, 3H), 11.29 (s, 1H). 13C-NMR (101 MHz, DMSO-d6) δ 155.6, 153.1, 152.1, 140.8, 140.7, 136.0, 129.5, 128.9, 128.5, 128.3.
- ∗
- 1-[6-(2-Fluorophenyl)pyridin-2-yl] guanidiniumtrifluoroacetate (1j). Following general method E and starting from 4j (166 mg, 0.39 mmol), 1j was obtained as a white solid (107 mg, 0.31 mmol, 81%). Purity ≥ 98%; mp = 199–200 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.10 (d, 1H, J = 8.3 Hz), 7.35–7.40 (m, 2H), 7.50–7.57 (m, 2H), 7.81 (td, 1H, J = 1.8 Hz, J = 7.8 Hz), 8.00 (t, 1H, J = 7.9 Hz), 8.60 (br s, 4H), 11.55 (s, 1H); 19F-NMR (376 MHz, CDCl3) δ ppm −111.7, −74.0; 13C-NMR (101 MHz, DMSO-d6) δ ppm 112.9, 117.1 (d, J = 22.2 Hz), 119.7 (d, J = 5.1 Hz), 125.6 (d, J = 3.7 Hz), 126.4 (d, J = 10.3 Hz), 130.9 (d, J = 2.2 Hz), 131.9 (d, J = 8.8 Hz), 140.8, 150.7 (d, J = 2.2 Hz), 152.4, 155.9, 158.9, 160.3 (q, J = 32.3 Hz), 161.4; HRMS (M + H)+ 231.1034 (calcd for C12H11FN4H+ 231.1041).
- ∗
- 1-[6-(2-Methylphenyl)pyridin-2-yl] guanidiniumtrifluoroacetate (1l). Following general method E and starting from 4l (104 mg, 0.24 mmol), 1l was obtained as a white solid (76 mg, 0.22 mmol, 92%). Purity ≥ 95%; mp = 168–169 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 2.32 (s, 3H), 7.08 (d, 1H, J = 8.2 Hz), 7.29–7.36 (m, 4H), 7.40 (d, 1H, J = 7.3 Hz), 7.97 (t, 1H, J = 7.9 Hz), 8.58 (br s, 4H), 11.57 (s, 1H); 13C-NMR (101 MHz, DMSO) δ ppm 20.4, 112.0, 120.0, 126.6, 129.1, 129.8, 131.3, 135.6, 139.4, 140.5, 152.1, 156.0, 156.7, 160.4 (q, J = 32.3 Hz); HRMS (M + H)+ 227.1282 (calcd for C13H14N4H+ 227.1291).
- ∗
- 1-[6-(2-Trifluoromethylphenyl)pyridin-2-yl] guanidinium trifluoroacetate (1m). Following general method E and starting from 4m (291 mg, 0.61 mmol), 1m was obtained as a white solid (176 mg, 0.45 mmol, 74%). Purity ≥ 98%; mp = 157–158 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.15 (d, 1H, J = 7.9 Hz), 7.29 (d, 1H, J = 7.5 Hz), 7.60 (d, 1H, J = 7.5 Hz), 7.71 (t, 1H, J = 7.5 Hz), 7.80 (t, 1H, J = 7.5 Hz), 7.89 (d, 1H, J = 7.9 Hz), 8.01 (t, 1H, J = 7.9 Hz), 8.55 (br s, 4H), 11.63 (s, 1H); 19F-NMR (376 MHz, CDCl3) δ ppm −74.0, −55.7; 13C-NMR (101 MHz, DMSO-d6) δ ppm 113.0, 119.8, 124.6 (q, J = 273.6), 127.1 (q, J = 5.1 Hz), 127.1 (q, J = 30.8 Hz), 129.9, 132.1, 133.2, 138.7, 140.1, 151.9, 155.0, 155.8, 160.5 (q, J = 32.3 Hz); HRMS (M + H)+ 281.1007 (calcd for C13H11F3N4H+ 281.1009).
- ∗
- 1-[6-(2,3-Dichlorophenyl)pyridin-2-yl] guanidinium trifluoroacetate (1o). Following general method E and starting from 4o (153 mg, 0.32 mmol), 1o was obtained as a white solid (75 mg, 0.19 mmol, 60%). Purity ≥ 98%; mp = 190–191 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.15 (d, 1H, J = 8.0 Hz), 7.41 (d, 1H, J = 8.0 Hz), 7.49 (t, 1H, J = 7.9 Hz), 7.56 (dd, 1H, J = 1.6 Hz, J = 7.9 Hz), 7.75 (dd, 1H, J = 1.6 Hz, J = 7.9 Hz), 8.02 (t, 1H, J = 8.0 Hz), 8.59 (br s, 4H), 11.69 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 113.3, 120.4, 129.0, 129.9, 130.5, 131.4, 133.1, 140.3, 140.7, 152.1, 153.6, 155.9, 160.5 (q, J = 33.0 Hz); HRMS (M + H)+ 281.0341 (calcd for C12H10Cl2N4H+ 281.0355).
- ∗
- 1-[6-(2,4-Dichlorophenyl)pyridin-2-yl] guanidiniumtrifluoroacetate (1p). Following general method E and starting from 4p (205 mg, 0.43 mmol), 1p was obtained as a white solid (95 mg, 0.24 mmol, 57%). Purity ≥ 98%; mp = 196–197 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.13 (d, 1H, J = 8.0 Hz), 7.43 (d, 1H, J = 8.0 Hz), 7.57 (dd, 1H, J = 2.0 Hz, J = 8.3 Hz), 7.64 (d, 1H, J = 8.3 Hz), 7.78 (d, 1H, J = 2.0 Hz), 8.01 (t, 1H, J = 8.0 Hz), 8.57 (br s, 4H), 11.62 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 113.2, 120.4, 128.4, 130.2, 132.6, 133.2, 134.8, 136.8, 140.7, 152.2, 152.8, 155.8, 160.5 (q, J = 33.0 Hz); HRMS (M + H)+ 281.0348 (calcd for C12H10Cl2N4H+ 281.0355).
- ∗
- 1-[6-(2,5-Dichlorophenyl)pyridin-2-yl] guanidiniumtrifluoroacetate (1q). Following general method E and starting from 4q (109 mg, 0.23 mmol), 1q was obtained as a white solid (81 mg, 0.20 mmol, 91%). Purity = 95%; mp = 151–152 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.15 (d, 1H, J = 8.0 Hz), 7.45 (d, 1H, J = 8.0 Hz), 7.57 (dd, 1H, J = 2.5 Hz, J = 8.6 Hz), 7.65 (d, 1H, J = 8.6 Hz), 7.69 (d, 1H, J = 2.5 Hz), 8.01 (t, 1H, J = 8.0 Hz), 8.32 (br s, 4H), 11.71 (s, 1H); 13C-NMR (101 MHz, DMSO) δ ppm 113.4, 120.6, 130.4, 130.8, 131.4, 132.4, 132.7, 139.5, 140.7, 152.1, 152.6, 155.7, 160.2 (q, J = 32.3 Hz); HRMS (M + H)+ 281.0343 (calcd for C12H10Cl2N4H+ 281.0355).
- ∗
- 1-[6-(Pyridin-4-yl)pyridin-2-yl] guanidiniumtrifluoroacetate (1r). Following general method E and starting from 4r (88 mg, 0.21 mmol), 1s was obtained as a white solid (70 mg, 0.21 mmol, 100%). Purity ≥ 98%; mp = 208–209 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.21 (d, 1H, J = 8.0 Hz), 7.96 (d, 1H, J = 8.0 Hz), 8.08 (t, 1H, J = 8.0 Hz), 8.15 (d, 2H, J = 6.3 Hz), 8.57 (br s, 4H), 8.84 (d, 2H, J = 6.3 Hz), 11.61 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 115.2, 118.1, 122.3, 141.4, 147.4, 148.7, 151.2, 152.9, 155.7, 159.7 (q, J = 33.7 Hz); HRMS (M + H)+ 214.1070 (calcd for C11H11N5H+ 214.1087).
- ∗
- 1-[6-(Pyridin-3-yl)pyridin-2-yl] guanidinium trifluoroacetate (1s). Following general method E and starting from crude 4s (20 mg, 0.048 mmol), 1s was obtained as a white hygroscopic solid (11 mg, 0.02 mmol, 52%). Purity ≥ 97%; 1H-NMR (400 MHz, CD3OD) δ 7.10 (d, J = 7.9 Hz, 1H), 7.74 (d, J = 7.4 Hz, 1H), 7.90–7.83 (m, 1H), 7.95 (t, J = 8.0 Hz, 1H), 8.73 (d, J = 8.0 Hz, 2H), 9.22 (s, 1H). LC/MS (M + H)+ = 214.062.
- ∗
- 1-(2′-Chloro-[2,3′-bipyridin]-6-yl) guanidiniumtrifluoroacetate (1t). Following general method E and starting from 4t (120 mg, 0.027 mmol), 1t was obtained as a white hygroscopic solid (85 mg, 0.18 mmol, 66%). Purity ≥ 98%;1H-NMR (500 MHz, CD3OD) δ 7.16 (d, J = 8.2 Hz, 1H), 7.54–7.49 (m, 1H), 7.57 (dd, J = 7.6, 4.8 Hz, 1H), 8.05–7.99 (m, 1H), 8.07 (dd, J = 7.6, 1.9 Hz, 1H), 8.49 (dd, J = 4.8, 1.9 Hz, 1H); 13C-NMR (126 MHz, MeOD) δ 112.71, 123.17, 123.62, 134.48, 140.02, 140.28, 148.37, 149.44, 151.75, 152.59, 155.75. HRMS (M + H)+ 248.0703 (calcd for C11H10ClN5H+ 248.0698).
- ∗
- 1-(2′-Methoxy-[2,3′-bipyridin]-6-yl) guanidiniumtrifluoroacetate (1u). Following general method E and starting from 4u (234 mg, 0.53 mmol), 1u was obtained as a white hygroscopic solid (70 mg, 0.37 mmol, 70%). Purity ≥ 97%;1H-NMR (400 MHz, MeOD) δ4.03 (s, 3H), 7.03 (dd, J = 8.2, 0.6 Hz, 1H), 7.14 (dd, J = 7.5, 5.0 Hz, 1H), 7.67 (dd, J = 7.8, 0.6 Hz, 1H), 7.94 (t, J = 8.0 Hz, 1H), 8.07 (dd, J = 7.5, 1.9 Hz, 1H), 8.26 (dd, J = 5.0, 1.9 Hz, 1H); 13C-NMR (101 MHz, MeOD) δ 52.7, 111.5, 117.2, 119.5, 121.6, 138.8, 139.6, 147.4, 151.3, 152.2, 156.1, 160.8. HRMS (M + H)+ 244.1198 (calcd for C12H13N5OH+ 244.1189).
- ∗
- 1-(6-Benzylpyridin-2-yl) guanidiniumtrifluoroacetate (17). Following general method E and starting from 16 (73 mg, 0.17 mmol), 17 was obtained as a white solid (56 mg, 0.17 mmol, 96%). Purity ≥ 98%; mp = 161–162 °C; 1H-NMR (500 MHz, DMSO-d6) δ ppm 4.10 (s, 4H), 6.87 (d, 1H, J = 7.8 Hz), 7.10 (d, 1H, J = 7.8 Hz), 7.22 (t, 1H, J = 6.7 Hz), 7.27–7.33 (m, 4H), 7.79 (t, 1H, J = 7.8 Hz), 8.41 (br s, 4H), 11.21 (s, 1H); 13C-NMR (125 MHz, DMSO) δ ppm 43.2, 111.0, 118.7, 126.8, 129.0, 129.5, 139.6, 140.6, 152.1, 155.8, 158.9, 160.0 (q, J = 31.8 Hz); HRMS (M + H)+ 227.1285 (calcd for C13H14N4H+ 227.1291).
4.1.7. General Method F: Reduction of Aromatic Nitriles to Amines Using the comPlex BH3.SMe2: Preparation of cpd. 36
- ∗
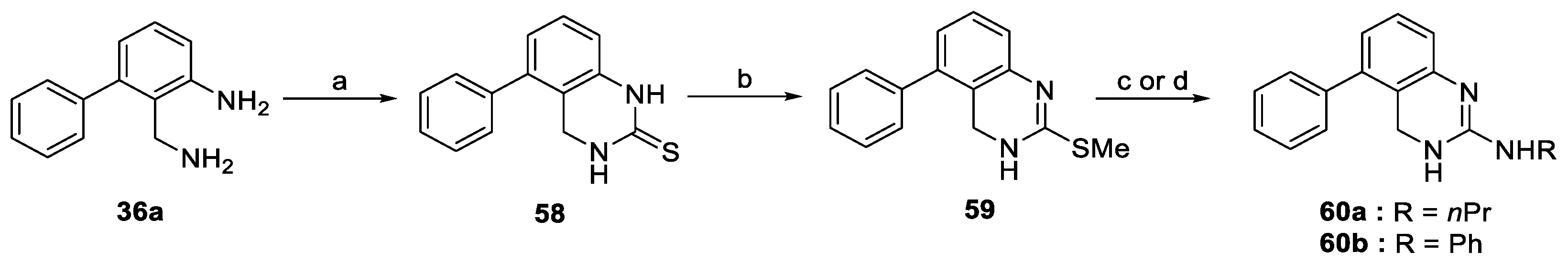
- 2-(Aminomethyl)-3-(4-chlorophenyl)aniline (36b). Following general method F and starting from 35b (100 mg, 0.44 mmol), 36b was obtained as a yellow solid (74 mg, 0.32 mmol, 73%). 1H-NMR (400 MHz, CDCl3) δ ppm 2.77 (br s, 4H), 3.78 (s, 2H), 6.61 (d, 1H, J = 7.8 Hz), 6.71 (d, 1H, J = 7.8 Hz), 7.10 (t, 1H, J = 7.8 Hz), 7.21 (d, 2H, J = 8.4 Hz), 7.36 (d, 2H, J = 8.4 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 40.6, 115.5, 119.9, 123.4, 127.6, 128.2, 130.4, 132.9, 140.5, 141.6, 147.4.
- ∗
- 2-(Aminomethyl)-3-(4-methoxyphenyl)aniline (36c). Following general method F and starting from 35c (100 mg, 0.45 mmol), 36c was obtained as a colorless oil (77 mg, 0.34 mmol, 76%). 1H-NMR (400 MHz, CDCl3) δ ppm 3.85 (s, 3H), 6.64 (dd, 1H, J = 0.9 Hz, J = 7.7 Hz), 6.69 (dd, 1H, J = 0.9 Hz, J = 7.7 Hz), 6.93 (d, 2H, J = 8.7 Hz), 7.09 (t, 1H, J = 7.7 Hz), 7.20 (d, 2H, J = 8.7 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 41.7, 55.3, 113.4, 119.1, 120.3, 127.5, 130.1, 134.4, 142.5, 147.2, 158.7.
- ∗
- 2-(Aminomethyl)-3-(3-chlorophenyl)aniline (36d). Following general method F and starting from 35d (100 mg, 0.44 mmol), 36d was obtained as a colorless oil (77 mg, 0.33 mmol, 75%). 1H-NMR (400 MHz, CDCl3) δ ppm 2.88 (br s, 4H), 3.78 (s, 2H), 6.61 (d, 1H, J = 7.7 Hz), 6.71 (d, 1H, J = 7.7 Hz), 7.10 (t, 1H, J = 7.7 Hz), 7.14–7.17 (m, 1H), 7.28–7.33 (m, 3H); 13C-NMR (101 MHz, CDCl3) δ ppm 40.6, 115.6, 119.8, 123.2, 127.0, 127.3, 127.7, 129.1, 129.2, 133.9, 141.4, 143.8, 147.3.
- ∗
- 2-(Aminomethyl)-3-(3-methoxyphenyl)aniline (36e). Following general method F and starting from 35e (100 mg, 0.45 mmol), 36e was obtained as a yellow oil (72 mg, 0.32 mmol, 71%). 1H-NMR (400 MHz, CDCl3) δ ppm 3.80 (s, 2H), 3.83 (s, 3H), 6.66 (dd, 1H, J = 1.0 Hz, J = 7.8 Hz), 6.70 (dd, 1H, J = 1.0 Hz, J = 7.8 Hz), 6.83–6.91 (m, 3H), 7.10 (t, 1H, J = 7.8 Hz), 7.31 (t, 1H, J = 7.9 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 40.6, 55.3, 112.3, 114.8, 115.2, 119.9, 121.6, 123.5, 127.5, 128.9, 142.7, 143.5, 147.2, 159.2.
- ∗
- 2-(Aminomethyl)-3-(2-chlorophenyl)aniline (36f). Following general method F and starting from 35f (200 mg, 0.87 mmol), 36f was obtained as a yellow solid (292 mg, 0.84 mmol, 96%). 1H-NMR (400 MHz, DMSO-d6 + D2O) δ ppm 6.47 (dd, 1H, J = 1.0 Hz, J = 7.8 Hz), 6.86 (dd, 1H, J = 1.0 Hz, J = 7.8 Hz), 7.17 (t, 1H, J = 7.8 Hz), 7.35–7.38 (m, 1H), 7.40–7.44 (m, 2H), 7.51–7.54 (m, 1H); 13C-NMR (101 MHz, DMSO) δ ppm 36.9, 115.9, 116.3, 127.7, 129.7, 129.9, 130.0, 132.1, 132.7, 139.4, 141.0, 148.0.
Miscellaneous: Reduction of Aromatic Nitrile with the Use of LiAlH4 and AlCl3: Preparation of cpd. 36g
- ▪
- 2-(Aminomethyl)-3-(2-methoxyphenyl)aniline (36g). In a two-neck round bottom-flask (oven dried and under argon), anhydrous THF (15.0 mL) was introduced and LiAlH4 (406 mg, 10.7 mmol, 12 equiv.) was added slowly followed by AlCl3 (357 mg, 2.67 mmol, 3 equiv.). The reaction mixture was cooled at 0 °C and stirred 5 min. A solution of 35g (200 mg, 0.89 mmol, 1 equiv.) in anhydrous THF (5.0 mL) was added dropwise. The resulting solution was stirred overnight at rt. Water was added dropwise followed by H2SO4 (6N). The solution was stirred 30 min, filtered and concentrated under vacuum. The residue was basified until pH 9 using NaOH. The resulting solid was filtered and washed with water, yielding to 36g as a brown solid (143 mg, 0.63 mmol, 70%).1H-NMR (400 MHz, CDCl3) δ ppm 2.83 (br s, 4H), 3.67 (s, 3H), 3.69 (s, 2H), 6.48–6.52 (m, 1H), 6.62 (d, 1H, J = 7.8 Hz), 6.85–6.88 (m, 1H), 6.91–6.95 (m, 1H), 7.09 (t, 2H, J = 7.0 Hz), 7.25 (t, 1H, J = 7.8 Hz); 13C-NMR (101 MHz, CDCl3) δ ppm 41.1, 55.6, 110.7, 114.2, 119.3, 120.7, 127.8, 128.5, 131.0, 131.3, 136.9, 138.8, 142.3, 156.5.
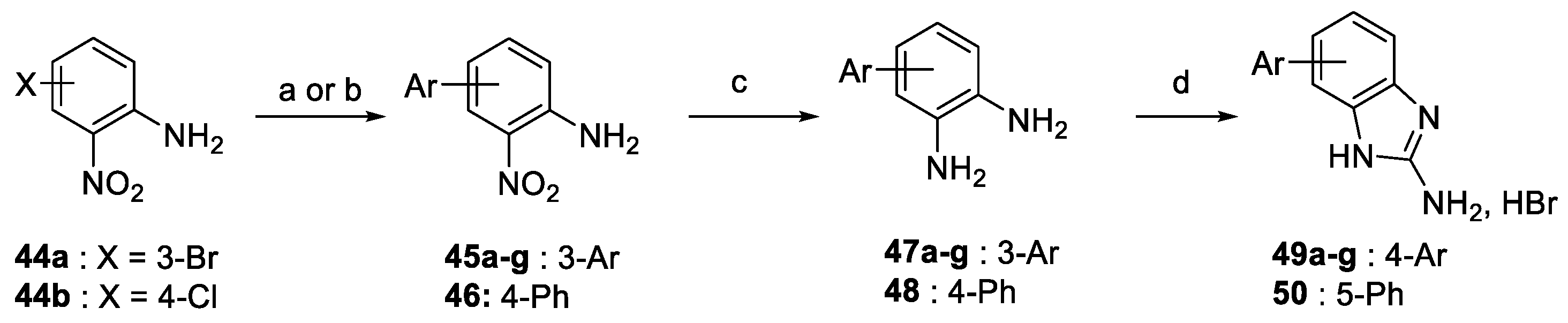
4.1.8. General Method G: Reduction of Aromatic Nitro Compounds Using Tin and HCl. Preparation of cpds. 47a–g
- ∗
- 3-Phenylbenzene-1,2-diamine (47a). Following general method G and starting from 45a (70 mg, 0.33 mmol), 47b was obtained as an orange oil (61 mg, 0.33 mmol, 100%).1H-NMR (400 MHz, DMSO-d6) δ ppm 5.45 (br s, 4H), 6.50 (d, 1H, J = 7.5 Hz), 6.57 (t, 1H, J = 7.5 Hz), 6.72 (d, 1H, J = 7.5 Hz), 7.32–7.47 (m, 5H). 13C-NMR (101 MHz, DMSO-d6) δ ppm 116.3, 118.3, 121.5, 127.3, 127.9, 129.2, 129.3, 132.7, 132.9, 140.4. Analytical data are consistent with the previously reported characterization [43].
- ∗
- 3-(4-Chlorophenyl)benzene-1,2-diamine (47b). Following general method G and starting from 45b (85 mg, 0.34 mmol), 47b was obtained as a white solid (51 mg, 0.23 mmol, 68%). 1H-NMR (400 MHz, DMSO-d6) δ ppm 4.12 (s, 2H), 4.61 (s, 2H), 6.33 (d, 1H, J = 7.5 Hz), 6.49 (t, 1H, J = 7.5 Hz), 6.57 (d, 1H, J = 7.5 Hz), 7.39 (d, 2H, J = 8.3 Hz), 7.47 (d, 2H, J = 8.3 Hz); 13C-NMR (101 MHz, DMSO-d6) δ ppm 114.6, 118.1, 119.2, 125.7, 129.1, 131.1, 131.7, 131.8, 136.0, 139.8. Analytical data are consistent with the previously reported characterization [45].
- ∗
- 3-(4-Methoxyphenyl)benzene-1,2-diamine (47c). Following general method G and starting from 45c (80 mg, 0.33 mmol), 47c was obtained as an orange oil (60 mg, 0.28 mmol, 86%). 1H-NMR (400 MHz, DMSO-d6) δ ppm 3.78 (s, 3H), 4.27 (br s, 4H), 6.33 (dd, 1H, J = 1.5 Hz, J = 7.7 Hz), 6.47 (t, 1H, J = 7.7 Hz), 6.54 (dd, 1H, J = 1.5 Hz, J = 7.7 Hz), 6.99 (d, 2H, J = 8.7 Hz), 7.29 (d, 2H, J = 8.7 Hz); 13C-NMR (101 MHz, DMSO-d6) δ ppm 55.5, 114.2, 114.6, 118.0, 119.4, 126.9, 130.3, 131.8, 133.0, 135.9, 158.5.
- ∗
- 3-(2-Chlorophenyl)benzene-1,2-diamine (47d). Following general method G and starting from 45d (193 mg, 0.78 mmol), 47d was obtained as an orange solid (172 mg, 0.78 mmol, 100%). 1H-NMR (400 MHz, DMSO-d6) δ ppm 3.86 (s, 2H), 4.60 (s, 2H), 6.24 (dd, 1H, J = 1.5 Hz, J = 7.5 Hz), 6.48 (t, 1H, J = 7.5 Hz), 6.58 (dd, 1H, J = 1.5 Hz, J = 7.5 Hz), 7.26–7.30 (m, 1H), 7.35–7.42 (m, 2H), 7.52–7.55 (m, 1H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 114.6, 117.6, 119.0, 124.8, 127.9, 129.4, 130.1, 132.2, 132.4, 135.8, 139.2.
- ∗
- 3-(2,3-Dichlorophenyl)benzene-1,2-diamine (47e). Following general method G and starting from 45e (137 mg, 0.48 mmol), 47e was obtained as an orange solid (122 mg, 0.48 mmol, 100%). 1H-NMR (400 MHz, DMSO-d6) δ ppm 5.01 (br s, 4H), 6.33 (dd, 1H, J = 1.1 Hz, J = 7.5 Hz), 6.51 (t, 1H, J = 7.5 Hz), 6.68 (dd, J = 1.1 Hz, J = 7.5 Hz), 7.24 (dd, 1H, J = 1.4 Hz, J = 7.9 Hz), 7.40 (t, 1H, J = 7.9 Hz), 6.62 (dd, 1H, J = 1.4 Hz, J = 7.8 Hz); 13C-NMR (101 MHz, DMSO-d6) δ ppm 116.1, 117.5, 120.3, 124.8, 128.8, 130.0, 131.1, 131.8, 132.6, 133.0, 133.5, 141.5.
- ∗
- 3-(2,5-Dichlorophenyl)benzene-1,2-diamine (47f). Following general method G and starting from 45f (90 mg, 0.32 mmol), 47f was obtained as an orange solid (80 mg, 0.32 mmol, 100%). 1H-NMR (400 MHz, DMSO-d6) δ ppm 4.00 (s, 2H), 4.63 (s, 2H), 6.23 (dd, 1H, J = 1.5 Hz, J = 7.5 Hz), 6.47 (t, 1H, J = 7.5 Hz), 6.58 (dd, 1H, J = 1.5 Hz, J = 7.5 Hz), 7.30 (d, 1H, J = 2.5 Hz), 7.43 (dd, 1H, J = 2.5 Hz, J = 8.7 Hz), 7.56 (d, 1H, J = 8.7 Hz); 13C-NMR (101 MHz, DMSO-d6) δ ppm 114.9, 117.5, 118.8, 123.4, 129.2, 131.7, 131.9, 132.1, 132.3, 132.4, 135.9, 141.2.
- ∗
- 3-(2-Methoxyphenyl)benzene-1,2-diamine (47g). Following general method G and starting from 45g (80 mg, 0.33 mmol), 47g was obtained as an orange solid (69 mg, 0.32 mmol, 98%). 1H-NMR (400 MHz, DMSO-d6) δ ppm 3.72 (s, 3H), 4.74 (br s, 4H), 6.32 (dd, 1H, J = 1.5 Hz, J = 7.5 Hz), 6.49 (t, 1H, J = 7.5 Hz), 6.60 (dd, 1H, J = 1.5 Hz, J = 7.5 Hz), 7.00 (t, 1H, J = 7.3 Hz), 7.07–7.11 (m, 2H), 7.24 (td, 1H, J = 1.9 Hz, J = 8.4 Hz); 13C-NMR (101 MHz, DMSO-d6) δ ppm 55.7, 112.0, 114.9, 117.8, 120.8, 121.1, 125.1, 129.0, 129.1, 131.7, 132.8, 134.6, 156.9.
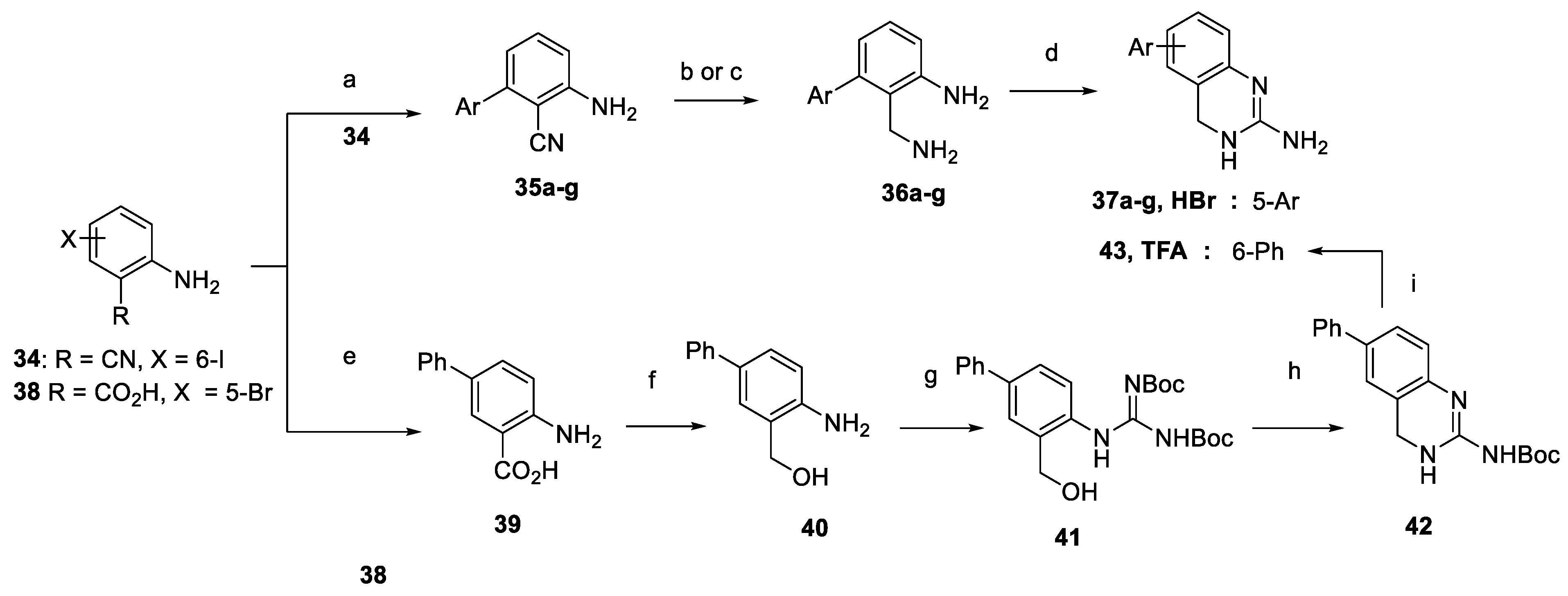
4.1.9. General Method H. Cyclisation of a Diamino Derivatives 36 and 47 Using BrCN: Preparation of Dihydroquinazolines 37 and Benzimidazoles 49
- ∗
- 5-(4-Chlorophenyl)-3,4-dihydroquinazolin-2-amine Hydrobromide (37b). Following general method H and starting from 36b (38 mg, 0.17 mmol), 37b was obtained as a white solid (44 mg, 0.13 mmol, 79%). Purity ≥ 98 %; mp = 196–197 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 4.36 (s, 2H), 7.05 (d, 1H, J = 7.5 Hz), 7.06 (d, 1H, J = 7.5 Hz), 7.36 (t, 1H, J = 7.5 Hz), 7.37 (d, 2H, J = 8.4 Hz), 7.54 (d, 2H, J = 8.4 Hz), 7.56 (s, 2H), 8.14 (s, 1H), 10.64 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 40.2, 115.5, 116.5, 125.8, 129.0, 129.1, 131.0, 133.3, 134.2, 137.7, 138.8, 153.1; HRMS (M + H)+ 258.0782 (calcd for C14H12ClN3H+ 258.0793).
- ∗
- 5-(4-Methoxyphenyl)-3,4-dihydroquinazolin-2-amine Hydrobromide (37c). Following general method H and starting from 36c (48 mg, 0.22 mmol), 37c was obtained as a white solid (51 mg, 0.16 mmol, 71%). Purity ≥ 98%; 1H-NMR (400 MHz, DMSO-d6) δ ppm 3.80 (s, 3H), 4.37 (s, 2H), 7.00–7.04 (m, 4H), 7.26 (d, 2H, J = 8.4 Hz), 7.33 (t, 1H, J = 7.8 Hz), 7.55 (s, 2H), 8.15 (s, 1H), 10.62 (s, 1H); 13C-NMR (101 MHz, DMSO) δ ppm 40.4, 55.7, 114.5, 114.8, 116.5, 125.9, 128.8, 130.3, 131.1, 134.1, 139.9, 153.0, 159.4; HRMS (M + H)+ 254.1281 (calcd for C15H15N3OH+ 254.1288).
- ∗
- 5-(3-Chlorophenyl)-3,4-dihydroquinazolin-2-amine Hydrobromide (37d). Following general method H and starting from 36d (15 mg, 0.065 mmol), 37d was obtained as a white solid (11 mg, 0.032 mmol, 49%). Purity ≥ 98%; mp = 228–230 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 4.38 (s, 2H), 7.07 (d, 2H, J = 7.8 Hz), 7.31 (t, 1H, J = 3.8 Hz), 7.38 (t, 1H, J = 7.8 Hz), 7.42 (s, 1H), 7.50–7.52 (m, 2H), 7.56 (s, 2H), 8.07 (s, 1H), 10.61 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 115.7, 116.5, 125.9, 127.9, 128.3, 128.8, 129.0, 130.9, 133.7, 134.2, 138.6, 141.0, 153.0; HRMS (M + H)+ 258.0780 (calcd for C14H12ClN3H+ 258.0793).
- ∗
- 5-(3-Methoxyphenyl)-3,4-dihydroquinazolin-2-amine Hydrobromide (37e). Following general method H and starting from 36e (19 mg, 0.081 mmol), 37e was obtained as a yellow solid (18 mg, 0.053 mmol, 65%). Purity ≥ 98%; mp = 198–199 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 3.79 (s, 3H), 4.38 (s, 2H), 6.86–6.89 (m, 2H), 6.99 (dd, 1H, J = 2.5 Hz, J = 8.0 Hz), 7.03–7.07 (m, 2H), 7.33–7.40 (m, 2H), 7.57 (s, 2H), 8.12 (s, 1H), 10.65 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 40.3, 55.7, 113.8, 114.7, 115.2, 116.4, 121.3, 125.8, 128.9, 130.1, 134.1, 140.0, 140.3, 153.1, 159.7; HRMS (M + H)+ 254.1280 (calcd for C15H15N3OH+ 254.1288).
- ∗
- 5-(2-Chlorophenyl)-3,4-dihydroquinazolin-2-amine Hydrobromide (37f). Following general method H and starting from 36f (62 mg, 0.27 mmol), 37f was obtained as a white solid (61 mg, 0.18 mmol, 68%). Purity ≥ 98%; mp = 178–179 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 4.07 (d, 1H, J = 14.7 Hz), 4.15 (d, 1H, J = 14.7 Hz), 6.96 (d, 1H, J = 7.9 Hz), 7.08 (d, 1H, J = 7.9 Hz), 7.32 (dd, 1H, J = 1.9 Hz, J = 6.9 Hz), 7.37 (t, 1H, J = 7.9 Hz), 7.42–7.50 (m, 2H), 7.58–7.61 (m, 3H), 8.11 (s, 1H), 10.69 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 40.1, 115.6, 115.7, 116.8, 125.8, 128.0, 128.9, 130.0, 130.6, 131.5, 132.3, 133.9, 137.3, 137.4, 152.8; HRMS (M + H)+ 258.0788 (calcd for C14H12ClN3H+ 258.0793).
- ∗
- 5-(2-Methoxyphenyl)-3,4-dihydroquinazolin-2-amine Hydrobromide (37g). Following general method H and starting from 36g (20 mg, 0.086 mmol), 37g was obtained as an orange solid (20 mg, 0.061 mmol, 70%). Purity ≥ 98%; mp = 226–229 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 3.37 (s, 3H), 4.05 (d, 1H, J = 14.6 Hz), 4.17 (d, 1H, J = 14.6 Hz), 6.94 (d, 1H, J = 7.5 Hz), 7.01 (d, 1H, J = 8.0 Hz), 7.05 (d, 1H, J = 7.5 Hz), 7.10–7.14 (m, 2H), 7.31 (t, 1H, J = 8.0 Hz), 7.42 (td, 1H, J = 1.8 Hz, J = 8.0 Hz), 7.55 (br s, 2H), 8.06 (br s, 1H), 10.60 (br s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 40.2, 55.9, 111.8, 114.9, 117.6, 121.1, 126.4, 127.4, 128.6, 130.2, 130.9, 133.7, 137.1, 153.0, 156.4; HRMS (M + H)+ 254.1281 (calcd for C15H15N3OH+ 254.1288).
- ∗
- 4-Phenyl-1H-benzo[d]imidazol-2-amine hydrobromide (49a). Following general method H and starting from 47a (153 mg, 0.83 mmol), 49a was obtained as a yellow solid (178 mg, 0.61 mmol, 74%). 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.27 (d, 1H, J = 7.7 Hz), 7.33 (t, 1H, J = 7.7 Hz), 7.39 (d, 1H, J = 7.7 Hz), 7.48 (t, 1H, J = 7.0 Hz), 7.54–7.61 (m, 4H), 8.06 (s, 2H), 12.50 (s, 2H). 13C-NMR (101 MHz, DMSO-d6) δ ppm 111.2, 123.6, 124.2, 125.8, 127.3, 128.6, 129.7, 130.8, 136.8, 151.7. Analytical data are consistent with previously reported characterization [46].
- ∗
- 4-(4-Chlorophenyl)-1H-benzo[d]imidazol-2-amine Hydrobromide (49b). Following general method H and starting from 47b (39 mg, 0.18 mmol), 49b was obtained as a white solid (29 mg, 0.09 mmol, 49%). Purity ≥ 98%; mp = 254–256 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.26 (dd, J = 0.9 Hz, J = 7.8 Hz), 7.33 (t, 1H, J = 7.8 Hz), 7.40 (dd, 1H, J = 0.9 Hz, J = 7.8 Hz), 7.61 (s, 4H), 8.12 (s, 2H), 12.54 (s, 2H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 111.5, 123.6, 124.2, 124.6, 127.5, 129.6, 130.6, 130.9, 133.4, 135.6, 151.7; HRMS (M + H)+ 244.0626 (calcd for C13H10ClN3H+ 244.0636).
- ∗
- 4-(4-Methoxyphenyl)-1H-benzo[d]imidazol-2-amine Hydrobromide (49c). Following general method H and starting from 47c (48 mg, 0.22 mmol), 49c was obtained as a white solid (51 mg, 0.16 mmol, 71%). Purity ≥ 98%; mp = 218–220 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 3.83 (s, 3H), 7.11 (d, 2H, J = 8.7 Hz), 7.22 (d, 1H, J = 7.2 Hz), 7.29 (t, 1H, J = 7.2 Hz), 7.35 (d, 1H, J = 7.2 Hz), 7.53 (d, 2H, J = 8.7 Hz), 8.05 (s, 2H), 12.46 (s, 2H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 55.8, 110.6, 115.1, 123.4, 124.2, 125.6, 127.2, 129.0, 129.8, 130.7, 131.6, 159.7; HRMS (M + H)+ 240.1124 (calcd for C14H13N3OH+ 240.1131).
- ∗
- 4-(2-Chlorophenyl)-1H-benzo[d]imidazol-2-amine Hydrobromide (49d). Following general method H and starting from 47d (50 mg, 0.23 mmol), 49d was obtained as an orange solid (35 mg, 0.11 mmol, 48%). Purity ≥ 98%; mp = 255–256 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.13 (d, 1H, J = 7.5 Hz), 7.32 (t, 1H, J = 7.5 Hz), 7.43–7.54 (m, 4H), 7.65 (d, 1H, J = 7.3 Hz), 8.15 (s, 2H), 12.54 (s, 2H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 111.7, 123.4, 123.7, 124.8, 128.1, 128.2, 130.2, 130.3, 130.7, 132.3, 132.9, 135.6, 151.4; HRMS (M + H)+ 244.0629 (calcd for C13H10ClN3H+ 244.0636).
- ∗
- 4-(2,3-Dichlorophenyl)-1H-benzo[d]imidazol-2-amine Hydrobromide (49e). Following general method H and starting from 47e (122 mg, 0.48mmol), 49e was obtained as an orange solid (53 mg, 0.15 mmol, 31%). Purity ≥ 98%; mp = 314–315 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.14 (d, 1H, J = 7.9 Hz), 7.32 (t, 1H, J = 7.9 Hz), 7.43 (dd, 1H, J = 1.5 Hz, J = 7.9 Hz), 7.46 (d, 1H, J = 7.9 Hz), 7.51 (t, 1H, J = 7.9 Hz), 7.78 (dd, 1H, J = 1.5 Hz, J = 7.9 Hz), 8.24 (s, 2H), 12.55 (s, 1H), 12.61 (s, 1H); 13C-NMR (101 MHz, DMSO) δ ppm 112.1, 123.1, 123.8, 124.5, 128.2, 129.0, 130.3, 131.0, 131.1, 131.3, 132.7, 138.1, 151.4; HRMS (M + H)+ 278.0235 (calcd for C13H9Cl2N3H+ 278.0246).
- ∗
- 4-(2,5-Dichlorophenyl)-1H-benzo[d]imidazol-2-amine Hydrobromide (49f). Following general method H and starting from 47f (80 mg, 0.32 mmol), 49f was obtained as a white solid (70 mg, 0.19 mmol, 61%). Purity ≥ 98%; mp = 202–205 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 7.15 (d, 1H, J = 7.8 Hz), 7.31 (t, 1H, J = 7.8 Hz), 7.45 (d, 1H, J = 7.8 Hz), 7.55–7.61 (m, 2H), 7.68 (d, 1H, J = 8.5 Hz), 8.26 (s, 2H), 12.65 (s, 2H); 13C-NMR (101 MHz, DMSO-d6) δ ppm 112.1, 122.0, 123.6, 124.7, 128.3, 130.4, 131.7, 131.8, 132.4, 137.4, 151.5; HRMS (M + H)+ 278.0240 (calcd for C13H9Cl2N3H+ 278.0246).
- ∗
- 4-(2-Methoxyphenyl)-1H-benzo[d]imidazol-2-amine Hydrobromide (49g). Following general method H and starting from 47g (55 mg, 0.26 mmol), 49g was obtained as an orange solid (44 mg, 0.14 mmol, 54%). Purity ≥ 95%; mp = 61–66 °C; 1H-NMR (400 MHz, DMSO-d6) δ ppm 3.77 (s, 3H), 7.09 (t, 1H, J = 7.4 Hz), 7.14 (d, 1H, J = 7.4 Hz), 7.19 (d, 1H, J = 7.9 Hz), 7.28 (t, 1H, J = 7.9 Hz), 7.33 (d, 1H, J = 7.4 Hz), 7.37 (d, 1H, J = 7.9 Hz), 7.47 (t, 1H, J = 7.4 Hz), 8.05 (s, 2H), 12.09 (s, 1H), 12.48 (s, 1H); 13C-NMR (101 MHz, DMSO) δ ppm 55.9, 110.9, 112.0, 121.1, 123.1, 123.7, 124.8, 125.3, 128.2, 129.9, 130.4, 131.3, 151.0, 156.7; HRMS (M + H)+ 240.1126 (calcd for C14H13N3OH+ 240.1131).
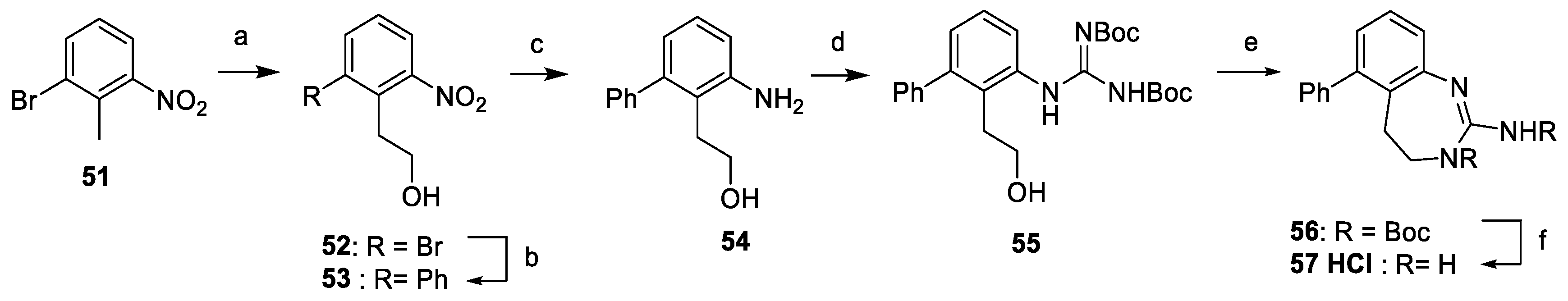
4.1.10. Preparation of Dihydrobenzodiazepine 57
- ∗
- (6-Bromo-2-nitrophenyl)ethanol (52). Paraformaldehyde (114 mg, 3.80 mmol, 1.0 equiv.), 4-bromo-6-nitrotoluene 51 (2.00 g, 9.26 mmol, 2.44 equiv.) and Triton-B (114 µL, 40% in methanol) was dissolved in DMSO (2.0 mL). The resulting mixture was heated overnight at 90 °C. After it was cooled, the reaction mixture was diluted with a saturated solution of NH4Cl. The aqueous phase was extracted twice with EtOAc. The organic layers were combined, washed with brine, dried over Na2SO4, filtered, concentrated and purified by silica gel column chromatography (hexane:EtOAc 3:1 to 1:2), yielding to 52 as a white solid (720 mg, 2.93 mmol, 77%). 1H-NMR (300 MHz, CDCl3) δ ppm 3.31 (t, 2H, J = 6.9 Hz), 3.97 (t, 2H, J = 6.9 Hz), 7.26 (t, 1H, J = 7.8 Hz), 7.75 (d, 1H, J = 7.8 Hz), 7.83 (d, 1H, J = 7.8 Hz).
- ∗
- (2-Nitro-6-phenylphenyl)ethanol (53). A microvawe vial under argon was charged with 52 (490 mg, 1.99 mmol, 1 equiv.), phenylboronic acid (267 mg, 2.19 mmol, 1.1 equiv.), Pd(OAc)2 (9 mg, 0.04 mmol, 20 mol%), K2CO3 (688 mg, 4.98 mmol, 2.5 equiv.) and TBAB (642 mg, 1.99 mmol, 1 equiv.) in water (2.2 mL). The vial was capped properly, flushed with argon and heated to 70°C for 3 h. After it was cooled, the reaction mixture was filtered through a pad of Celite®, washed with EtOAc, concentrated under vacuum and purified by silica gel column chromatography (hexane:EtOAc 2:1), yielding to 53 a white solid (441 mg, 1.81 mmol, 91%). 1H-NMR (300 MHz, CDCl3 ) δ ppm 1.39 (t, 1H, J = 5.9 Hz), 3.11 (t, 1H, J = 6.9 Hz), 3.64 (q, 1H, J = 6.5 Hz), 7.27–7.31 (m, 2H), 7.42–7.49 (m, 5H), 7.81 (d, 1H, J =5 7.5 Hz).
- ∗
- (2-Amino-6-phenylphenyl)ethanol (54). Following general method H and starting from 53 (420 mg, 1.73 mmol), 54 was obtained as a purple solid (367 mg, 1.73 mmol, 100%). 1H-NMR (400 MHz, CDCl3) δ ppm 2.79 (t, 2H, J = 6.4 Hz), 3.75 (t, 2H, J = 6.4 Hz), 4.02 (br s, 2H), 6.70 (d, 1H, J = 7.6 Hz), 6.75 (d, 1H, J = 7.6 Hz), 7.10 (t, 1H, J = 7.6 Hz), 7.27–7.43 (m, 5H).
- ∗
- {2-[2,3-Di(tert-butoxycarbonyl)guanidino]-6-phenylphenyl}ethanol (55). Following general method A and starting from 54 (100 mg, 0.47 mmol), 55 was obtained as a white solid (210 mg, 0.46 mmol, 98%). 1H-NMR (300 MHz, CDCl3) δ ppm 1.49 (s, 9H), 1.57 (s, 9H), 2.87 (t, 2H, J = 6.6 Hz), 3.61 (t, 2H, J = 6.6 Hz), 7.07 (d, 1H, J = 7.5 Hz), 7.26–7.44 (m, 6H), 7.76 (d, 1H, J = 7.5 Hz), 10.25 (s, 1H), 11.75 (s, 1H).
- ∗
- 6-Phenyl-3,4-dihydrobenzodiazepin-2-amine Hydrochloride (57). Compound 55 (105 mg, 0.23 mmol, 1 equiv.) was dissolved in THF (5.0 mL), and PPh3 (121 mg, 0.46 mmol, 2 equiv.) and DIAD (89.0 µL, 0.46 mmol, 2 equiv.) were added. The resulting mixture was stirred at rt for 1.5 h. The solution was concentrated under vacuum and purified by silica gel column chromatography (hexane:EtOAc 3:1). The intermediate 56 was diluted in a solution of HCl (4.0 N) in dioxane. The reaction mixture was stirred overnight at rt. The solution was concentrated under vacuum, yielding to 57 as a white solid (31 mg, 0.11 mmol, 49%). Purity ≥ 98 %; mp = 207–209 °C; 1H-NMR (500 MHz, DMSO-d6) δ ppm 3.21 (t, 2H, J = 8.1 Hz), 4.03 (t, 2H, J = 8.1 Hz), 7.16 (t, 1H, J = 4.6 Hz), 7.37–7.39 (m, 2H), 7.41 (m, 1H), 7.49 (m, 4H), 8.02 (br s, 4H); 13C-NMR (125 MHz, DMSO-d6) δ ppm 27.8, 51.2, 114.0, 124.9, 128.1, 128.5, 128.6, 129.1, 131.4, 139.2, 139.5, 141.3, 154.6; HRMS (M + H)+ 238.1331 (calcd for C15H15N3H+ 238.1339).
4.2. Biology
4.2.1. High-Throughput Screening and Assays for Hit Validation
4.2.2. In Vitro Evaluation of the Effect of MSK1 Inhibitors
4.2.3. Cytotoxicity Test
4.2.4. Evaluation of the In Vivo Toxicity of MSK1 Inhibitors
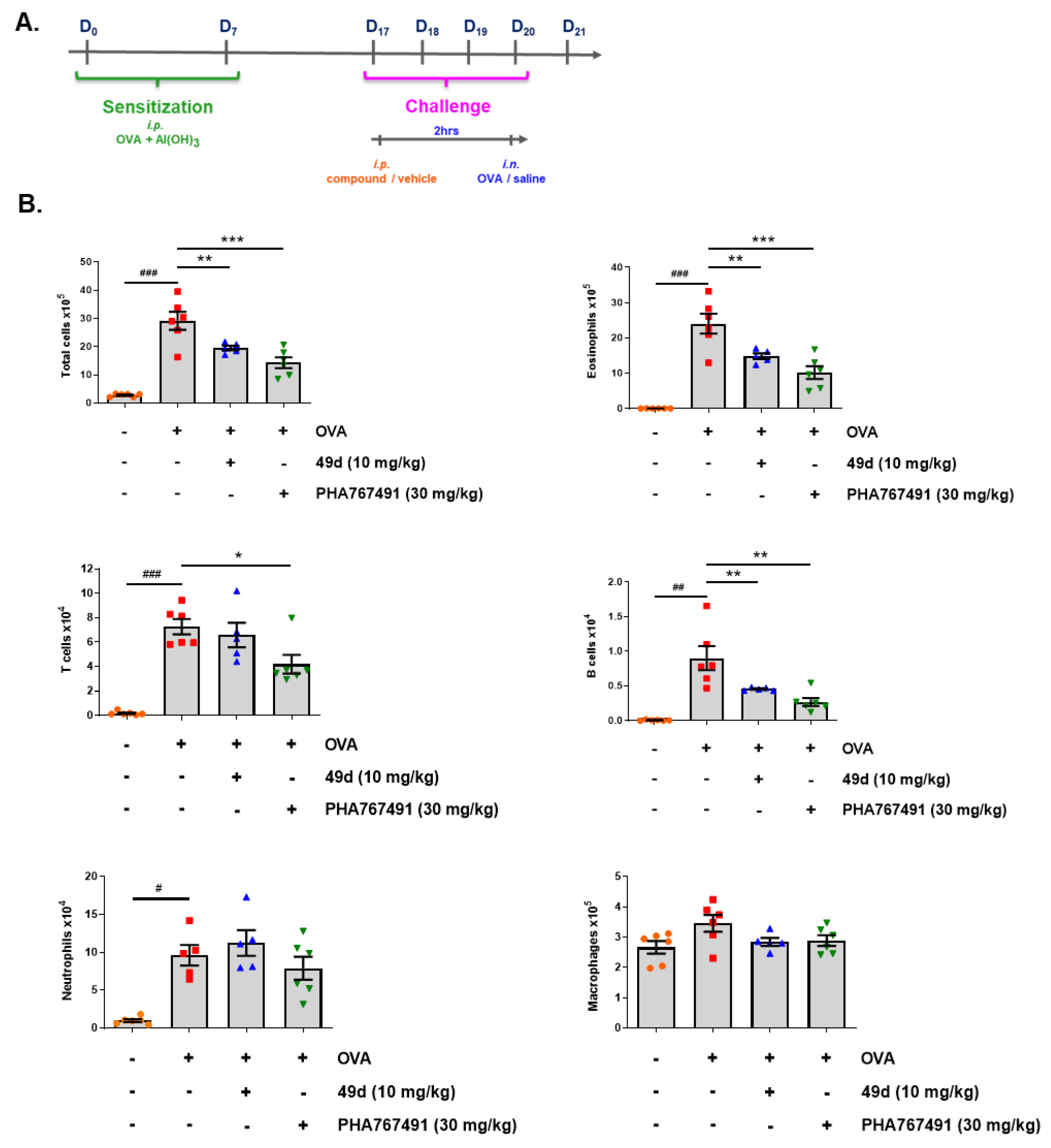
4.2.5. Evaluation of Compound Activity in an Asthma Model in Mice
4.2.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deak, M.; Clifton, A.D.; Lucocq, L.M.; Alessi, D.R. Mitogen- and Stress-Activated Protein Kinase-1 (MSK1) Is Directly Activated by MAPK and SAPK2/P38, and May Mediate Activation of CREB. EMBO J. 1998, 17, 4426–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reber, L.; Vermeulen, L.; Haegeman, G.; Frossard, N. Ser276 Phosphorylation of NF-KB P65 by MSK1 Controls SCF Expression in Inflammation. PLoS ONE 2009, 4, e4393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staples, C.J.; Owens, D.M.; Maier, J.V.; Cato, A.C.B.; Keyse, S.M. Cross-Talk between the P38alpha and JNK MAPK Pathways Mediated by MAP Kinase Phosphatase-1 Determines Cellular Sensitivity to UV Radiation. J. Biol. Chem. 2010, 285, 25928–25940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, L.; De Wilde, G.; Van Damme, P.; Vanden Berghe, W.; Haegeman, G. Transcriptional Activation of the NF-KappaB P65 Subunit by Mitogen- and Stress-Activated Protein Kinase-1 (MSK1). EMBO J. 2003, 22, 1313–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abécassis, L.; Rogier, E.; Vazquez, A.; Atfi, A.; Bourgeade, M.-F. Evidence for a Role of MSK1 in Transforming Growth Factor-Beta-Mediated Responses through P38alpha and Smad Signaling Pathways. J. Biol. Chem. 2004, 279, 30474–30479. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.S.C.; Cohen, P. MSK1 Is Required for CREB Phosphorylation in Response to Mitogens in Mouse Embryonic Stem Cells. FEBS Lett. 2000, 482, 44–48. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Fujita, J.; Kokubu, F.; Huang, S.-K.; Homma, T.; Matsukura, S.; Adachi, M.; Hizawa, N. IL-17F-Induced IL-11 Release in Bronchial Epithelial Cells via MSK1-CREB Pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L804–L810. [Google Scholar] [CrossRef] [Green Version]
- Marchand, C.; Favier, J.; Sirois, M.G. Role of MSK1 in the Signaling Pathway Leading to VEGF-Mediated PAF Synthesis in Endothelial Cells. J. Cell. Biochem. 2006, 98, 1095–1105. [Google Scholar] [CrossRef]
- Mayo, L.D.; Kessler, K.M.; Pincheira, R.; Warren, R.S.; Donner, D.B. Vascular Endothelial Cell Growth Factor Activates CRE-Binding Protein by Signaling through the KDR Receptor Tyrosine Kinase. J. Biol. Chem. 2001, 276, 25184–25189. [Google Scholar] [CrossRef] [Green Version]
- Teng, H.; Ballim, R.D.; Mowla, S.; Prince, S. Phosphorylation of Histone H3 by Protein Kinase C Signaling Plays a Critical Role in the Regulation of the Developmentally Important TBX2 Gene. J. Biol. Chem. 2009, 284, 26368–26376. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.; Omran, E.; Xu, N.; Liu, L. The Specific Mitogen- and Stress-Activated Protein Kinase MSK1 Inhibitor SB-747651A Modulates Chemokine-Induced Neutrophil Recruitment. Int J. Mol. Sci. 2017, 18, 2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naqvi, S.; Macdonald, A.; McCoy, C.E.; Darragh, J.; Reith, A.D.; Arthur, J.S.C. Characterization of the Cellular Action of the MSK Inhibitor SB-747651A. Biochem. J. 2012, 441, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otkjaer, K.; Kragballe, K.; Johansen, C.; Funding, A.T.; Just, H.; Jensen, U.B.; Sørensen, L.G.; Nørby, P.L.; Clausen, J.T.; Iversen, L. IL-20 Gene Expression Is Induced by IL-1beta through Mitogen-Activated Protein Kinase and NF-KappaB-Dependent Mechanisms. J. Invest. Dermatol. 2007, 127, 1326–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reber, L.L.; Daubeuf, F.; Nemska, S.; Frossard, N. The AGC Kinase Inhibitor H89 Attenuates Airway Inflammation in Mouse Models of Asthma. PLoS ONE 2012, 7, e49512. [Google Scholar] [CrossRef] [Green Version]
- Seidel, P.; Merfort, I.; Hughes, J.M.; Oliver, B.G.G.; Tamm, M.; Roth, M. Dimethylfumarate Inhibits NF-{kappa}B Function at Multiple Levels to Limit Airway Smooth Muscle Cell Cytokine Secretion. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L326–L339. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and Mechanism of Action of Some Commonly Used Protein Kinase Inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Anderson, D.R.; Meyers, M.J.; Vernier, W.F.; Mahoney, M.W.; Kurumbail, R.G.; Caspers, N.; Poda, G.I.; Schindler, J.F.; Reitz, D.B.; Mourey, R.J. Pyrrolopyridine Inhibitors of Mitogen-Activated Protein Kinase-Activated Protein Kinase 2 (MK-2). J. Med. Chem. 2007, 50, 2647–2654. [Google Scholar] [CrossRef]
- Tachibana, E.; Harada, T.; Shibuya, M.; Saito, K.; Takayasu, M.; Suzuki, Y.; Yoshida, J. Intra-Arterial Infusion of Fasudil Hydrochloride for Treating Vasospasm Following Subarachnoid Haemorrhage. Acta Neurochir. 1999, 141, 13–19. [Google Scholar] [CrossRef]
- Tanaka, K.; Minami, H.; Kota, M.; Kuwamura, K.; Kohmura, E. Treatment of Cerebral Vasospasm with Intra-Arterial Fasudil Hydrochloride. Neurosurgery 2005, 56, 214–223. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Bollenbach, M.; Salvat, E.; Daubeuf, F.; Wagner, P.; Yalcin, I.; Humo, M.; Letellier, B.; Becker, L.J.; Bihel, F.; Bourguignon, J.-J.; et al. Phenylpyridine-2-Ylguanidines and Rigid Mimetics as Novel Inhibitors of TNFα Overproduction: Beneficial Action in Models of Neuropathic Pain and of Acute Lung Inflammation. Eur. J. Med. Chem. 2018, 147, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, S.; Zheng, J.-F.; Zhou, J. Room-Temperature Suzuki–Miyaura Coupling of Heteroaryl Chlorides and Tosylates. Eur. J. Org. Chem. 2012, 2012, 6248–6259. [Google Scholar] [CrossRef]
- Alijevic, O.; Hammoud, H.; Vaithia, A.; Trendafilov, V.; Bollenbach, M.; Schmitt, M.; Bihel, F.; Kellenberger, S. Heteroarylguanidines as Allosteric Modulators of ASIC1a and ASIC3 Channels. ACS Chem. Neurosci. 2018, 9, 1357–1365. [Google Scholar] [CrossRef] [Green Version]
- Manetsch, R.; Zheng, L.; Reymond, M.T.; Woggon, W.-D.; Reymond, J.-L. A Catalytic Antibody against a Tocopherol Cyclase Inhibitor. Chemistry 2004, 10, 2487–2506. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T.; Westermann, E. Phosphane-Free Palladium-Catalyzed Coupling Reactions: The Decisive Role of Pd Nanoparticles. Angew. Chem. Int. Ed. 2000, 39, 165–168. [Google Scholar] [CrossRef]
- Grosso, J.A.; Nichols, D.E.; Nichols, M.B.; Yim, G.K. Synthesis and Adrenergic Blocking Effects of 2-(Alkylamino)-3,4-Dihydroquinazolines. J. Med. Chem. 1980, 23, 1261–1264. [Google Scholar] [CrossRef]
- Cook, J.; Zusi, F.C.; Hill, M.D.; Fang, H.; Pearce, B.; Park, H.; Gallagher, L.; McDonald, I.M.; Bristow, L.; Macor, J.E.; et al. Design and Synthesis of a Novel Series of (1’S,2R,4’S)-3H-4’-Azaspiro[Benzo[4,5]Imidazo[2,1-b]Oxazole-2,2’-Bicyclo[2.2.2]Octanes] with High Affinity for the A7 Neuronal Nicotinic Receptor. Bioorg. Med. Chem. Lett. 2017, 27, 5002–5005. [Google Scholar] [CrossRef]
- Kelly, B.; O’Donovan, D.H.; O’Brien, J.; McCabe, T.; Blanco, F.; Rozas, I. Pyridin-2-Yl Guanidine Derivatives: Conformational Control Induced by Intramolecular Hydrogen-Bonding Interactions. J. Org. Chem. 2011, 76, 9216–9227. [Google Scholar] [CrossRef] [Green Version]
- Grant, S.K. Therapeutic Protein Kinase Inhibitors. Cell. Mol. Life Sci. 2009, 66, 1163–1177. [Google Scholar] [CrossRef]
- Patterson, H.; Nibbs, R.; McInnes, I.; Siebert, S. Protein Kinase Inhibitors in the Treatment of Inflammatory and Autoimmune Diseases. Clin. Exp. Immunol. 2014, 176, 1–10. [Google Scholar] [CrossRef]
- Lin, S.-C.; Shi, L.-S.; Ye, Y.-L. Advanced Molecular Knowledge of Therapeutic Drugs and Natural Products Focusing on Inflammatory Cytokines in Asthma. Cells 2019, 8, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goplen, N.; Gorska, M.M.; Stafford, S.J.; Rozario, S.; Guo, L.; Liang, Q.; Alam, R. A Phosphosite Screen Identifies Autocrine TGF-Beta-Driven Activation of Protein Kinase R as a Survival-Limiting Factor for Eosinophils. J. Immunol. 2008, 180, 4256–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Park, S.H.; Park, S.-Y.; Moon, U.Y.; Lee, B.D.; Yoon, S.H.; Lee, J.-G.; Baek, S.J.; Yoon, J.-H. Epigallocatechin-3-Gallate Inhibits Interleukin-1beta-Induced MUC5AC Gene Expression and MUC5AC Secretion in Normal Human Nasal Epithelial Cells. J. Nutr. Biochem. 2008, 19, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Song, K.S.; Lee, W.-J.; Chung, K.C.; Koo, J.S.; Yang, E.J.; Choi, J.Y.; Yoon, J.-H. Interleukin-1 Beta and Tumor Necrosis Factor-Alpha Induce MUC5AC Overexpression through a Mechanism Involving ERK/P38 Mitogen-Activated Protein Kinases-MSK1-CREB Activation in Human Airway Epithelial Cells. J. Biol. Chem. 2003, 278, 23243–23250. [Google Scholar] [CrossRef] [Green Version]
- Caivano, M.; Cohen, P. Role of Mitogen-Activated Protein Kinase Cascades in Mediating Lipopolysaccharide-Stimulated Induction of Cyclooxygenase-2 and IL-1 Beta in RAW264 Macrophages. J. Immunol. 2000, 164, 3018–3025. [Google Scholar] [CrossRef] [Green Version]
- Eliopoulos, A.G.; Dumitru, C.D.; Wang, C.-C.; Cho, J.; Tsichlis, P.N. Induction of COX-2 by LPS in Macrophages Is Regulated by Tpl2-Dependent CREB Activation Signals. EMBO J. 2002, 21, 4831–4840. [Google Scholar] [CrossRef] [Green Version]
- Reyskens, K.M.S.E.; Arthur, J.S.C. Emerging Roles of the Mitogen and Stress Activated Kinases MSK1 and MSK2. Front. Cell Dev. Biol. 2016, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Beck, I.M.E.; Vanden Berghe, W.; Gerlo, S.; Bougarne, N.; Vermeulen, L.; De Bosscher, K.; Haegeman, G. Glucocorticoids and Mitogen- and Stress-Activated Protein Kinase 1 Inhibitors: Possible Partners in the Combat against Inflammation. Biochem. Pharmacol. 2009, 77, 1194–1205. [Google Scholar] [CrossRef] [Green Version]
- Vettorazzi, S.; Bode, C.; Dejager, L.; Frappart, L.; Shelest, E.; Klaßen, C.; Tasdogan, A.; Reichardt, H.M.; Libert, C.; Schneider, M.; et al. Glucocorticoids Limit Acute Lung Inflammation in Concert with Inflammatory Stimuli by Induction of SphK1. Nat. Commun. 2015, 6, 7796. [Google Scholar] [CrossRef] [Green Version]
- Brasier, A.R. The Nuclear Factor-ΚB–Interleukin-6 Signalling Pathway Mediating Vascular Inflammation. Cardiovasc. Res. 2010, 86, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-H.; Rieke, R.D. 2-Pyridyl and 3-Pyridylzinc Bromides: Direct Preparation and Coupling Reaction. Tetrahedron 2010, 66, 3135–3146. [Google Scholar] [CrossRef]
- Xu, P.; Zhu, Y.-M.; Wang, F.; Wang, S.-Y.; Ji, S.-J. Mn(III)-Mediated Cascade Cyclization of 3-Isocyano-[1,1′-Biphenyl]-2-Carbonitrile with Arylboronic Acid: Construction of Pyrrolopyridine Derivatives. Org. Lett. 2019, 21, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Spinks, D.; Ong, H.B.; Mpamhanga, C.P.; Shanks, E.J.; Robinson, D.A.; Collie, I.T.; Read, K.D.; Frearson, J.A.; Wyatt, P.G.; Brenk, R.; et al. Design, Synthesis and Biological Evaluation of Novel Inhibitors of Trypanosoma Brucei Pteridine Reductase 1. Chem. Med. Chem. 2011, 6, 302–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cee, V.J.; Frohn, M.; Lanman, B.A.; Golden, J.; Muller, K.; Neira, S.; Pickrell, A.; Arnett, H.; Buys, J.; Gore, A.; et al. Discovery of AMG 369, a Thiazolo[5,4-b]Pyridine Agonist of S1P1 and S1P5. ACS Med. Chem. Lett. 2011, 2, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, T.; Imoto, M.; Takeda, M.; Nakai, T.; Mihara, M.; Mizuno, T.; Nomoto, A.; Ogawa, A. Regioselective Radical Arylation of Aromatic Diamines with Arylhydrazines. Synthesis 2017, 49, 1623–1631. [Google Scholar] [CrossRef] [Green Version]
- Mpamhanga, C.P.; Spinks, D.; Tulloch, L.B.; Shanks, E.J.; Robinson, D.A.; Collie, I.T.; Fairlamb, A.H.; Wyatt, P.G.; Frearson, J.A.; Hunter, W.N.; et al. One Scaffold, Three Binding Modes: Novel and Selective Pteridine Reductase 1 Inhibitors Derived from Fragment Hits Discovered by Virtual Screening. J. Med. Chem. 2009, 52, 4454–4465. [Google Scholar] [CrossRef] [PubMed] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | MSK1 a | ||||
|---|---|---|---|---|---|
| % Inhibition | IC50 | ||||
| Entry | Cpd | R | 10 µM | 1 µM | µM |
| 1 | 14 | 3-Ph | 11 ± 5 | 1 ± 5 | nd |
| 2 | 13 | 4-Ph | 5 ± 1 | −4 ± 1 | nd |
| 3 | 12 | 5-Ph | −11 ± 1 | −10 ± 2 | nd |
| 4 | 25 | 5-(CH2)2Ph | 6 ± 4 | 2 ± 4 | nd |
| 5 | 1a | 6-Ph | 42 ± 9 | 4 ± 4 | 17.9 ± 3.9 |
| 6 | 17 | 6-Bn | −2 ± 3 | −2 ± 1 | nd |
| 7 | 24 | 6-(CH2)2Ph | −8 ± 1 | −4 ± 1 | nd |
| 8 | H89 | - | 93 ± 10 | 80 ± 8 | 0.25 ± 0.06 |
| 9 | Fasudil | - | 82 ± 10 | 50 ± 10 | 1.74 ± 0.16 |
| 10 | PHA767491 | - | 94 ± 4 | 72 ± 5 | 0.62 ± 0.13 |
 | MSK1 a | ||||
|---|---|---|---|---|---|
| % Inhibition | IC50 | ||||
| Entry | Cpd | R | 10 µM | 1 µM | µM |
| 1 | 1a | H | 42 ± 9 | 4 ± 4 | 17.9 ± 3.9 |
| 2 | 1b | 4-Cl | 23 ± 1 | −4 ± 5 | nd |
| 3 | 1c | 4-CF3 | −1 ± 5 | −6 ± 2 | nd |
| 4 | 1d | 4-OMe | −3 ± 4 | −5 ± 3 | nd |
| 5 | 1e | 3-Cl | 45 ± 5 | 0 ± 4 | 9.7 ± 1.0 |
| 6 | 1f | 3-OMe | 16 ± 4 | −7 ± 4 | nd |
| 7 | 1j | 2-F | 45 ± 9 | 6 ± 3 | 5.0 ± 0.5 |
| 8 | 1k | 2-Cl | 95 ± 8 | 62 ± 8 | 0.6 ± 0.1 |
| 9 | 1l | 2-Me | 48 ± 15 | 4 ± 8 | 3.5 ± 0.3 |
| 10 | 1m | 2-CF3 | 38 ± 9 | 4 ± 2 | 8.1 ± 1.2 |
| 11 | 1n | 2-OMe | 66 ± 12 | 16 ± 3 | 2.3 ± 0.3 |
| 12 | 1o | 2,3-Cl2 | 71 ± 13 | 7 ± 3 | 2.3 ± 0.3 |
| 13 | 1p | 2,4-Cl2 | 31 ± 7 | 2 ± 2 | ns |
| 14 | 1q | 2,5-Cl2 | 84 ± 4 | 23 ± 6 | 0.9 ± 0.1 |
| 15 | 1i | 2-Cl, 3-CF3 | 62 ± 15 | 10 ± 5 | 8.6 ± 0.9 |
 | MSK1 a | ||||
|---|---|---|---|---|---|
| % inhibition | IC50 | ||||
| Entry | Cpd | Het | 10 µM | 1 µM | µM |
| 1 | 1g |  | 33 ± 8 | 4 ± 3 | ns |
| 2 | 1h |  | 1 ± 6 | −7 ± 5 | nd |
| 3 | 1r |  | 18 ± 2 | 1 ± 4 | nd |
| 4 | 1s |  | 70 ± 7 | 9 ± 4 | 5.8 ± 0.6 |
| 5 | 1t |  | 69 ± 11 | 10 ± 6 | 12.6 ± 4.0 |
| 6 | 1u |  | 42 ± 7 | 3 ± 7 | ns |
| 7 | 31 |  | 16 ± 4 | −7 ± 4 | nd |
 | MSK1 a | ||||||
|---|---|---|---|---|---|---|---|
| % Inhibition | IC50 | ||||||
| Entry | Cpd | Position | R1 | R2 | 10 µM | 1 µM | µM |
| 1 | 37a | 5 | H | H | 44 ± 1 | −19 ± 1 | 15.8 ± 3.2 |
| 2 | 37b | 5 | 4-Cl | H | 0 ± 7 | −9 ± 2 | nd |
| 3 | 37c | 5 | 4-OMe | H | −5 ± 5 | −7 ± 4 | nd |
| 4 | 37d | 5 | 3-Cl | H | 10 ± 7 | −5 ± 4 | nd |
| 5 | 37e | 5 | 3-OMe | H | 3 ± 7 | −6 ± 4 | nd |
| 6 | 37f | 5 | 2-Cl | H | 38 ± 4 | 0 ± 2 | 16.4 ± 4.0 |
| 7 | 37g | 5 | 2-OMe | H | −5 ± 1 | −14 ± 3 | nd |
| 8 | 60a | 5 | H | n-Pr | 2 ± 3 | 0 ± 1 | nd |
| 9 | 60b | 5 | H | Ph | 1 ± 2 | −2 ± 1 | nd |
| 10 | 43 | 6 | H | H | 9 ± 6 | 7 ± 9 | nd |
 | MSK1 a | ||||||
|---|---|---|---|---|---|---|---|
| % Inhibition | IC50 | ||||||
| Entry | Cpd | Position | R1 | R2 | 10 µM | 1 µM | µM |
| 1 | 49a | 4 | H | H | 46 ± 6 | 5 ± 3 | 3.6 ± 0.5 |
| 2 | 49b | 4 | 4-Cl | H | 49 ± 9 | −3 ± 6 | nd |
| 3 | 49c | 4 | 4-OMe | H | 25 ± 5 | −2 ± 6 | nd |
| 4 | 49d | 4 | 2-Cl | H | 71 ± 7 | 21 ± 5 | 1.6 ± 0.1 |
| 5 | 49e | 4 | 2,3-Cl2 | H | 75 ± 8 | 20 ± 3 | 3.2 ± 0.2 |
| 6 | 49f | 4 | 2,5-Cl2 | H | 61 ± 5 | 6 ± 3 | 6.8 ± 0.8 |
| 7 | 49g | 4 | 2-OMe | H | 39 ± 7 | −3 ± 5 | nd |
| 8 | 74a | 4 | 2-Cl | (CH2)2-Ph | 41 ± 6 | −6 ± 3 | nd |
| 9 | 74b | 4 | 2-Cl | (CH2)2-OH | 49 ± 13 | −2 ± 4 | nd |
| 10 | 74c | 4 | 2-Cl | (CH2)3-NHCOPh | 37 ± 5 | −4 ± 2 | nd |
| 11 | 74d | 4 | 2-Cl | (CH2)4-Ph | 5 ± 3 | 1 ± 3 | nd |
| 12 | 50 | 5 | H | H | 4 ± 4 | −7 ± 5 | nd |
| 13 | 75 | 7 | 2-Cl | (CH2)2-Ph | 1 ± 2 | −6 ± 2 | nd |
| Il-6 a | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Group | R | Cpd | % Inhibition | IC50 (µM) | Cell Viability | ||
| 10 µM | 30 µM | 10 µM | 30 µM | |||||
| 1 |  | H | 1a | 17 ± 7 | 60 ± 4 | 16.3 ± 6.1 | 122 ± 14 | 103 ± 5 |
| 2 | 2-Cl | 1j | 42 ± 4 | T | nd | 100 ± 8 | 41 ± 9 | |
| 3 | 2,5-Cl2 | 1q | T | T | nd | 31 ± 19 | 13 ± 1 | |
| 4 |  | H | 37a | 18 ± 7 | 23 ± 6 | ns | 122 ± 14 | 103 ± 5 |
| 5 | Cl | 37f | 17 ± 5 | T | nd | 100 ± 8 | 41 ± 9 | |
| 6 |  | H | 49a | 12 ± 4 | 22 ± 1 | ns | 93 ± 17 | 91 ± 15 |
| 7 | Cl | 49d | 39 ± 15 | 57 ± 2 | 13.9 ± 9.7 | 85 ± 13 | 76 ± 12 | |
| 8 | Reference compounds | H89 | 38 ± 5 | 96 ± 1 | 10.5 ± 5.1 | 96 ± 4 | 104 ± 3 | |
| Fasudil | 38 ± 6 | 81 ± 6 | 8.5 ± 5.1 | 95 ± 3 | 92 ± 3 | |||
| PHA767491 | 97 ± 1 | 98 ± 2 | 1.0 ± 0.1 | 95 ± 9 | 87 ± 7 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bollenbach, M.; Nemska, S.; Wagner, P.; Camelin, G.; Daubeuf, F.; Obrecht, A.; Villa, P.; Rognan, D.; Bihel, F.; Bourguignon, J.-J.; et al. Design, Synthesis and Biological Evaluation of Arylpyridin-2-yl Guanidine Derivatives and Cyclic Mimetics as Novel MSK1 Inhibitors. An Application in an Asthma Model. Molecules 2021, 26, 391. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020391
Bollenbach M, Nemska S, Wagner P, Camelin G, Daubeuf F, Obrecht A, Villa P, Rognan D, Bihel F, Bourguignon J-J, et al. Design, Synthesis and Biological Evaluation of Arylpyridin-2-yl Guanidine Derivatives and Cyclic Mimetics as Novel MSK1 Inhibitors. An Application in an Asthma Model. Molecules. 2021; 26(2):391. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020391
Chicago/Turabian StyleBollenbach, Maud, Simona Nemska, Patrick Wagner, Guillaume Camelin, François Daubeuf, Adeline Obrecht, Pascal Villa, Didier Rognan, Frédéric Bihel, Jean-Jacques Bourguignon, and et al. 2021. "Design, Synthesis and Biological Evaluation of Arylpyridin-2-yl Guanidine Derivatives and Cyclic Mimetics as Novel MSK1 Inhibitors. An Application in an Asthma Model" Molecules 26, no. 2: 391. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020391