
Heterologous Expression of Pseudouridimycin and Description of the Corresponding Minimal Biosynthetic Gene Cluster


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
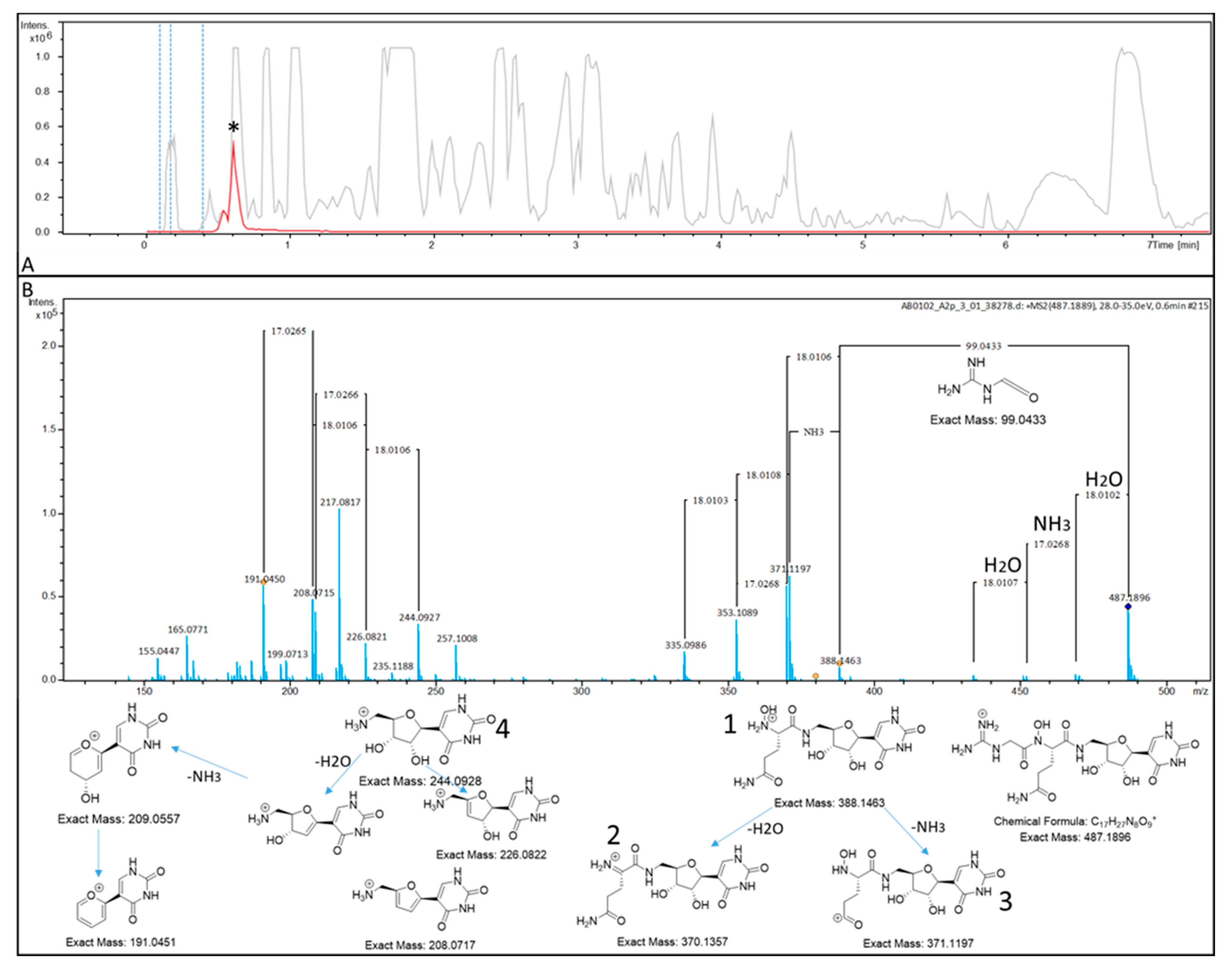
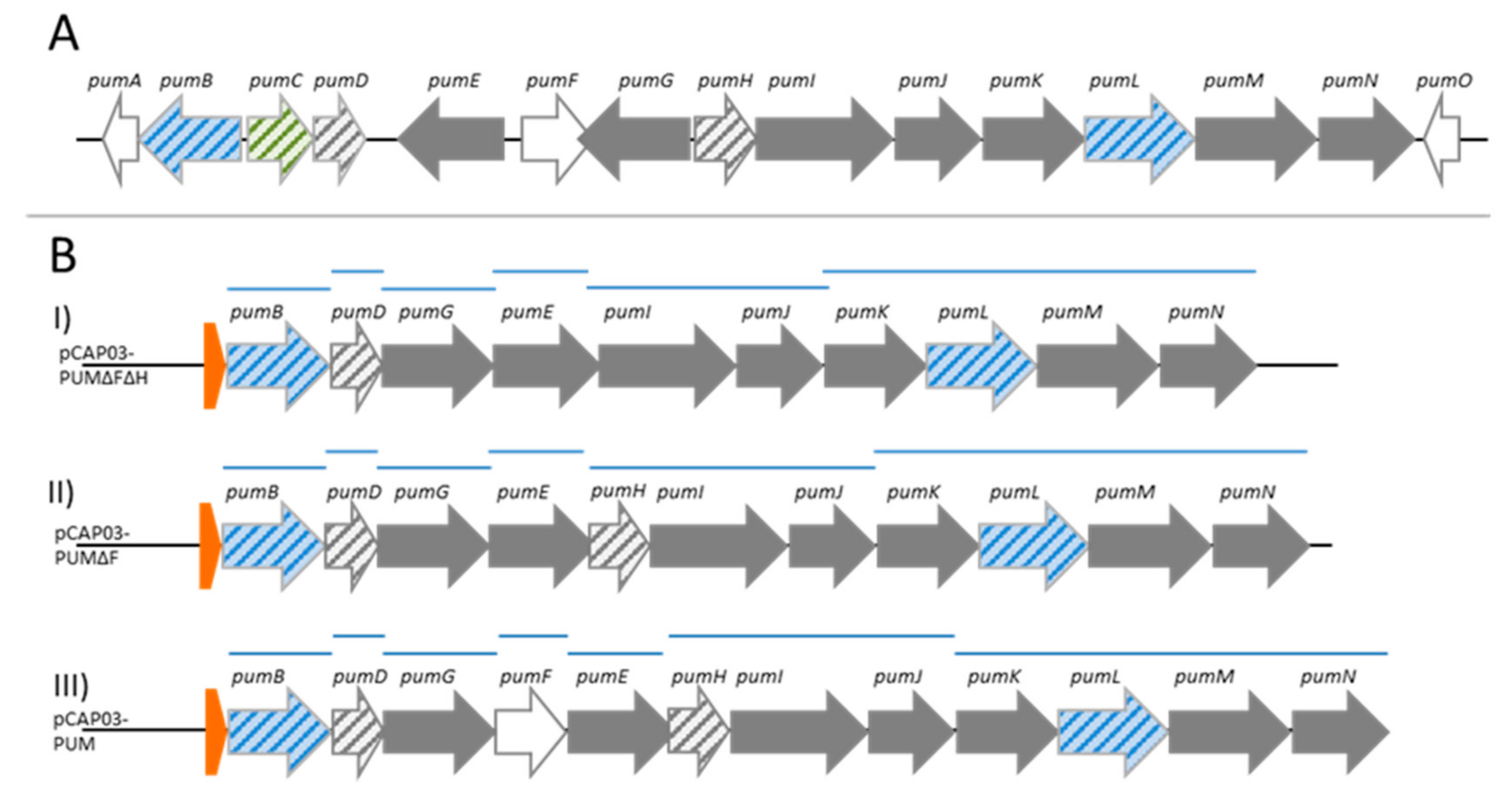
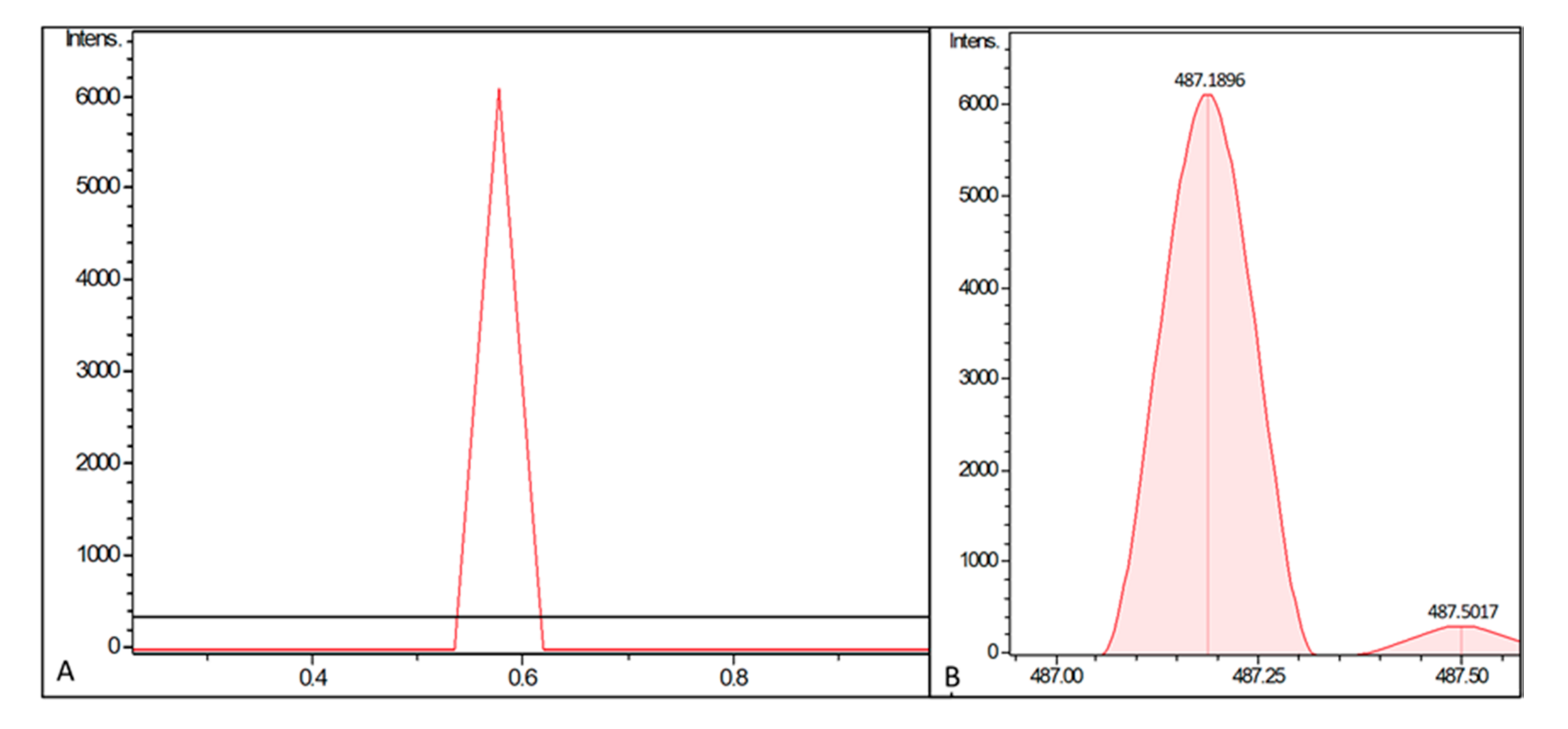
2. Results
3. Discussion
4. Materials and Methods
4.1. Cultivation of Bacteria
4.2. UHPLC–MS Analysis of PUM
4.3. Sequence Homology Analysis
4.4. General Molecular Biology Techniques
4.5. Construction of pGEM-teasy_Apra-ermE* and pGEM-teasy_Apra-tcp830
4.6. Construction of the Integrative Streptomyces PUM Expression Plasmids pCAP03-PUMΔFΔH_ermE*/tcp830, pCAP03-PUMΔF_ermE*/tcp830 and pCAP03-PUM_ermE*/tcp830
4.7. Heterologous Expression of the PUM BGC
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Maffioli, S.I.; Zhang, Y.; Degen, D.; Carzaniga, T.; Del Gatto, G.; Serina, S.; Monciardini, P.; Mazzetti, C.; Guglierame, P.; Candiani, G.; et al. Antibacterial Nucleoside-Analog Inhibitor of Bacterial RNA Polymerase. Cell 2017, 169, 1240–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, L.C.; Lockwook, D.N.J. Leprosy now: Epidemiology, progress, challenges, and research gaps. Lancet Infect. Dis. 2011, 11, 464–470. [Google Scholar] [CrossRef]
- Srivastava, A.; Talaue, M.; Liu, S.; Degen, D.; Ebright, R.Y.; Sineva, E.; Chakraborty, A.; Druzhinin, S.Y.; Chatterjee, S.; Mukhopadhyay, J.; et al. New Target for Inhibition of Bacterial Rnapolymerase: “Switch Region”. Curr. Opin. Microbiol. 2011, 14, 532–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäberle, T.F.; Schiefer, A.; Schmitz, A.; König, G.M.; Hoerauf, A.; Pfarr, K. Corallopyronin A—A promising antibiotic for treatment of filariasis. Int. J. Med. Microbiol. 2014, 304, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Schäberle, T.F.; Schmitz, A.; Zocher, G.; Schiefer, A.; Kehraus, S.; Neu, E.; Roth, M.; Vassylyev, D.G.; Stehle, T.; Bierbaum, G.; et al. Insights into Structure-Activity Relationships of Bacterial RNA Polymerase Inhibiting Corallopyronin Derivatives. J. Nat. Prod. 2015, 78, 2505–2509. [Google Scholar] [CrossRef] [PubMed]
- Krome, A.K.; Becker, T.; Kehraus, S.; Schiefer, A.; Steinebach, C.; Aden, T.; Frohberger, S.J.; López Mármol, A.; Kapote, D.; Jansen, R.; et al. Solubility and Stability Enhanced Oral Formulationsfor the Anti-Infective Corallopyronin A. Pharmaceutics 2020, 12, 1105. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, J.; Das, K.; Ismail, S.; Koppstein, D.; Jang, M.; Hudson, B.; Sarafianos, S.; Tuske, S.; Patel, J.; Jansen, R.; et al. The RNA polymerase “switch region” is a target for inhibitors. Cell 2008, 135, 295–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belogurov, G.A.; Vassylyeva, M.N.; Sevostyanova, A.; Appleman, J.R.; Xiang, A.X.; Lira, R.; Webber, S.E.; Klyuyev, S.; Nudler, E.; Artsimovitch, I.; et al. Transcription inactivation through local refolding of the RNA polymerase structure. Nature 2009, 457, 332–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosio, M.; Gaspari, E.; Iorio, M.; Pessina, S.; Medema, M.; Bernasconi, A.; Simone, M.; Maffioli, S.I.; Ebright, R.H.; Donadio, S. Analysis of the Pseudouridimycin Biosynthetic Pathway Provides Insights into the Formation of C-nucleoside Antibiotics. Cell Chem. Biol. 2018, 25, 540–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenqvist, P.; Palmu, K.; Prajapati, R.K.; Yamada, K.; Niemi, J.; Belogurov, G.A.; Metsä-Ketelä, M.; Virta, P. Characterization of C-nucleoside Antimicrobials from Streptomyces albus DSM 40763: Strepturidin is Pseudouridimycin. Sci. Rep. 2019, 9, 8935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Escribano, J.P.; Bibb, M.J. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 2011, 4, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cousin, C.; Derouiche, A.; Shi, L.; Pagot, Y.; Poncet, S.; Mijakovic, I. Protein-serine/threonine/tyrosine kinases in bacterial signaling and regulation. FEMS Microbiol. Lett. 2013, 346, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Draelos, M.M.; Thanapipatsiri, A.; Sucipto, H.; Yokoyama, K. Cryptic phosphorylation in nucleoside natural product biosynthesis. Nat. Chem. Biol. 2020. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Hopwood, D.A.; Kieser, T.; Bibb, M.; Buttner, M.; Chater, K. Practical Streptomyces Genetics; John Innes Foundation: Norwich, UK, 2000. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Siegl, T.; Tokovenko, B.; Myronovskyi, M.; Luzhetskyy, A. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab. Eng. 2013, 19, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Dangel, V.; Westrich, L.; Smith, M.C.M.; Heide, L.; Gust, B. Use of an inducible promoter for antibiotic production in a heterologous host. Appl. Microbiol. Biotechnol. 2010, 87, 261–269. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Böhringer, N.; Patras, M.A.; Schäberle, T.F. Heterologous Expression of Pseudouridimycin and Description of the Corresponding Minimal Biosynthetic Gene Cluster. Molecules 2021, 26, 510. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020510
Böhringer N, Patras MA, Schäberle TF. Heterologous Expression of Pseudouridimycin and Description of the Corresponding Minimal Biosynthetic Gene Cluster. Molecules. 2021; 26(2):510. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020510
Chicago/Turabian StyleBöhringer, Nils, Maria A. Patras, and Till F. Schäberle. 2021. "Heterologous Expression of Pseudouridimycin and Description of the Corresponding Minimal Biosynthetic Gene Cluster" Molecules 26, no. 2: 510. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020510