
iTRAQ-BASED Proteomic Analysis of the Mechanism of Fructose on Improving Fengycin Biosynthesis in Bacillus Amyloliquefaciens

 ,
,
Abstract
:
1. Introduction
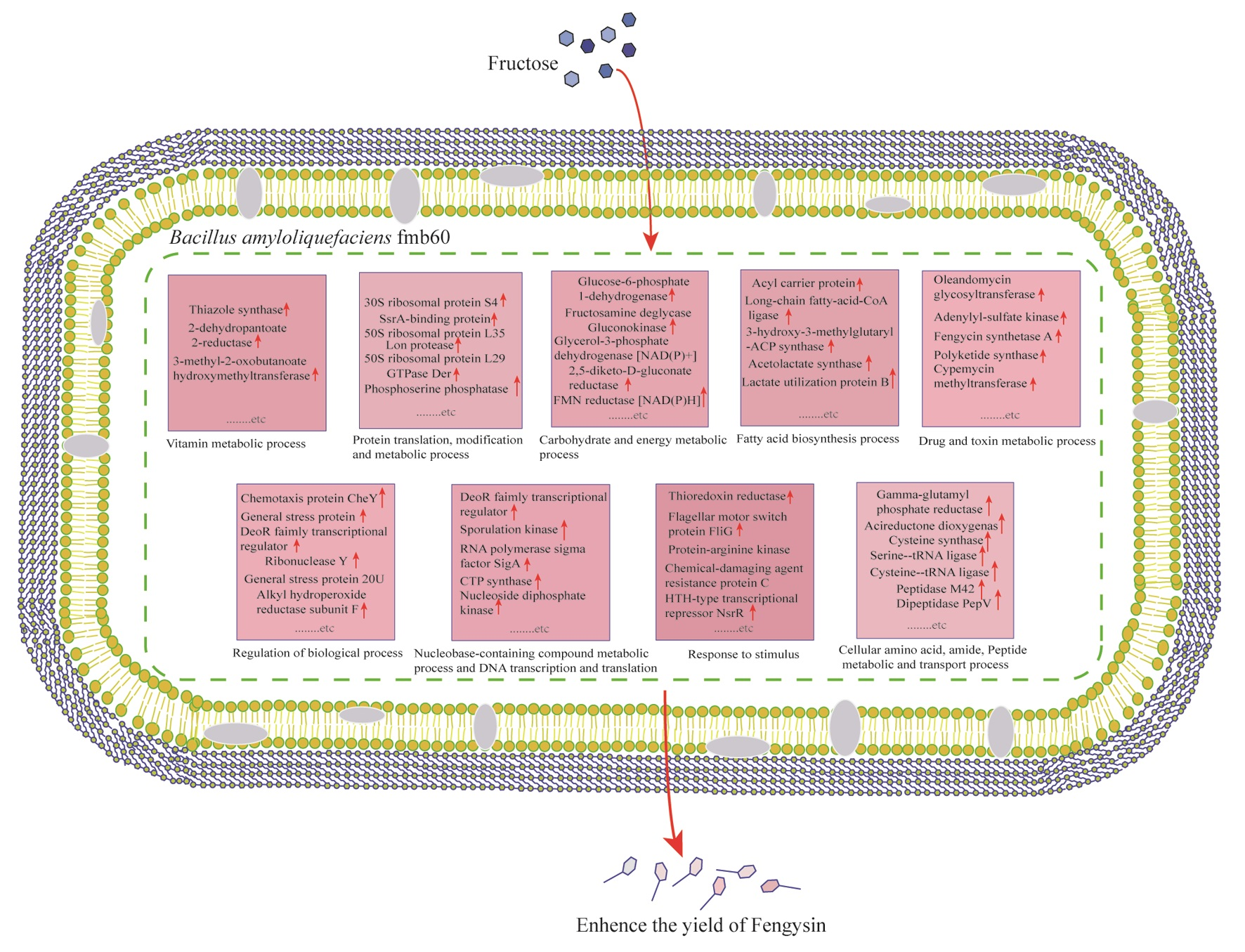
2. Results
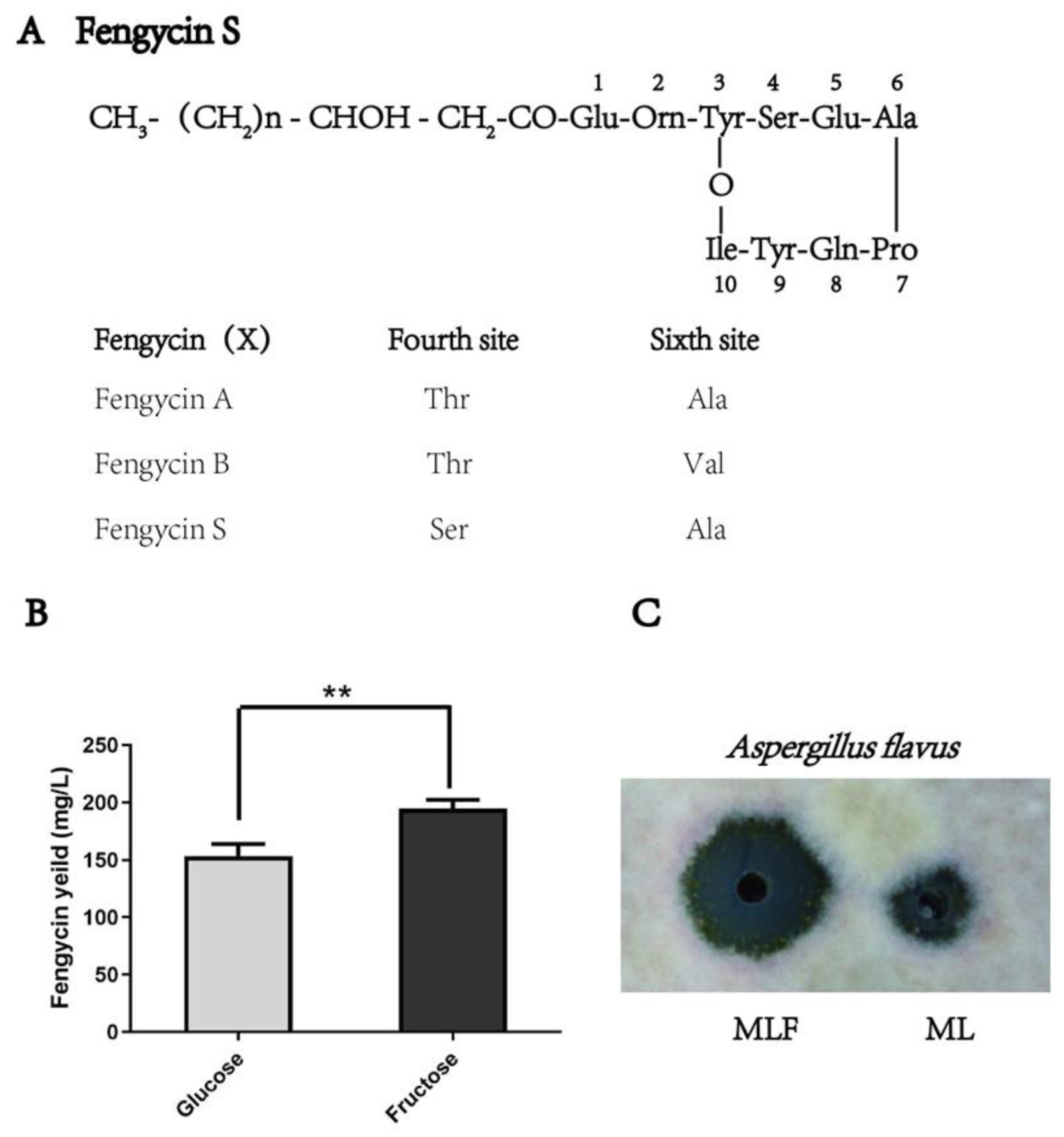
2.1. Effect of Fructose on the Yield of Fengycin
2.2. Analysis of Primary Data and Identification of Protein
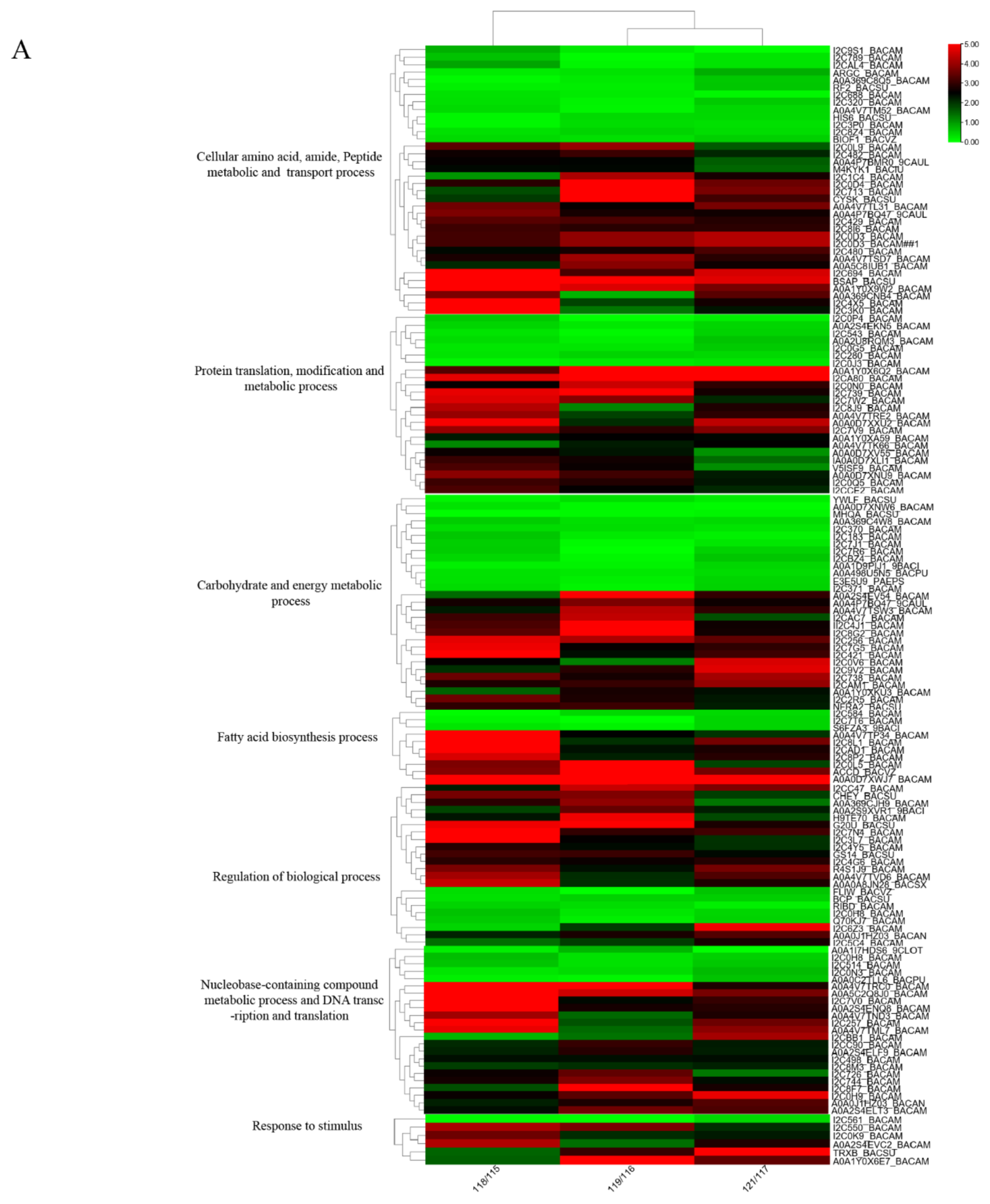
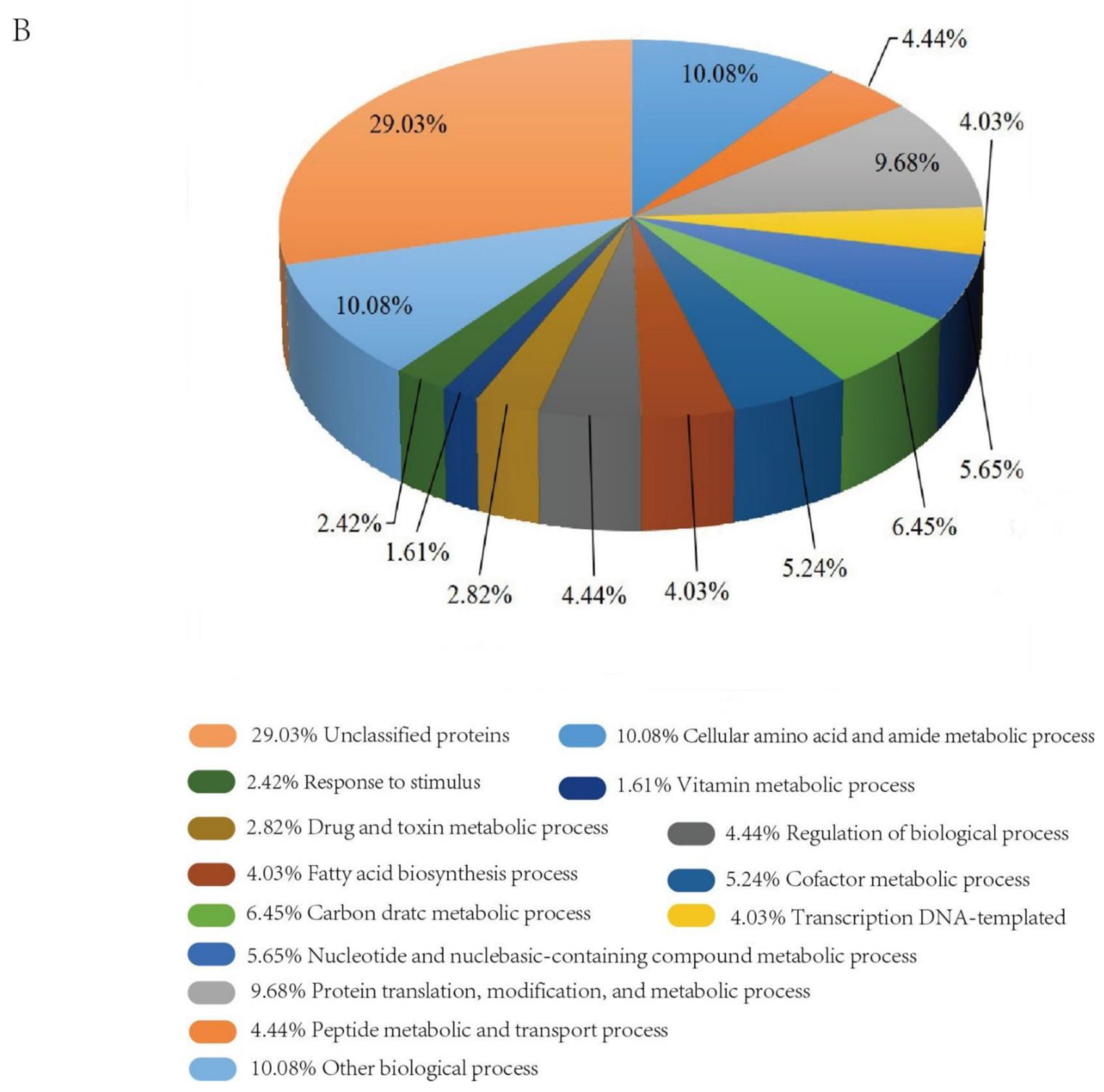
2.3. Functional Categories of Differentially Expressed Proteins
2.3.1. Cellular Amino Acid, Amide, Peptide Metabolic and Transport Process
2.3.2. Protein Translation, Modification, and Metabolic Process
2.3.3. Carbohydrate and Energy Metabolic Process
2.3.4. Fatty Acid Biosynthesis Process
2.3.5. Regulation of Biological Process
2.3.6. Drug and Toxin Metabolic Process
2.3.7. Vitamin Metabolic Process
2.3.8. Nucleobase-Containing Compound Metabolic Process and DNA Transcription and Translation
2.3.9. Response to Stimulus
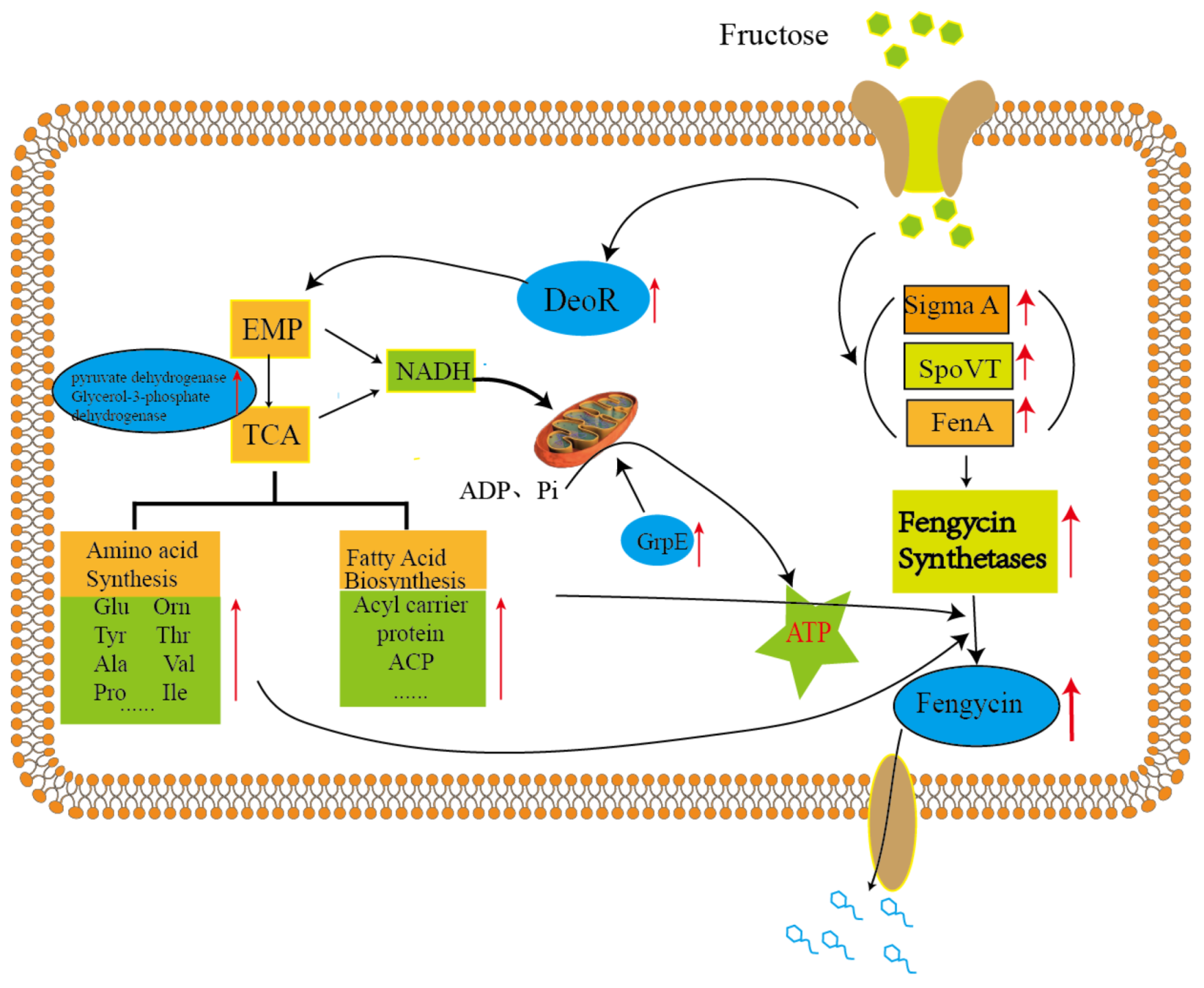
3. Discussion
4. Materials and Methods
4.1. Microorganisms and Fermentation
4.2. Preparation Sample Preparation and LC-MS/MS Analysis
4.3. Extraction of RNA and the Quantitative Real-Time PCR (qRT-PCR)
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stein, T. Bacillus subtilis antibiotics: Structures, syntheses and specific functions. Mol. Microbiol. 2005, 56, 845–857. [Google Scholar] [CrossRef]
- Cleveland, J.; Montville, T.J.; Nes, I.F.; Chikindas, M.L. Bacteriocins: Safe, natural antimicrobials for food preservation. Int. J. Food Microbiol. 2001, 71, 1–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S.; Liu, L.; Wu, J. Acetoin production enhanced by manipulating carbon flux in a newly isolated Bacillus amyloliquefaciens. Bioresour. Technol. 2013, 130, 256–260. [Google Scholar] [CrossRef]
- Koumoutsi, A.; Chen, X.-H.; Henne, A.; Liesegang, H.; Hitzeroth, G.; Franke, P.; Vater, J.; Borriss, R. Structural and Functional Characterization of Gene Clusters Directing Nonribosomal Synthesis of Bioactive Cyclic Lipopeptides in Bacillus amyloliquefaciens Strain FZB42. J. Bacteriol. 2004, 186, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Ishihara, A.; Nakajima, H. Isolation of anteiso-C17, iso-C17, iso-C16, and iso-C15 Bacillomycin D from Bacillus amyloliquefaciens SD-32 and Their Antifungal Activities against Plant Pathogens. J. Agric. Food Chem. 2014, 62, 1469–1476. [Google Scholar] [CrossRef]
- Kim, P.; Chung, K.C. Production of an antifungal protein for control of Colletotrichum lagenarium by Bacillus amyloliquefaciens MET0908. FEMS Microbiol. Lett. 2004, 234, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Li, B.; Zhang, N.; Waseem, R.; Shen, Q.; Huang, Q. Production of Bacillomycin- and Macrolactin-Type Antibiotics by Bacillus amyloliquefaciens NJN-6 for Suppressing Soilborne Plant Pathogens. J. Agric. Food Chem. 2012, 60, 2976–2981. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sun, L.; Wang, Y.; Lei, X.; Xu, D. Modeling Antimicrobial Activity of Lipopeptides from Bacillus amyloliquefaciens ES-2 against Shewanella putrefaciens in Shrimp Meat Using a Response Surface Method. J. Food Prot. 2012, 75, 1855–1858. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Bie, X.; Lu, Z.; Lv, F.; Tao, Y.; Qu, X. Effects of fengycin from Bacillus subtilis fmbJ on apoptosis and necrosis in Rhizopus stolonifer. J. Microbiol. 2014, 52, 675–680. [Google Scholar] [CrossRef]
- Yin, H.; Guo, C.; Wang, Y.; Liu, D.; Lv, Y.; Lv, F.; Lu, Z. Fengycin inhibits the growth of the human lung cancer cell line 95D through reactive oxygen species production and mitochondria-dependent apoptosis. Anti-Cancer Drugs 2013, 24, 587–598. [Google Scholar] [CrossRef]
- Yánez-Mendizábal, V.; Usall, J.; Viñas, I.; Casals, C.; Marín, S.; Solsona, C.; Teixidó, N. Potential of a new strain of Bacillus subtilis CPA-8 to control the major postharvest diseases of fruit. Biocontrol Sci. Technol. 2011, 21, 409–426. [Google Scholar] [CrossRef]
- Hu, L.B.; Shi, Z.Q.; Zhang, T.; Yang, Z.M. Fengycin antibiotics isolated from B-FS01 culture inhibit the growth of Fusarium moniliforme Sheldon ATCC 38932. FEMS Microbiol. Lett. 2007, 272, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toure, Y.; Ongena, M.; Jacques, P.; Guiro, A.; Thonart, P. Role of lipopeptides produced by Bacillus subtilis GA1 in the reduction of grey mould disease caused by Botrytis cinerea on apple. J. Appl. Microbiol. 2004, 96, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, Y.; Lin, F.; Lu, Z.; Lu, Y. CodY, ComA, DegU and Spo0A controlling lipopeptides biosynthesis in Bacillus amyloliquefaciens fmbJ. J. Appl. Microbiol. 2021, 131, 1289–1304. [Google Scholar] [CrossRef]
- Lu, H.; Qian, S.; Muhammad, U.; Jiang, X.; Han, J.; Lu, Z. Effect of fructose on promoting fengycin biosynthesis in Bacillus amyloliquefaciens fmb-60. J. Appl. Microbiol. 2016, 12, 11653–11664. [Google Scholar]
- Zeng, X.; Saxild, H.H. Identification and characterization of a DeoR-specific operator sequence essential for induction of dra-nupC-pdp operon expression in Bacillus subtilis. J. Bacteriol. 1999, 181, 1719–1727. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.-Z.; Zheng, Q.-W.; Wei, T.; Zhang, Z.-Q.; Zhao, C.-F.; Zhong, H.; Xu, Q.-Y.; Lin, J.-F.; Guo, L.-Q. Isolation and Characterization of Fengycins Produced by Bacillus amyloliquefaciens JFL21 and Its Broad-Spectrum Antimicrobial Potential against Multidrug-Resistant Foodborne Pathogens. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Piewngam, P.; Zheng, Y.; Nguyen, T.H.; Dickey, S.W.; Joo, H.-S.; Villaruz, A.E.; Glose, K.A.; Fisher, E.L.; Hunt, R.; Li, B.; et al. Pathogen elimination by probiotic Bacillus via signalling interference. Nature 2018, 562, 532–537. [Google Scholar] [CrossRef]
- Hu, J.; Zheng, M.; Dang, S.; Shi, M.; Zhang, J.; Li, Y. Biocontrol Potential of Bacillus amyloliquefaciens LYZ69 Against Anthracnose of Alfalfa (Medicago sativa). Phytopathology 2021, PHYTO09200385R. [Google Scholar] [CrossRef]
- Kang, B.R.; Park, J.S.; Jung, W.-J. Antifungal evaluation of fengycin isoforms isolated from Bacillus amyloliquefaciens PPL against Fusarium oxysporum f. sp. lycopersici. Microb. Pathog. 2020, 149, 104509. [Google Scholar] [CrossRef]
- Hueck, C.J.; Hillen, W. Catabolite repression in Bacillus subtilis: A global regulatory mechanism for the gram-positive bacteria? Mol. Microbiol. 1995, 15, 395–401. [Google Scholar] [CrossRef]
- Zeng, X.; Galinier, A.; Saxild, H.H. Catabolite repression of dra-nupC-pdp operon expression in Bacillus subtilis. Microbiology 2000, 146, 2901–2908. [Google Scholar] [CrossRef] [Green Version]
- Fujita, Y.; Miwa, Y.; Galinier, A.; Deutscher, J. Specific recognition of the Bacillus subtilis gnt cis-acting catabolite-responsive element by a protein complex formed between CcpA and seryl-phosphorylated HPr. Mol. Microbiol. 1995, 17, 953–960. [Google Scholar] [CrossRef]
- Gu, Y.; Lv, X.; Liu, Y.; Li, J.; Du, G.; Chen, J.; Rodrigo, L.-A.; Liu, L. Synthetic redesign of central carbon and redox metabolism for high yield production of N-acetylglucosamine in Bacillus subtilis. Metab. Eng. 2018, 51, 59–69. [Google Scholar] [CrossRef]
- Akram, M. Citric Acid Cycle and Role of its Intermediates in Metabolism. Cell Biophys. 2013, 68, 475–478. [Google Scholar] [CrossRef]
- Harrison, C. GrpE, a nucleotide exchange factor for DnaK. Cell Stress Chaperon. 2003, 8, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Alarcon, D.A.; Nandi, M.; Carpena, X.; Fita, I.; Loewen, P.C. Structure of glycerol-3-phosphate dehydrogenase (GPD1) from Saccharomyces cerevisiae at 2.45 Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1279–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Lee, K.H.; Kim, T.Y.; Lee, S.Y. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc. Natl. Acad. Sci. USA 2007, 104, 7797–7802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Lee, S.Y. Metabolic pathways and fermentative production of L-aspartate family amino acids. Biotechnol. J. 2010, 5, 560–577. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Koumoutsi, A.; Scholz, R.; Schneider, K.; Vater, J.; Süssmuth, R.; Piel, J.; Borriss, R. Genome analysis of Bacillus amyloliquefaciens FZB42 reveals its potential for biocontrol of plant pathogens. J. Biotechnol. 2009, 140, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Stöver, A.G.; Driks, A. Control of Synthesis and Secretion of the Bacillus subtilis Protein YqxM. J. Bacteriol. 1999, 181, 7065–7069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Gao, P.; Zhao, S.; Bie, X.; Lu, Z.; Zhang, C.; Lv, F. iTRAQ-based proteomic analysis of LI-F type peptides produced by Paenibacillus polymyxa JSa-9 mode of action against Bacillus cereus. J. Proteom. 2017, 150, 130–140. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Protein ID | Protein Name | Gene | Primers |
|---|---|---|---|---|
| 1 | I2C429_BACAM | Gamma-glutamyl phosphate reductase | proA | F-GCGCTCGTCAGTGTCATTTA |
| R-GGTTTGGCTGTTTCGTCAAT | ||||
| 2 | I2C0D4_BACAM | Serine--tRNA ligase | serS | F-AACCTGCCGATCAATTATGC |
| R-ATCACCGGTACACATGCTCA | ||||
| 3 | A0A0D7XLI1_BACAM | 50S ribosomal protein L23 | rplW | F-TAAGCGCCCCGTCATTACT |
| R-TTCTGCGACGGCTAGTCATA | ||||
| 4 | A0A0D7XV55_BACAM | 30S ribosomal protein S4 | rpsD | F-ATTCCGCACATTGTTTGACA |
| R-CCGATTGTTTGACCAGGTTT | ||||
| 5 | I2C4J1_BACAM | Pyruvate dehydrogenase E1 component subunit alpha | pdhA | F-CGCTTGAACAGGACGACTTT |
| R-GTCTGTGCGGATGATTGCTT | ||||
| 6 | I2C7G5_BACAM | Glucose-6-phosphate 1-dehydrogenase | zwf | F-AGTCATCCGGTTTGCGAATG |
| R-TTCGCTGCGGATTTCTTCTG | ||||
| 7 | I2C738_BACAM | Glycerol-3-phosphate dehydrogenase [NAD(P)+] | gpsA | F-ACAAAAGGGGTCCATCTCGT |
| R-CAGCCATTTTACGGTAGCCC | ||||
| 8 | I2C8P2_BACAM | Long-chain fatty-acid-CoA ligase | lcfA | F-TGAGGAGCTGGCAGGTAAAA |
| R-GGTATGACGACAATTCCCGC | ||||
| 9 | I2C8L1_BACAM | Acetolactate synthase | ilvB | F-ACTGGTTTTATGCCGTCACG |
| R-CCCTGAAGGCGTTTGTTACC | ||||
| 10 | CHEY_BACSU | Chemotaxis protein CheY | CheY | F-GGAAGCTGAAAATGGAGCAC |
| R-TGATTGCTGTCCCATAGCAG | ||||
| 11 | H9TE70_BACAM | Fengycin synthetase A | fen A | F-GGAATCACAGAGCAGCATGA |
| R-TAAGCAGATGGTCGGCTTCT | ||||
| 12 | I2C3L7_BACAM | Thiazole synthase | thiG | F-AAAGCTTCAGGGCTTTGTGA |
| R-CACTCCGAGCTCCTCAAGAC | ||||
| 13 | I2C0H9_BACAM | AbrB family transcriptional regulator, stage V sporulation protein T | spoVT | F-GTGACGCGTATATTGCCGTT |
| R-GAAAAGATGACCACGGCACC | ||||
| 14 | TRXB_BACSU | Thioredoxin reductase | trxB | F-AACAGGCTGCGACATTTGAG |
| R-CCGGAAATCACGACATGCTT | ||||
| 15 | A0A1Y0X6E7_BACAM | Chemical-damaging agent resistance protein C | terD | F-ATGGTGCTTGTCATGAACGG |
| R-TCTGTATCCGCGTCCATCTC | ||||
| 16 | I2C550_BACAM | flagellar motor switch protein FliG | fliG | F-TAAATTACGCCCGCCAAGTG |
| R-TGCGTAAAGGAGGAGGACAG | ||||
| 17 | Housekeeping gene | gatB_Yqey | F-AGCTGGTCGTGAAGACCTTG | |
| R-CGGCATAACAGCAGTCATCA | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; Li, R.; Yang, P.; Luo, W.; Chen, S.; Bilal, M.; Xu, H.; Gu, C.; Liu, S.; Zhao, Y.; et al. iTRAQ-BASED Proteomic Analysis of the Mechanism of Fructose on Improving Fengycin Biosynthesis in Bacillus Amyloliquefaciens. Molecules 2021, 26, 6309. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26206309
Lu H, Li R, Yang P, Luo W, Chen S, Bilal M, Xu H, Gu C, Liu S, Zhao Y, et al. iTRAQ-BASED Proteomic Analysis of the Mechanism of Fructose on Improving Fengycin Biosynthesis in Bacillus Amyloliquefaciens. Molecules. 2021; 26(20):6309. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26206309
Chicago/Turabian StyleLu, Hedong, Ruili Li, Panping Yang, Weibo Luo, Shunxian Chen, Muhammad Bilal, Hai Xu, Chengyuan Gu, Shuai Liu, Yuping Zhao, and et al. 2021. "iTRAQ-BASED Proteomic Analysis of the Mechanism of Fructose on Improving Fengycin Biosynthesis in Bacillus Amyloliquefaciens" Molecules 26, no. 20: 6309. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26206309