
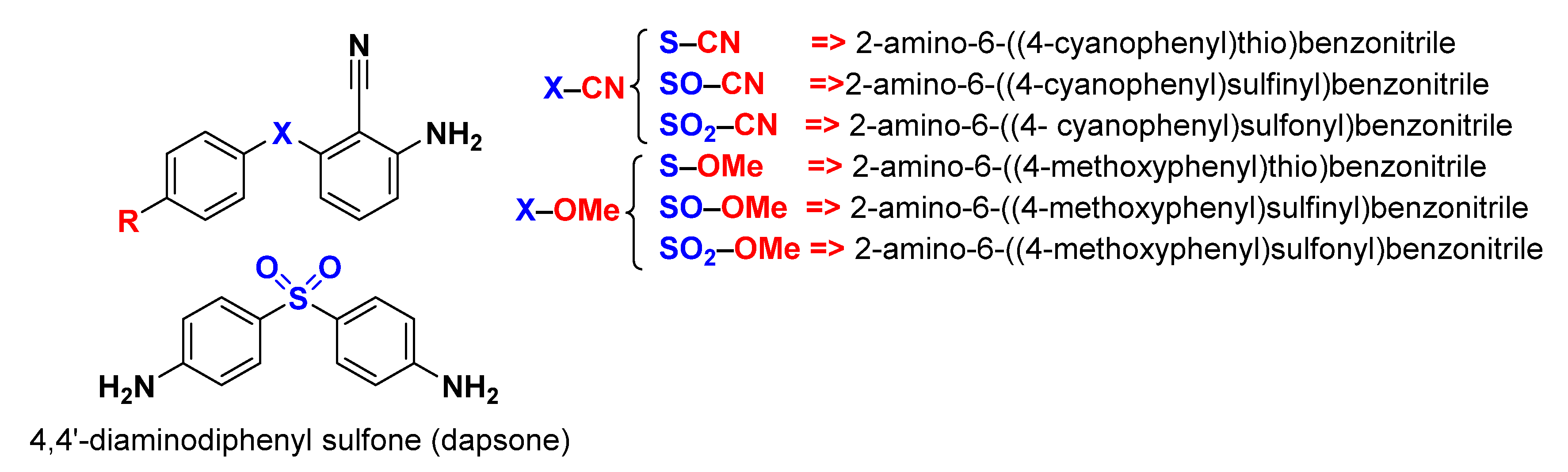
Diaryl Sulfide Derivatives as Potential Iron Corrosion Inhibitors: A Computational Study

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Computational Details

{kind=link}
{kind=link}
{kind=link}
| Descriptor | Mathematical Form |
|---|---|
| Ionization potential (I) | I = −EHOMO |
| Electron affinity (A) | A = −ELUMO |
| Energy gap (ΔE) | ΔE = ELUMO −EHOMO |
| Electronegativity (χ) [23,24] | |
| Chemical hardness (η) [23,24] | |
| Global electrophilicity index (ω) [25] | |
| The number of electrons transferred (ΔN) from the inhibitor to the iron surface. The work function of metal is the work function of iron [26] | |
| The energy associated with a backing donation |
3. Results and Discussion
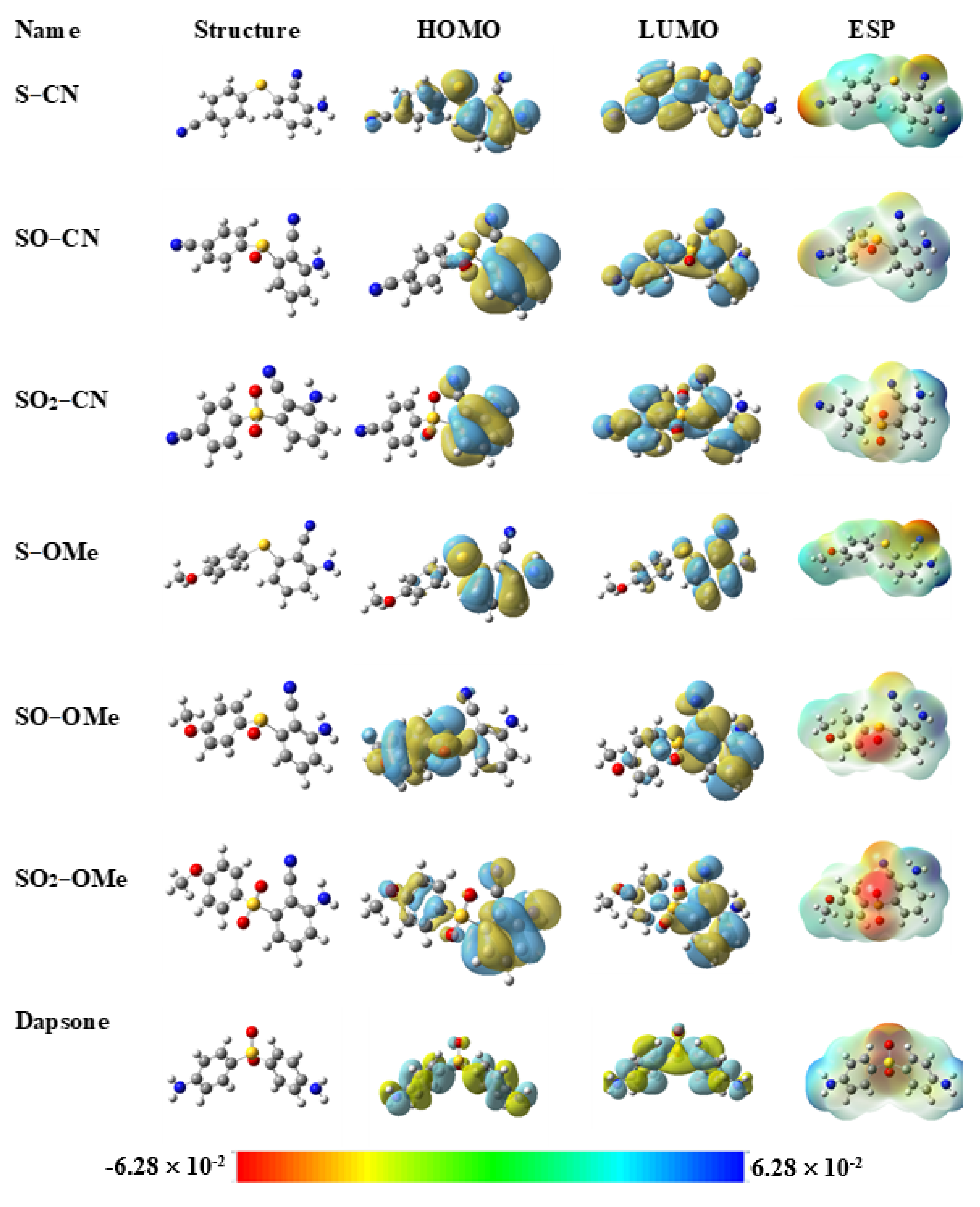
3.1. Quantum Chemical Study
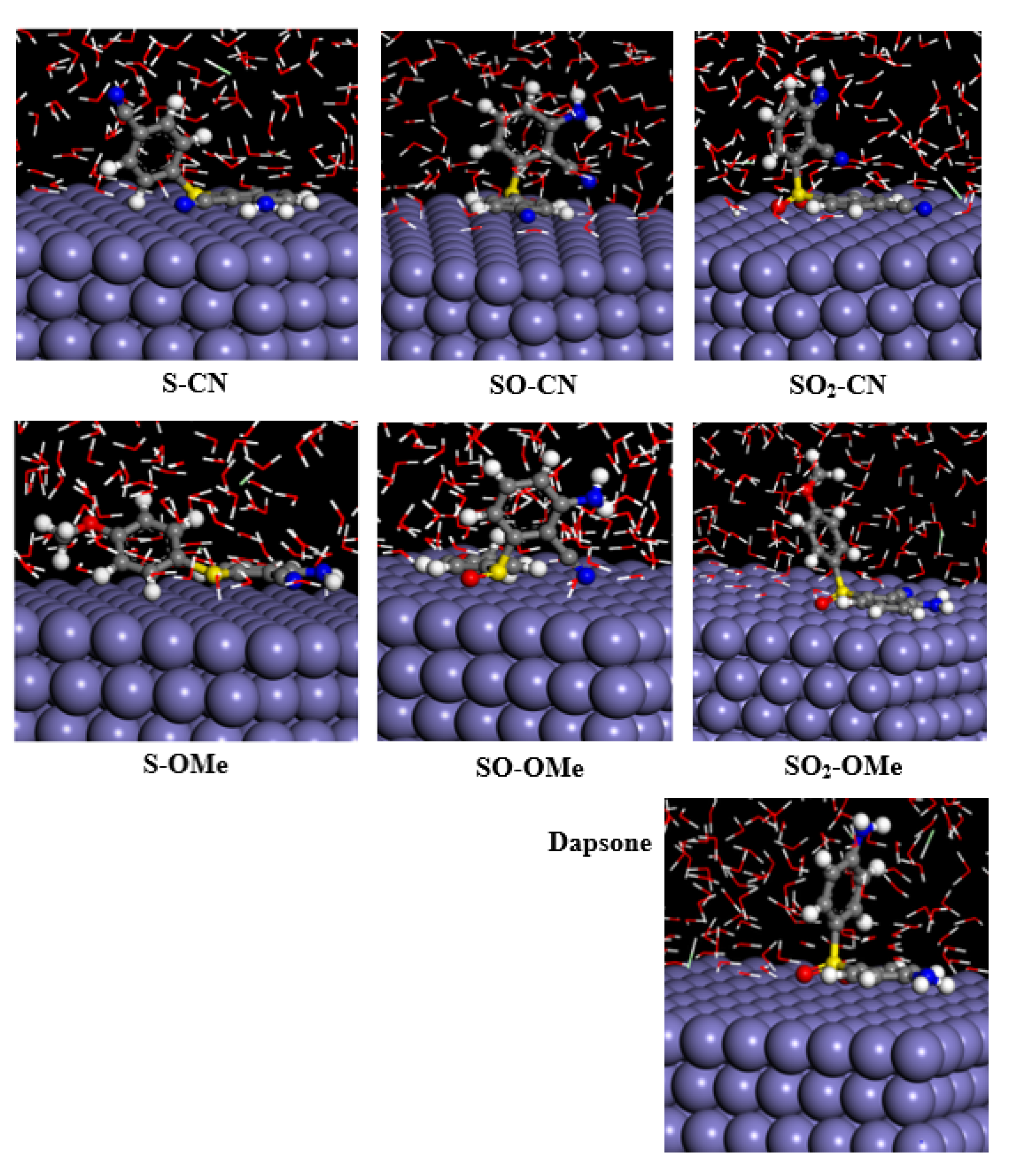
3.2. Monte Carlo (MC) Simulations
4. Conclusions
- The DFT geometry of the studied molecules is not flat and has a bent molecular shape, which leads to incomplete molecular adsorption on the surface.
- Two factors control the adsorption mode on the iron surface: the extent of coplanarity of aryl moiety with the X-group and the value of individual negative atomic charges.
- The dipole moment of the studied molecules correlates well with their adsorption ability. A molecule with a lower dipole moment has better adsorption on the iron surface.
- Although the X–CN molecules are more reactive and less rigid than X–OMe molecules, the latter have a stronger ability for adsorption because of the high electron-donating ability of the methoxy group.
- Based on the adsorption study, all the studied compounds, except for SO–CN, show higher inhibition efficiency than dapsone. Accordingly, we nominate these inhibitors as effective iron surface corrosion inhibitors.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaya, S.; Banerjee, P.; Saha, S.K.; Tüzün, B.; Kaya, C. Theoretical evaluation of some benzotriazole and phospono derivatives as aluminum corrosion inhibitors: DFT and molecular dynamics simulation approaches. RSC Adv. 2016, 6, 74550–74559. [Google Scholar] [CrossRef]
- Kabanda, M.M.; Obot, I.B.; Ebenso, E.E. Computational study of some amino acid derivatives as potential corrosion inhibitors for different metal surfaces and in different media. Int. J. Electrochem. Sci. 2013, 8, 10839–10850. [Google Scholar]
- Kandemirli, S.G.; Yilmazer, M.I.; Saracoglu, M.; Kandemirli, F. Theoretical B3LYP study of contrast agent metrizoate. Int. J. Chem. Technol. 2020, 4, 60–70. [Google Scholar] [CrossRef]
- Geerlings, P.; Proft, F.D.; Langenaeker, W. Conceptual Density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Ebenso, E.E.; Arslan, T.; Kandemirli, F.; Caner, N.; Love, I. Quantum chemical studies of some rhodanine azosulpha drugs as corrosion inhibitors for mild steel in acidic medium. Int. J. Quantum Chem. 2010, 110, 1003–1018. [Google Scholar] [CrossRef]
- Danaee, I.; RameshKumar, S.; RashvandAvei, M.; Vijayan, M. Electrochemical and quantum chemical studies on corrosion inhibition performance of 2,2′-(2-hydroxyethylimino)bis[N-(alphaalpha-dimethylphenethyl)-N-methylacetamide] on mild steel corrosion in 1 M HCl Solution. Mater. Res. 2020, 23, e20180610. [Google Scholar] [CrossRef]
- Sachin, H.P.; Moinuddin Khan, M.H.; Raghavendra, S.; Bhujangaiah, N.S. L-Dopa as corrosion inhibitor for mild steel in mineral acid medium. Open Electrochem. J. 2009, 1, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Dohare, P.; Chauhan, D.; Sorour, A.; Quraishi, M. DFT and experimental studies on the inhibition potentials of expired tramadol drug on mild steel corrosion in hydrochloric acid. Mater. Discov. 2017, 9, 30–41. [Google Scholar] [CrossRef]
- Al-Fahemia, J.H.; Abdallaha, M.; Elshafie; Gad, A.M.; Jahdaly, B. Experimental and theoretical approach studies for melatonin drug as safely corrosion inhibitors for carbon steel using DFT. J. Mol. Liq. 2016, 222, 1157–1163. [Google Scholar] [CrossRef]
- EL-Haddad, M.N.; Fouda, A.S.; Hassan, A.F. Data from chemical, electrochemical and quantum chemical studies for interaction between cephapirin drug as an eco-friendly corrosion inhibitor and carbon steel surface in acidic medium. Chem. Data Collect. 2019, 22, 100251. [Google Scholar] [CrossRef]
- Abdelaal, M.S.; Abdel-Wahab, A.A.; Assaf, F.H. Inhipition der korrosion von zinn und cadmium H2SO4 losungen durch verschiedene organische sulfone, sulfoxide und sulfide. Metalloberflaeche 1980, 34, 323–327. [Google Scholar]
- Trabanelli, G.; Zucchi, F.; Gullini, G.; Carassiti, V. Correlation of the Structure and the Inhibitive Action of Some Sulphoxides. Br. Corros. J. 1969, 4, 212–215. [Google Scholar] [CrossRef]
- James, J.P.; Lalgudi, R.S. Corrosion Inhibiting Coating Additive. U.S. Patent Application No. 16/347,247, 22 August 2019. [Google Scholar]
- Monzo, J.; Garcia-Anton, J.; Guinon, J.L. Study of corrosion on copper strips by mixtures of mercaptans, sulphides and disulphides with elemental sulphur in the ASTM D-130 test by means of electron microscopy (SEM) and energy dispersive X-ray (EDX). J. Anal. Chem. 1992, 343, 593–596. [Google Scholar] [CrossRef]
- Singh, A.; Singh, A.K.; Quraishi, M.A. Dapsone: A novel corrosion inhibitor for mild steel in acid media. Open Corros. J. 2010, 2, 43–51. [Google Scholar]
- Singh, P.; Chauhan, D.S.; Chauhan, S.S.; Singh, G.; Quraishi, M.A. Chemically modified expired Dapsone drug as environmentally benign corrosion inhibitor for mild steel in sulphuric acid useful for industrial pickling process. J. Mol. Liq. 2019, 286, 110903. [Google Scholar] [CrossRef]
- Zhang, R.; Ding, H.; Pu, X.; Qian, Z.; Xiao, Y. Recent advances in the synthesis of sulfides, sulfoxides and sulfones via C-S bond construction from non-halide substrates. Catalysts 2020, 10, 1339. [Google Scholar] [CrossRef]
- Chan, J.H.; Hong, J.S.; Hunter, R.N., III; Orr, G.F.; Cowan, J.R.; Sherman, D.B.; Sparks, S.M.; Reitter, B.E.; Andrews, C.W., III; Hazen, R.J.; et al. 2-Amino-6-arylsulfonylbenzonitriles as non-nucleoside reverse transcriptase inhibitors of HIV-1. J. Med. Chem. 2001, 44, 1866–1882. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases—the evolution of a chemical concept, Coord. Chem. Rev. 1990, 100, 403–425. [Google Scholar] [CrossRef]
- Dewar, M.J.; Thiel, W. Ground states of molecules. 38. The MNDO method. Approximations and parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: New York, NY, USA, 1960. [Google Scholar]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef]
- Kokalj, A. On the HSAB based estimate of charge transfer between adsorbates and metal surfaces. Chem. Phys. 2012, 393, 1–12. [Google Scholar] [CrossRef]
- Rubinstein, R.Y.; Ridder, A.; Vaisman, R. Fast Sequential Monte Carlo Methods for Counting and Optimization; John Wiley & Sons: New York, NY, USA, 2014. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Dassault Systèmes. Dassault Systèmes BIOVIA, Materials Studio, 17.1.0.48; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
- Sun, H.; Ren, P.; Fried, J.R. The COMPASS force field: Parameterization and validation for phosphazenes. Comput. Theor. Polym. Sci. 1998, 8, 229–246. [Google Scholar] [CrossRef]
- Guo, L.; Qi, C.; Zheng, X.; Zhang, R.; Shen, X.; Kaya, S. Toward understanding the adsorption mechanism of large size organic corrosion inhibitors on an Fe(110) surface using the DFTB method. RSC Adv. 2017, 7, 29042–29050. [Google Scholar] [CrossRef] [Green Version]
- Fouda, A.S.; Ismail, M.A.; EL-ewady, G.Y.; Abousalem, A.S. Evaluation of 4-amidinophenyl-2,2′-bithiophene and its aza-analogue as novel corrosion inhibitors for CS in acidic media: Experimental and theoretical study. J. Mol. Liq. 2017, 240, 372–388. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, E. Exploring Chemistry with Electronic Structure Methods, 3rd ed.; Gaussian, Inc.: Wallingford, CT, USA, 2015. [Google Scholar]
- Pearson, R.G. Absolute electronegativity and hardness: Application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]
- Lagren´ee, M.; Mernari, B.; Chaibi, N.; Traisnel, M.; Vezin, H.; Bentiss, F. Investigation of the inhibitive effect of substituted oxadiazoles on the corrosion of mild steel in HCl medium. Corros. Sci. 2001, 43, 951–962. [Google Scholar] [CrossRef]
- Quraishi, M.A.; Sardar, R. Hector bases—A new class of heterocyclic corrosion inhibitors for mild steel in acid solutions. J. Appl. Electrochem. 2003, 33, 1163–1168. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Saleh, D.I.; El-Hendawy, M.M.; Fallatah, A.M.; Mersal, G.A.M.; Boukherroub, R.; Wysocka, J.; Ryl, J.; Amin, M.A. Efficacious alkaline copper corrosion inhibition by a mixed ligand copper(II) complex of 2,2′-bipyridine and glycine: Electrochemical and theoretical studies. ChemElectroChem 2021, 8, 2052–2064. [Google Scholar] [CrossRef]
- Khalil, N. Quantum chemical approach of corrosion inhibition. Electrochim. Acta 2003, 48, 2635–2640. [Google Scholar] [CrossRef]



| S–CN | SO–CN | SO2–CN | S–OMe | SO–OMe | SO2–OMe | Dapsone | |
|---|---|---|---|---|---|---|---|
| CM–S; CD–S (Å) * | 1.79; 1.79 | 1.84; 1.84 | 1.81; 1.82 | 1.79; 1.79 | 1.82; 1.84 | 1.79; 1.82 | 1.79; 1.79 |
| <CM–S–CD (°) | 104 | 97 | 105 | 104 | 98 | 105 | 106 |
| <CM–CM–S–CD (°) | 139 | 97 | 82 | 89 | 88 | 84 | 90 |
| <CD–CD–S–CM (°) | 142 | 83 | 72 | 2 | 82 | 73 | 90 |
| Molecule | EHOMO (eV) | ELUMO (eV) | ∆E (eV) | DM. (D) | MV (cm3/mol) | η (eV) | χ (eV) | ω (eV) | ∆Eb-d (eV) | ∆N (e) |
|---|---|---|---|---|---|---|---|---|---|---|
| S−CN | −6.28 | −1.85 | 4.43 | 12.27 | 214 | 2.22 | −4.07 | 3.73 | −0.56 | 2.06 |
| SO−CN | −6.39 | −2.00 | 4.39 | 14.41 | 206 | 2.19 | −4.19 | 4.01 | −0.55 | 2.11 |
| SO2−CN | −6.48 | −2.29 | 4.19 | 12.89 | 195 | 2.10 | −4.39 | 4.59 | −0.53 | 2.25 |
| S−OMe | −6.19 | −1.75 | 4.44 | 6.09 | 186 | 2.22 | −3.97 | 3.54 | −0.56 | 2.04 |
| SO−OMe | −6.35 | −1.92 | 4.43 | 9.84 | 210 | 2.21 | −4.13 | 3.86 | −0.55 | 2.08 |
| SO2−OMe | −6.43 | −2.14 | 4.29 | 10.47 | 161 | 2.15 | −4.28 | 4.28 | −0.54 | 2.17 |
| Dapsone | −5.93 | −1.00 | 4.93 | 6.38 | 175 | 2.47 | −3.47 | 2.44 | −0.62 | 1.73 |
| Total Energy | Adsorption Energy | Rigid Adsorption Energy | Deformation Energy | dEad/dNi | |||
|---|---|---|---|---|---|---|---|
| Inh | H2O | HCl | |||||
| S–CN | −3791 | −3826 | −4023 | 197 | −148 | −11 | −9 |
| SO–CN | −3760 | −3802 | −3993 | 191 | −135 | −12 | −8 |
| SO2–CN | −3799 | −3845 | −4040 | 195 | −162 | −14 | −7 |
| S–OMe | −3823 | −3851 | −4046 | 195 | −146 | −12 | −9 |
| SO–OMe | −3825 | −3858 | −4054 | 196 | −125 | −13 | −9 |
| SO2–OMe | −3808 | −3845 | −4040 | 196 | −151 | −12 | −7 |
| Dapsone | −3795 | −3816 | −4007 | 191 | −137 | −11 | −7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Hendawy, M.M.; Kamel, A.M.; Mohamed, M.M.A.; Boukherroub, R.; Ryl, J.; Amin, M.A. Diaryl Sulfide Derivatives as Potential Iron Corrosion Inhibitors: A Computational Study. Molecules 2021, 26, 6312. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26206312
El-Hendawy MM, Kamel AM, Mohamed MMA, Boukherroub R, Ryl J, Amin MA. Diaryl Sulfide Derivatives as Potential Iron Corrosion Inhibitors: A Computational Study. Molecules. 2021; 26(20):6312. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26206312
Chicago/Turabian StyleEl-Hendawy, Morad M., Asmaa M. Kamel, Mahmoud M. A. Mohamed, Rabah Boukherroub, Jacek Ryl, and Mohammed A. Amin. 2021. "Diaryl Sulfide Derivatives as Potential Iron Corrosion Inhibitors: A Computational Study" Molecules 26, no. 20: 6312. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26206312