
Hybrid Chelator-Based PSMA Radiopharmaceuticals: Translational Approach

 , ,
, ,
Abstract
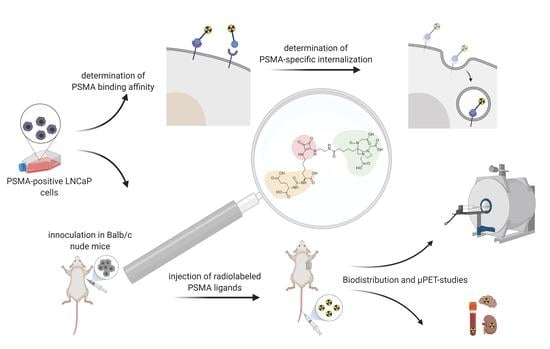
:
1. Introduction
2. Results
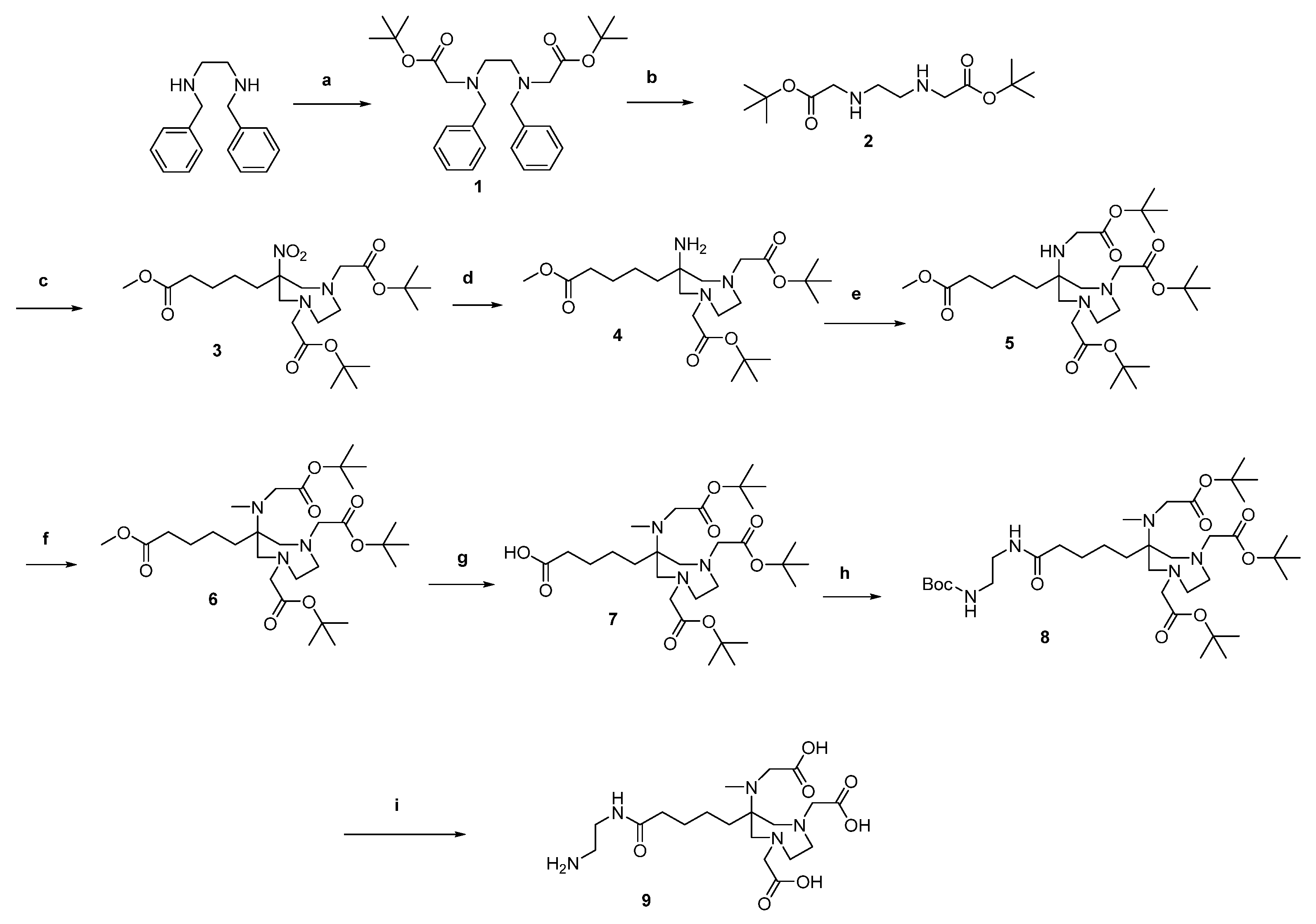
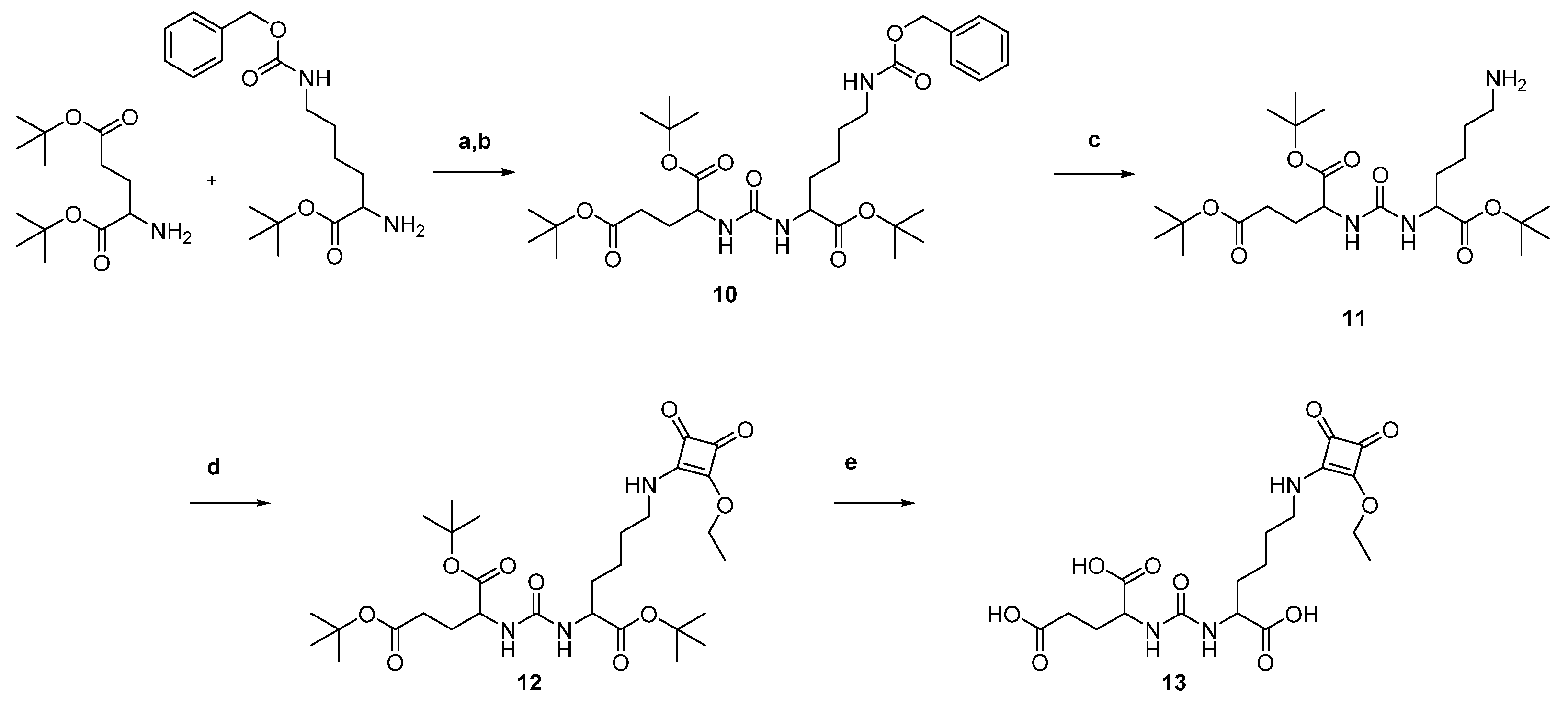
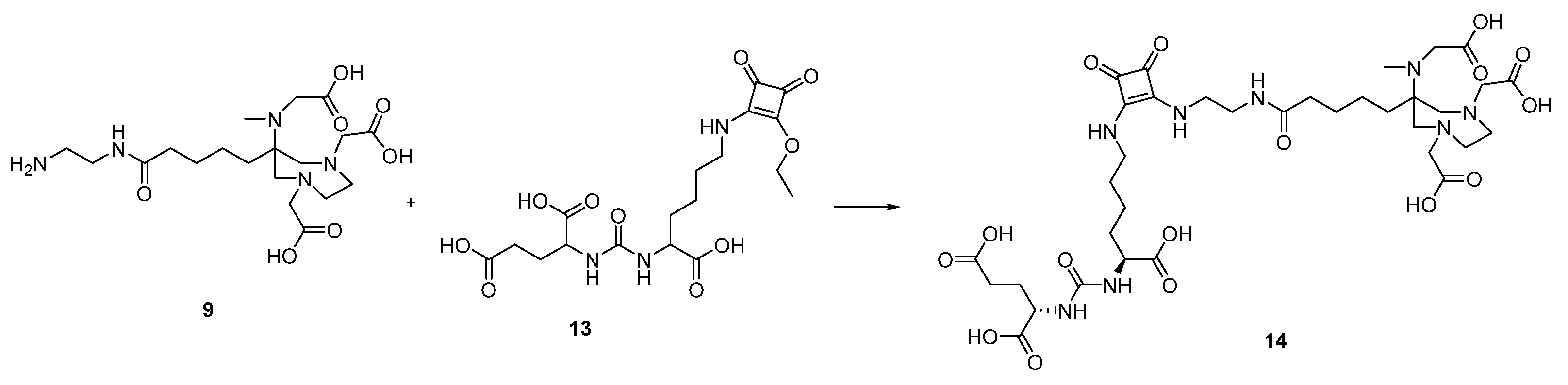
2.1. Organic Synthesis
2.1.1. Synthesis of AAZTA5.SA.KuE Was according to Literature
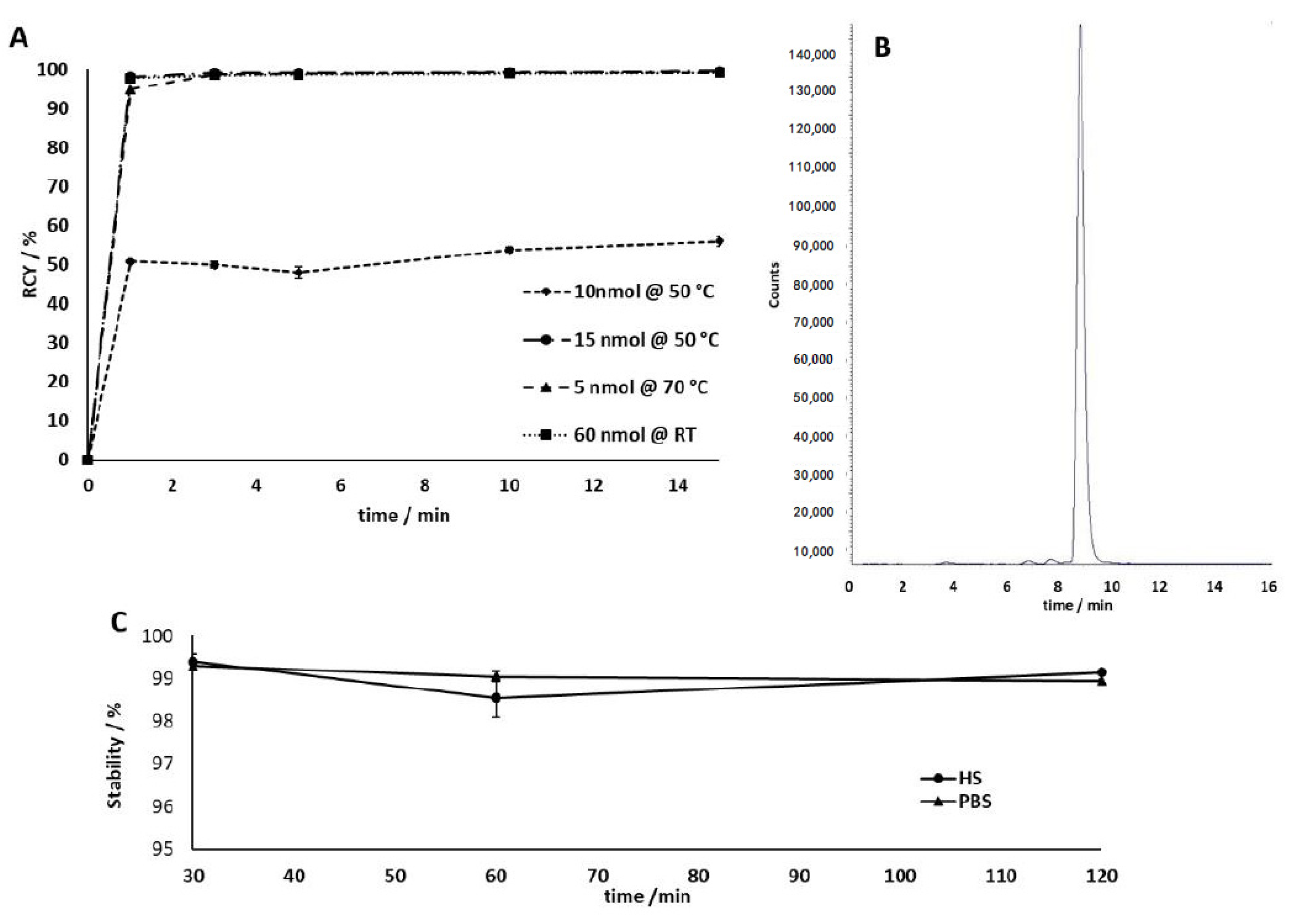
2.1.2. Radiolabeling
2.2. In Vitro Studies
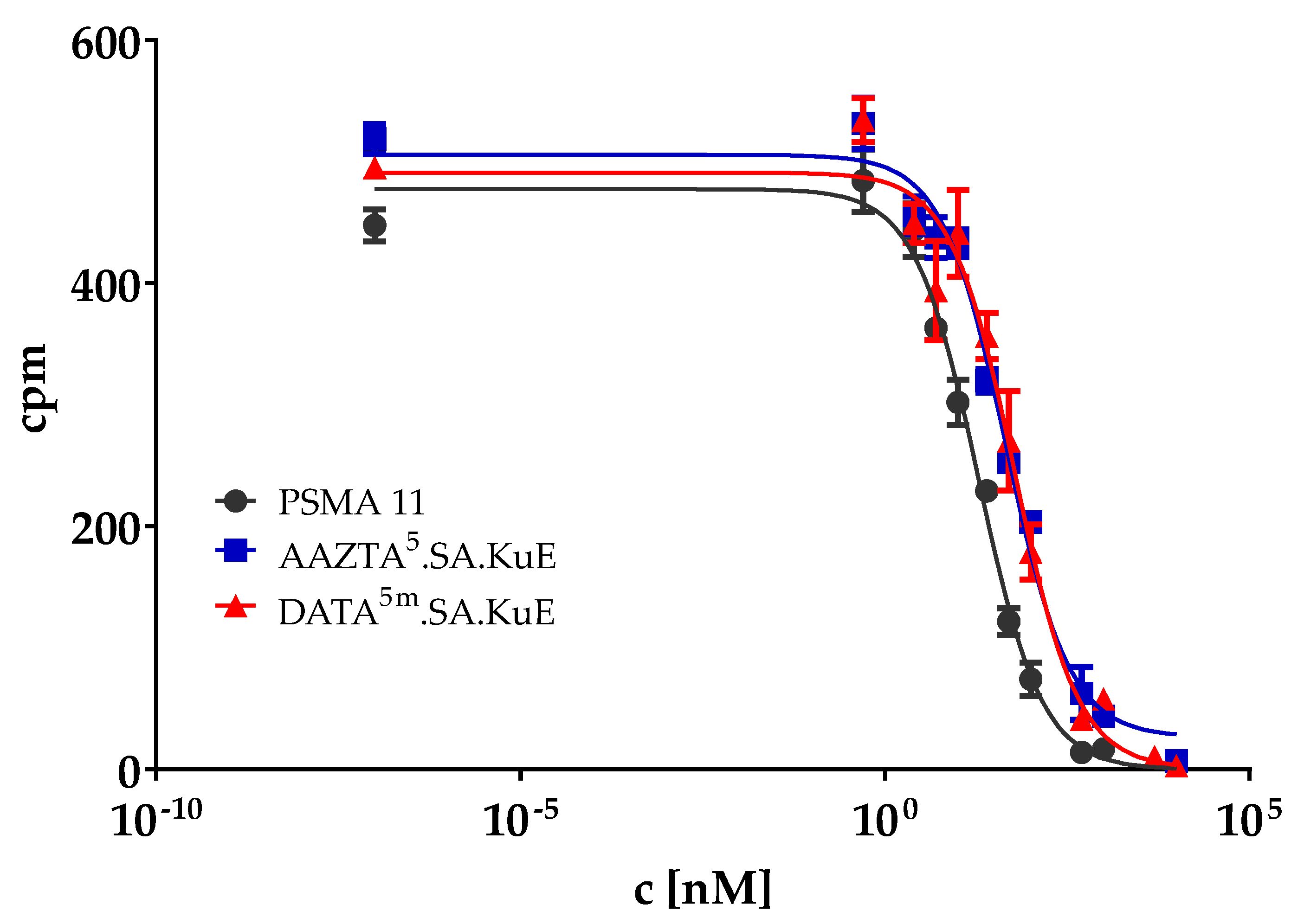
2.2.1. PSMA Binding Affinity
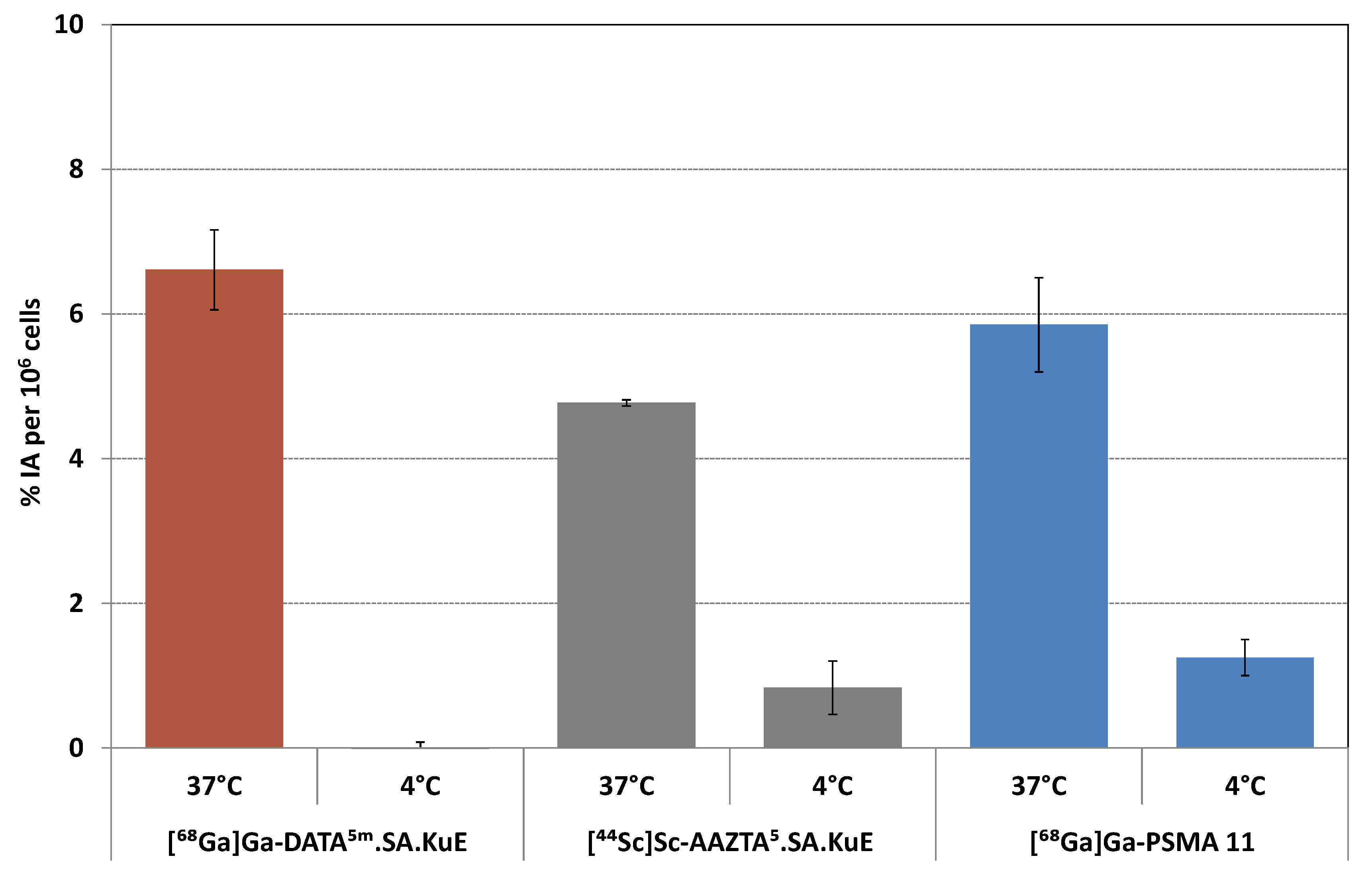
2.2.2. Internalization Ratio
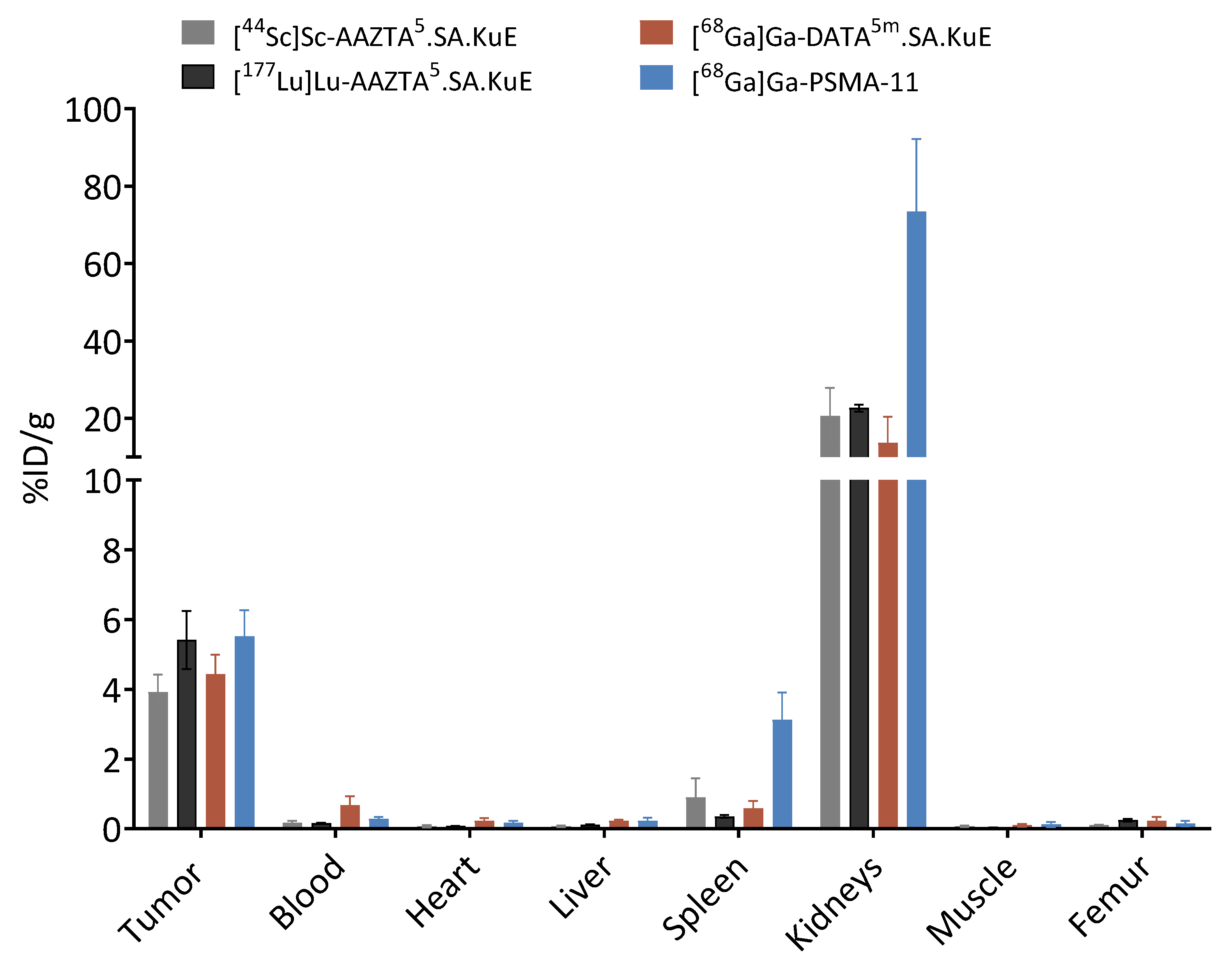
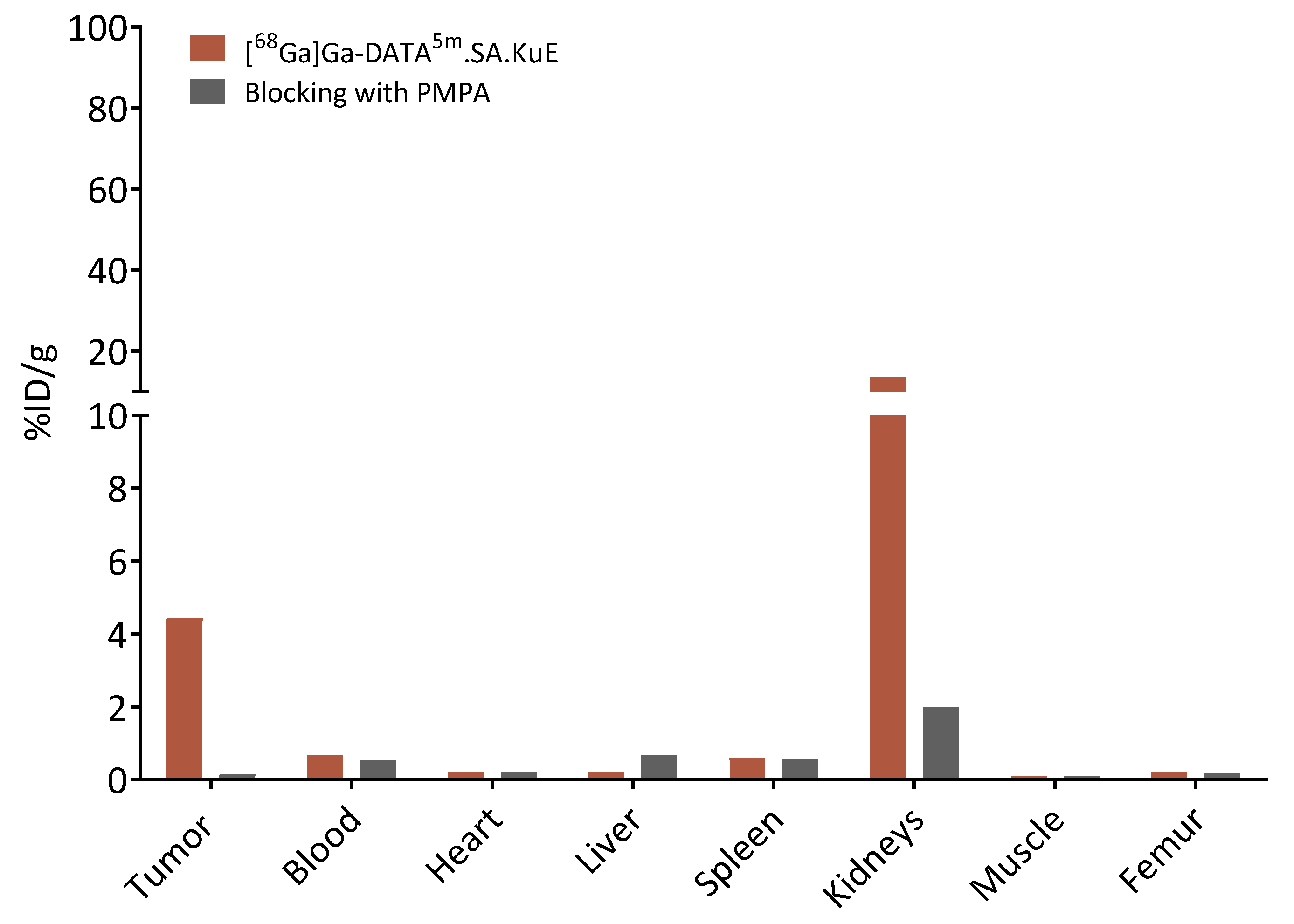
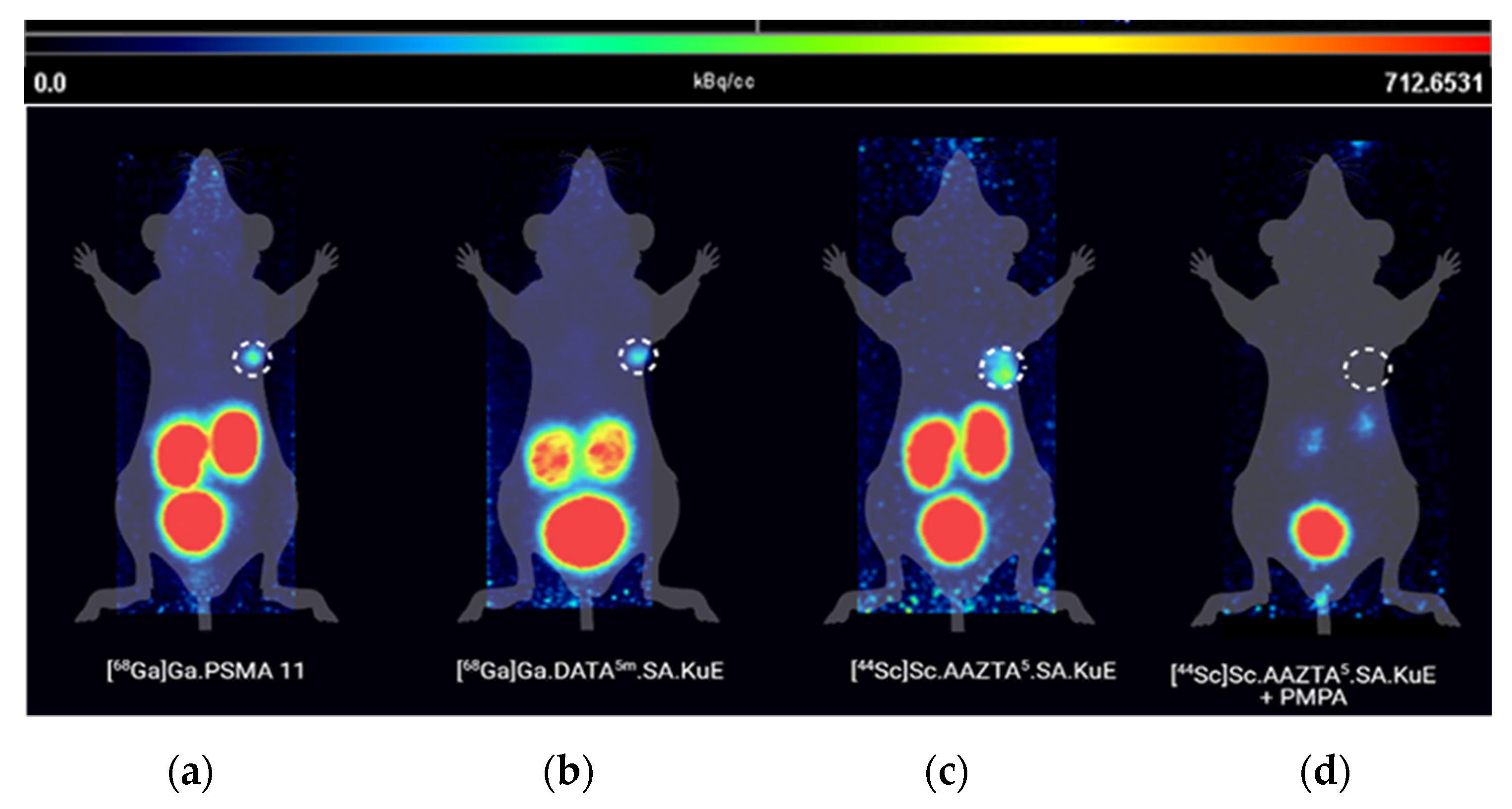
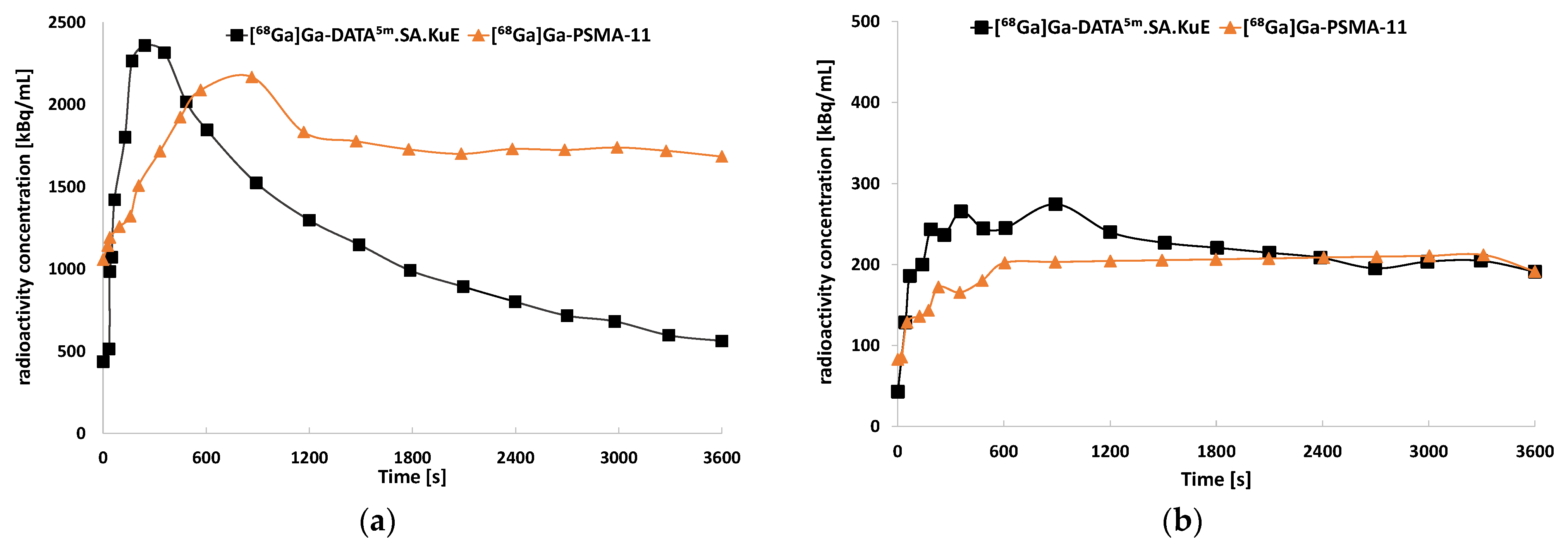
2.3. Animal Studies
3. Discussion
4. Materials and Methods
4.1. General
4.2. Organic Synthesis
4.3. Radiolabeling
4.4. In Vitro Stability Studies
4.5. In Vitro Binding Affinity
4.6. Internalization Ratio
4.7. Animal Studies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chang, S.S.; Heston, W.D.W. The clinical role of prostate-specific membrane antigen (PSMA). Urol. Oncol. 2002, 7, 7–12. [Google Scholar] [CrossRef]
- Chang, S.S.; Bander, N.H.; Heston, W.D.W. Biology of PSMA as a Diagnostic and Therapeutic Target. In Management of Prostate Cancer; Klein, E.A., Ed.; Humana Press: Totowa, NJ, USA, 2004; pp. 609–630. [Google Scholar]
- Gourni, E.; Henriksen, G.; Gamez, P.; Caballero, A.B. Metal-based PSMA radioligands. Molecules 2017, 22, 523. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar] [PubMed]
- Perner, S.; Hofer, M.D.; Kim, R.; Shah, R.B.; Li, H.; Möller, P.; Hautmann, R.E.; Gschwend, J.E.; Kuefer, R.; Rubin, M.A. Prostate-specific membrane antigen expression as a predictor of prostate cancer progression. Hum. Pathol. 2007, 38, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Prostate Cancer Statistics|World Cancer Research Fund International. Available online: https://www.wcrf.org/dietandcancer/prostate-cancer-statistics/ (accessed on 19 May 2021).
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef]
- Kopka, K.; Benešová, M.; Bařinka, C.; Haberkorn, U.; Babich, J. Glu-ureido-based inhibitors of prostate-specific membrane antigen: Lessons learned during the development of a novel class of low-molecular-weight theranostic radiotracers. J. Nucl. Med. 2017, 58, 17S–26S. [Google Scholar] [CrossRef] [Green Version]
- Benesová, M.; Schäfer, M.; Bauder-Wüst, U.; Afshar-Oromieh, A.; Kratochwil, C.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Preclinical evaluation of a tailor-made DOTA-conjugated PSMA inhibitor with optimized linker moiety for imaging and endoradiotherapy of prostate cancer. J. Nucl. Med. 2015, 56, 914–920. [Google Scholar] [CrossRef] [Green Version]
- Valstar, M.H.; de Bakker, B.S.; Steenbakkers, R.J.H.M.; de Jong, K.H.; Smit, L.A.; Klein Nulent, T.J.W.; van Es, R.J.J.; Hofland, I.; de Keizer, B.; Jasperse, B.; et al. The tubarial salivary glands: A potential new organ at risk for radiotherapy. Radiother. Oncol. 2021, 154, 292–298. [Google Scholar] [CrossRef]
- Rupp, N.J.; Umbricht, C.A.; Pizzuto, D.A.; Lenggenhager, D.; Töpfer, A.; Müller, J.; Muehlematter, U.J.; Ferraro, D.A.; Messerli, M.; Morand, G.B.; et al. First clinicopathologic evidence of a non-PSMA-related uptake mechanism for 68Ga-PSMA-11 in salivary glands. J. Nucl. Med. 2019, 60, 1270–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afshar-Oromieh, A.; Malcher, A.; Eder, M.; Eisenhut, M.; Linhart, H.G.; Hadaschik, B.A.; Holland-Letz, T.; Giesel, F.L.; Kratochwil, C.; Haufe, S.; et al. Pet imaging with a [68ga]gallium-labelled psma ligand for the diagnosis of prostate cancer: Biodistribution in humans and first evaluation of tumour lesions. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.S.; Hicks, R.J.; Maurer, T.; Eiber, M. Prostate-specific membrane antigen PET: Clinical utility in prostate cancer, normal patterns, pearls, and pitfalls. Radiographics 2018, 38, 200–217. [Google Scholar] [CrossRef] [Green Version]
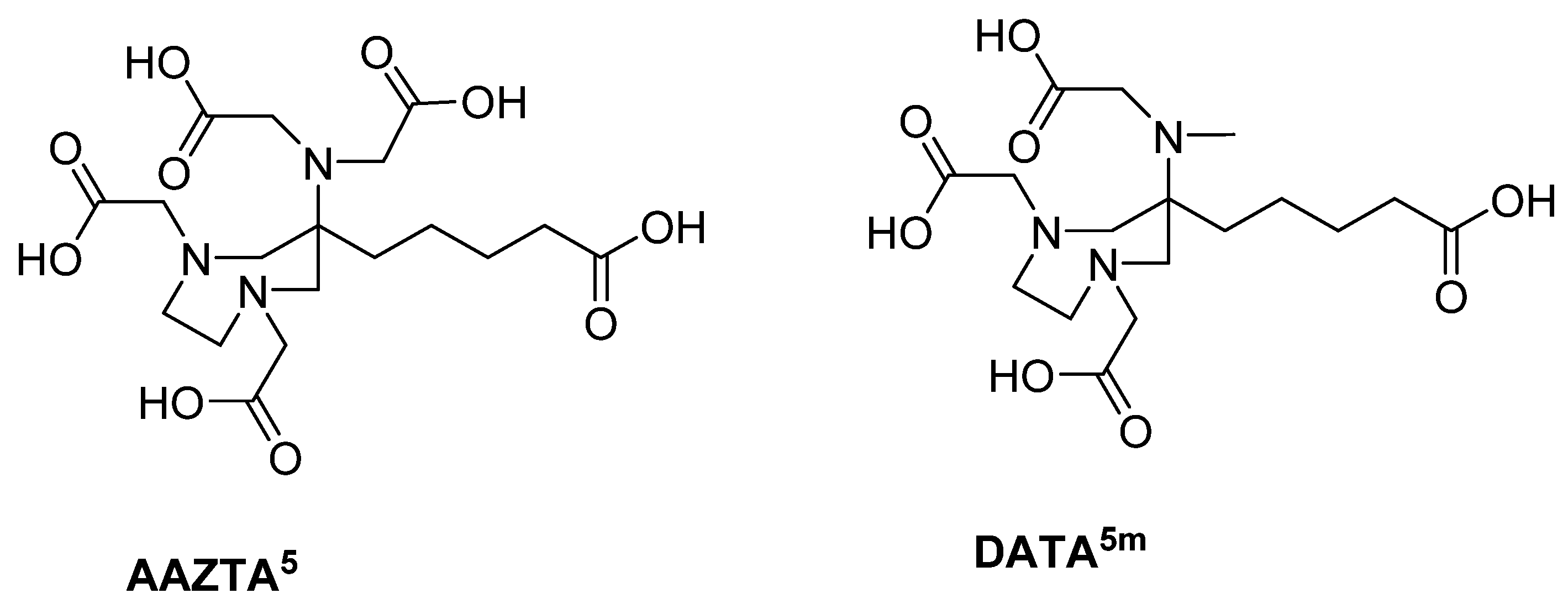
- Farkas, E.; Nagel, J.; Waldron, B.P.; Parker, D.; Tóth, I.; Brücher, E.; Rösch, F.; Baranyai, Z. Equilibrium, Kinetic and Structural Properties of Gallium(III) and Some Divalent Metal Complexes Formed with the New DATAm and DATA5m Ligands. Chem. Eur. J. 2017, 23, 10358–10371. [Google Scholar] [CrossRef] [Green Version]
- Greifenstein, L.; Grus, T.; Nagel, J.; Sinnes, J.P.; Rösch, F. Synthesis and labeling of a squaric acid containing PSMA-inhibitor coupled to AAZTA5 for versatile labeling with 44Sc, 64Cu, 68Ga and 177Lu. Appl. Radiat. Isot. 2020, 156, 108867. [Google Scholar] [CrossRef] [PubMed]
- Baranyai, Z.; Uggeri, F.; Giovenzana, G.B.; Bényei, A.; Brücher, E.; Aime, S. Equilibrium and kinetic properties of the lanthanoids(III) and various divalent metal complexes of the heptadentate ligand AAZTA. Chem. Eur. J. 2009, 15, 1696–1705. [Google Scholar] [CrossRef] [PubMed]
- Seemann, J.; Waldron, B.P.; Roesch, F.; Parker, D. Approaching “kit-type” labelling with 68Ga: The DATA chelators. Chem. Med. Chem. 2015, 10, 1019–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldron, B.P.; Parker, D.; Burchardt, C.; Yufit, D.S.; Zimny, M.; Roesch, F. Structure and stability of hexadentate complexes of ligands based on AAZTA for efficient PET labelling with gallium-68. Chem. Comm. 2013, 49, 579–581. [Google Scholar] [CrossRef]
- Nagy, G.; Szikra, D.D.; Trencsényi, G.; Fekete, A.; Garai, I.; Giani, A.M.; Negri, R.; Masciocchi, N.; Maiocchi, A.; Uggeri, F.; et al. AAZTA: An Ideal Chelating Agent for the Development of44Sc PET Imaging Agents. Angew. Chem. Int. Ed. Engl. 2017, 56, 2118–2122. [Google Scholar] [CrossRef]
- Aime, S.; Calabi, L.; Cavallotti, C.; Gianolio, E.; Giovenzana, G.B.; Losi, P.; Maiocchi, A.; Palmisano, G.; Sisti, M.; Giuria, V.P.; et al. [Gd-AAZTA]-: A new structural entry for an improved generation of MRI contrast agents. Inorg. Chem. 2004, 43, 7588–7590. [Google Scholar] [CrossRef]
- Tsionou, M.I.; Knapp, C.E.; Foley, C.A.; Munteanu, C.R.; Cakebread, A.; Imberti, C.; Eykyn, T.R.; Young, J.D.; Paterson, B.M.; Blower, P.J.; et al. Comparison of macrocyclic and acyclic chelators for gallium-68 radiolabelling. RSC Adv. 2017, 7, 49586–49599. [Google Scholar] [CrossRef] [Green Version]
- Benešová, M.; Bauder-Wüst, U.; Schäfer, M.; Klika, K.D.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Linker Modification Strategies to Control the Prostate-Specific Membrane Antigen (PSMA)-Targeting and Pharmacokinetic Properties of DOTA-Conjugated PSMA Inhibitors. J. Med. Chem. 2016, 59, 1761–1775. [Google Scholar] [CrossRef]
- Zhang, A.X.; Murelli, R.P.; Barinka, C.; Michel, J.; Cocleaza, A.; Jorgensen, W.L.; Lubkowski, J.; Spiegel, D.A. A Remote arene-binding site on prostate specific membrane antigen revealed by antibody-recruiting small molecules. J. Am. Chem Soc. 2010, 132, 12711–12716. [Google Scholar] [CrossRef] [Green Version]
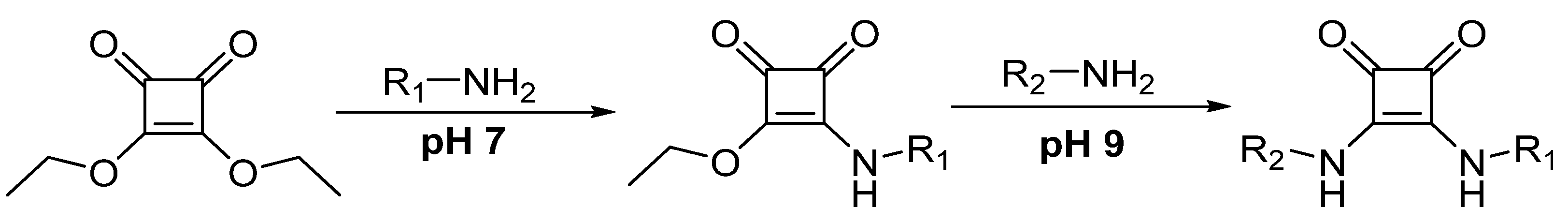
- Tietze, L.F.; Arlt, M.; Beller, M.; Gliisenkampb, K.; Jahdeb, E.; Rajewskyb, M.F.; Gl üsenkamp, K.-H.; Jähde, E.; Rajewsky, M.F. Anticancer Agents, 15. Squaric Acid Diethyl Ester: A New Coupling Reagent for the Formation of Drug Biopolymer Conjugates. Synthesis of Squaric Acid Ester Amides and Diamides. Chem Ber. 1991, 124, 1215–1221. [Google Scholar] [CrossRef]
- Wurm, F.R.; Klok, H.A. Be squared: Expanding the horizon of squaric acid-mediated conjugations. Chem Soc. Rev. 2013, 42, 8220–8236. [Google Scholar] [CrossRef]
- Greifenstein, L.; Engelbogen, N.; Lahnif, H.; Sinnes, J.P.J.-P.; Bergmann, R.; Bachmann, M.; Rösch, F. Synthesis, Labeling and Preclinical Evaluation of a Squaric Acid Containing PSMA Inhibitor Labeled with 68Ga: A Comparison with PSMA-11 and PSMA-617. Chem. Med. Chem 2020, 15, 695–704. [Google Scholar] [CrossRef]
- Grus, T.; Lahnif, H.; Klasen, B.; Moon, E.-S.; Greifenstein, L.; Roesch, F. Squaric Acid-Based Radiopharmaceuticals for Tumor Imaging and Therapy. Bioconj. Chem. 2021, 32, 1223–1231. [Google Scholar] [CrossRef]
- Prohens, R.; Portell, A.; Font-Bardia, M.; Bauzá, A.; Frontera, A. Experimental and theoretical study of aromaticity effects in the solid state architecture on squaric acid derivatives. Cryst. Growth Des. 2014, 14, 2578–2587. [Google Scholar] [CrossRef]
- Quiñonero, D.; Frontera, A.; Ballester, P.; Deyà, P.M. A theoretical study of aromaticity in squaramide and oxocarbons. Tetrahedron Lett. 2000, 41, 2001–2005. [Google Scholar] [CrossRef]
- Dingels, C.; Wurm, F.; Wagner, M.; Klok, H.A.; Frey, H. Squaric acid mediated chemoselective PEGylation of proteins: Reactivity of single-step-activated α-amino poly(ethylene glycol)s. Chem. Eur. J. 2012, 18, 16828–16835. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Rajasekaran, A.K.; Moy, P.; Xia, Y.; Kim, S.; Navarro, V.; Rahmati, R.; Bander, N.H. Constitutive and Antibody-induced Internalization of Prostate-specific Membrane Antigen. Cancer Res. 1998, 58, 4055–4060. [Google Scholar]
- Winter, G.; Vogt, A.; Jiménez-Franco, L.D.; Rinscheid, A.; Yousefzadeh-Nowshahr, E.; Solbach, C.; Beer, A.J.; Glatting, G.; Kletting, P. Modelling the internalisation process of prostate cancer cells for PSMA-specific ligands. Nucl. Med. Biol. 2019, 72–73, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.F.; Tays, K.L.; Maclin, K.M.; Ko, Y.S.; Li, W.; Vitharana, D.; Tsukamoto, T.; Stoermer, D.; Lu, X.C.; Wozniak, K.; et al. Design and Pharmacological Activity of Phosphinic Acid Based NAALADase Inhibitors. J. Med. Chem. 2001, 44, 4170–4175. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.F.; Cole, D.C.; Slusher, B.S.; Stetz, S.L.; Ross, L.E.; Donzanti, B.A.; Trainor, D.A. Design, Synthesis, and Biological Activity of a Potent Inhibitor of the Neuropeptidase N-Acetylated α-Linked Acidic Dipeptidase. J. Med. Chem. 1996, 39, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Rais, R.; Rojas, C.; Wozniak, K.; Wu, Y.; Zhao, M.; Tsukamoto, T.; Rudek, M.A.; Slusher, B.S. Bioanalytical method for evaluating the pharmacokinetics of the GCP-II inhibitor 2-phosphonomethyl pentanedioic acid (2-PMPA). J. Pharm Biomed. Anal. 2014, 88, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Killock, D. PSMA PET–CT improves staging. Nat. Rev. Clin. Oncol. 2020, 17, 337. [Google Scholar] [CrossRef]
- Langbein, T.; Chaussé, G.; Baum, R.P. Salivary gland toxicity of PSMA radioligand therapy: Relevance and preventive strategies. J. Nucl. Med. 2018, 59, 1172–1173. [Google Scholar] [CrossRef] [Green Version]
- Sinnes, J.-P.; Bauder-Wüst, U.; Schäfer, M.; Moon, E.S.; Kopka, K.; Rösch, F. 68Ga, 44Sc and 177Lu-labeled AAZTA5-PSMA-617: Synthesis, radiolabeling, stability and cell binding compared to DOTA-PSMA-617 analogues. EJNMMI Radiopharm. Chem. 2020, 5, 28. [Google Scholar] [CrossRef]
- Ghiani, S.; Hawala, I.; Szikra, D.; Trencsényi, G.; Baranyai, Z.; Nagy, G.; Vágner, A.; Stefania, R.; Pandey, S.; Maiocchi, A. Synthesis, radiolabeling, and pre-clinical evaluation of [ 44 Sc]Sc-AAZTA conjugate PSMA inhibitor, a new tracer for high-efficiency imaging of prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2351–2362. [Google Scholar] [CrossRef]
- Eppard, E.; de la Fuente, A.; Benešová, M.; Khawar, A.; Bundschuh, R.A.; Gärtner, F.C.; Kreppel, B.; Kopka, K.; Essler, M.; Rösch, F. Clinical translation and first in-human use of [44Sc]Sc-PSMA-617 for pet imaging of metastasized castrate-resistant prostate cancer. Theranostics 2017, 7, 4359–4369. [Google Scholar] [CrossRef]
- Schäfer, M.; Bauder-Wust, U.; Leotta, K. A dimerized urea-based inhibitor of the prostate-specific membrane antigen for 68Ga-PET imaging of prostate cancer. EJNMMI Res. 2012, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eppard, E.; Wuttke, M.; Nicodemus, P.L.; Rösch, F. Ethanol-based post-processing of generator-derived 68Ga Toward kit-type preparation of 68Ga-radiopharmaceuticals. J. Nucl. Med. 2014, 55, 1023–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eder, M.; Schäfer, M.; Bauder-Wüst, U.; Hull, W.E.; Wängler, C.; Mier, W.; Haberkorn, U.; Eisenhut, M. 68Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconj. Chem. 2012, 23, 688–697. [Google Scholar] [CrossRef]
- Askoxylakis, V.; Mier, W.; Zitzmann, S.; Ehemann, V.; Zhang, J.; Krämer, S.; Beck, C.; Schwab, M.; Eisenhut, M.; Haberkorn, U. Characterization and development of a peptide (p160) with affinity for neuroblastoma cells. J. Nucl. Med. 2006, 47, 981–988. [Google Scholar] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 [nM] |
|---|---|
| DATA5m.SA.KuE | 51.1 ± 5.5 |
| AAZTA5.SA.KuE | 52.1 ± 4.0 |
| PSMA-11 | 26.2 ± 2.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lahnif, H.; Grus, T.; Pektor, S.; Greifenstein, L.; Schreckenberger, M.; Rösch, F. Hybrid Chelator-Based PSMA Radiopharmaceuticals: Translational Approach. Molecules 2021, 26, 6332. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216332
Lahnif H, Grus T, Pektor S, Greifenstein L, Schreckenberger M, Rösch F. Hybrid Chelator-Based PSMA Radiopharmaceuticals: Translational Approach. Molecules. 2021; 26(21):6332. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216332
Chicago/Turabian StyleLahnif, Hanane, Tilmann Grus, Stefanie Pektor, Lukas Greifenstein, Mathias Schreckenberger, and Frank Rösch. 2021. "Hybrid Chelator-Based PSMA Radiopharmaceuticals: Translational Approach" Molecules 26, no. 21: 6332. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216332