
Mesoporous Poly(melamine-co-formaldehyde) Particles for Efficient and Selective Phosphate and Sulfate Removal

 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
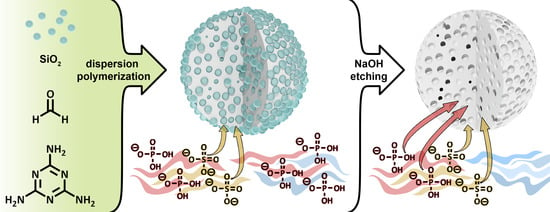
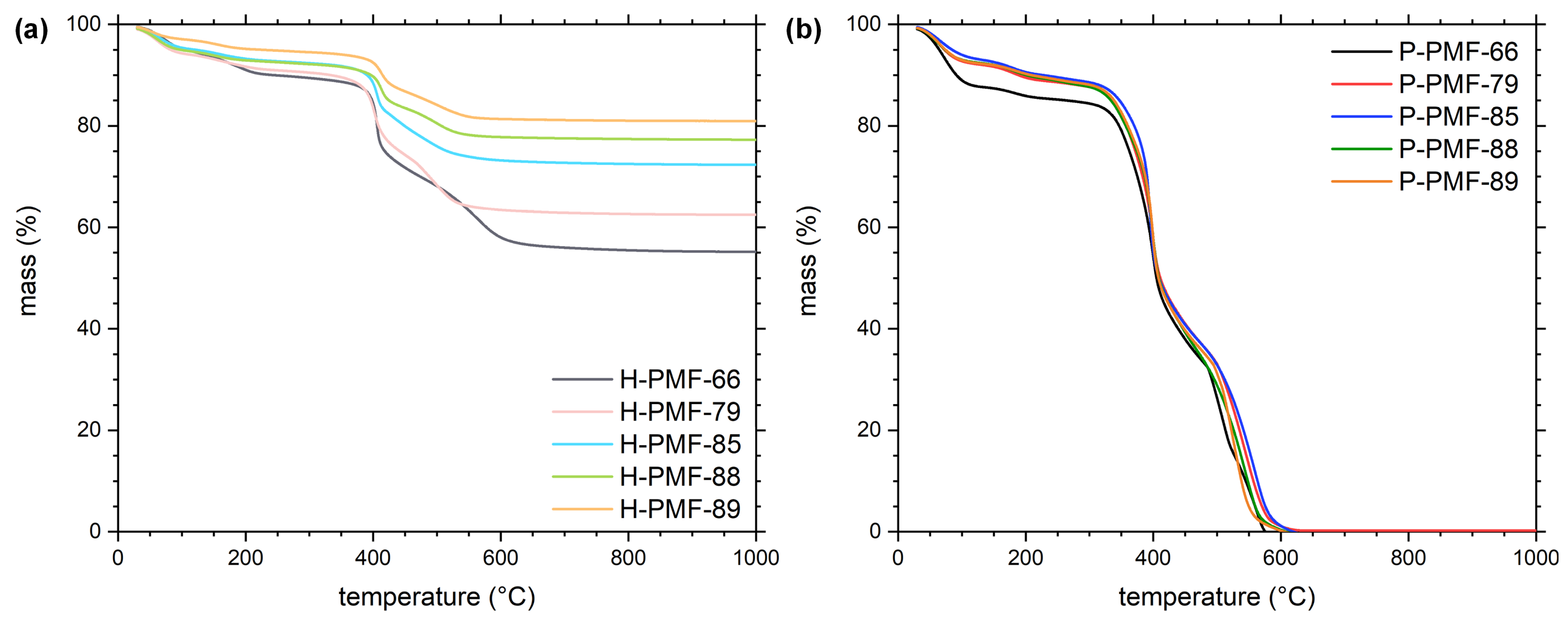
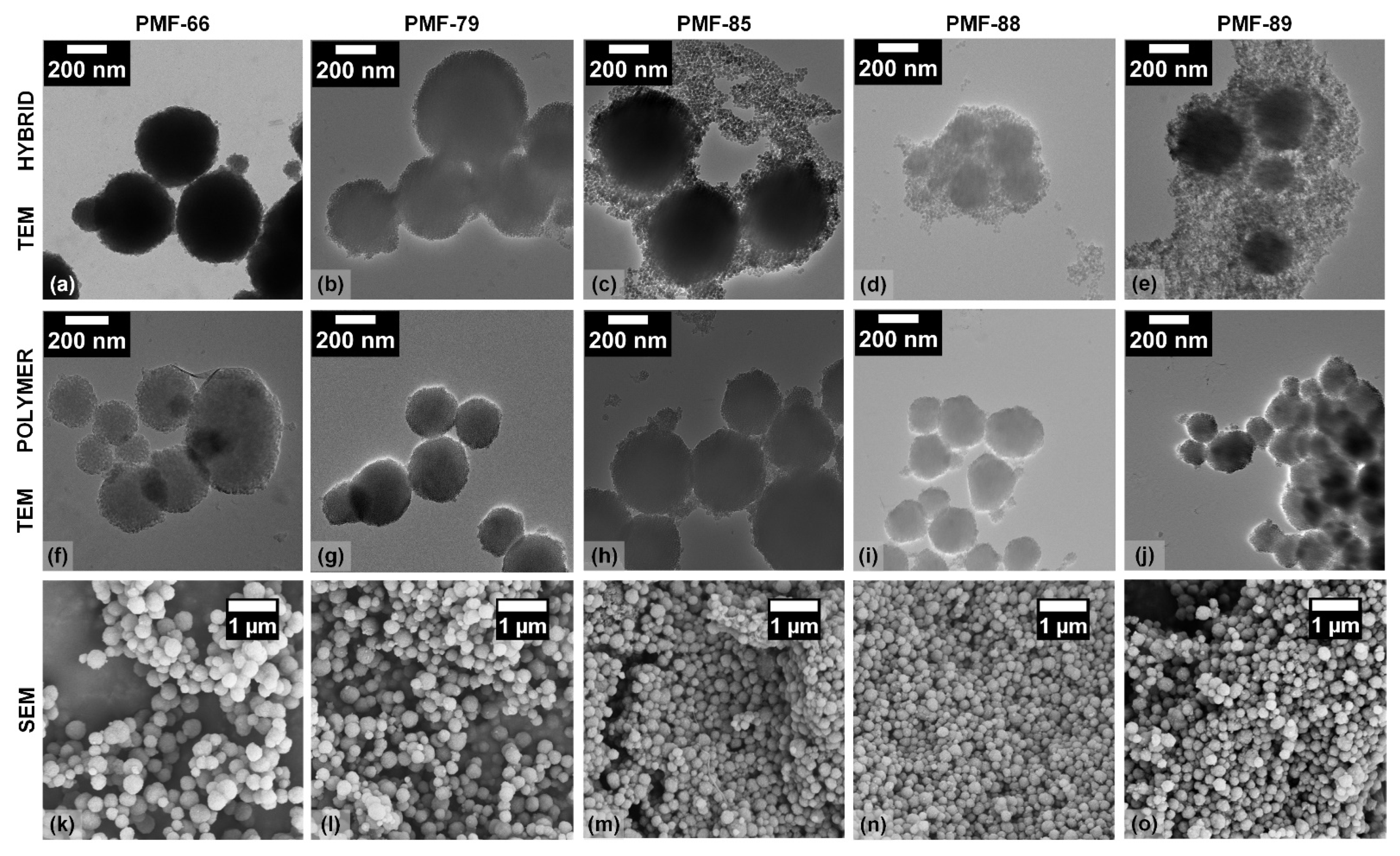
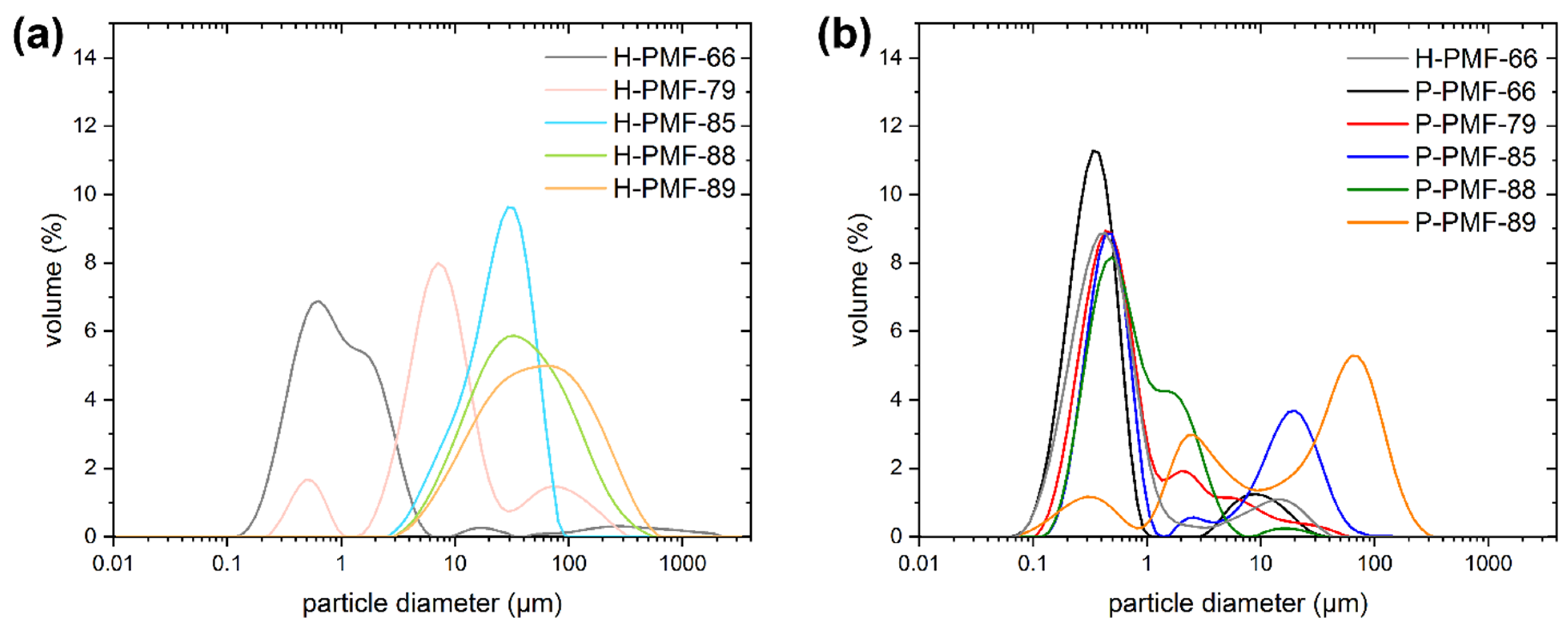
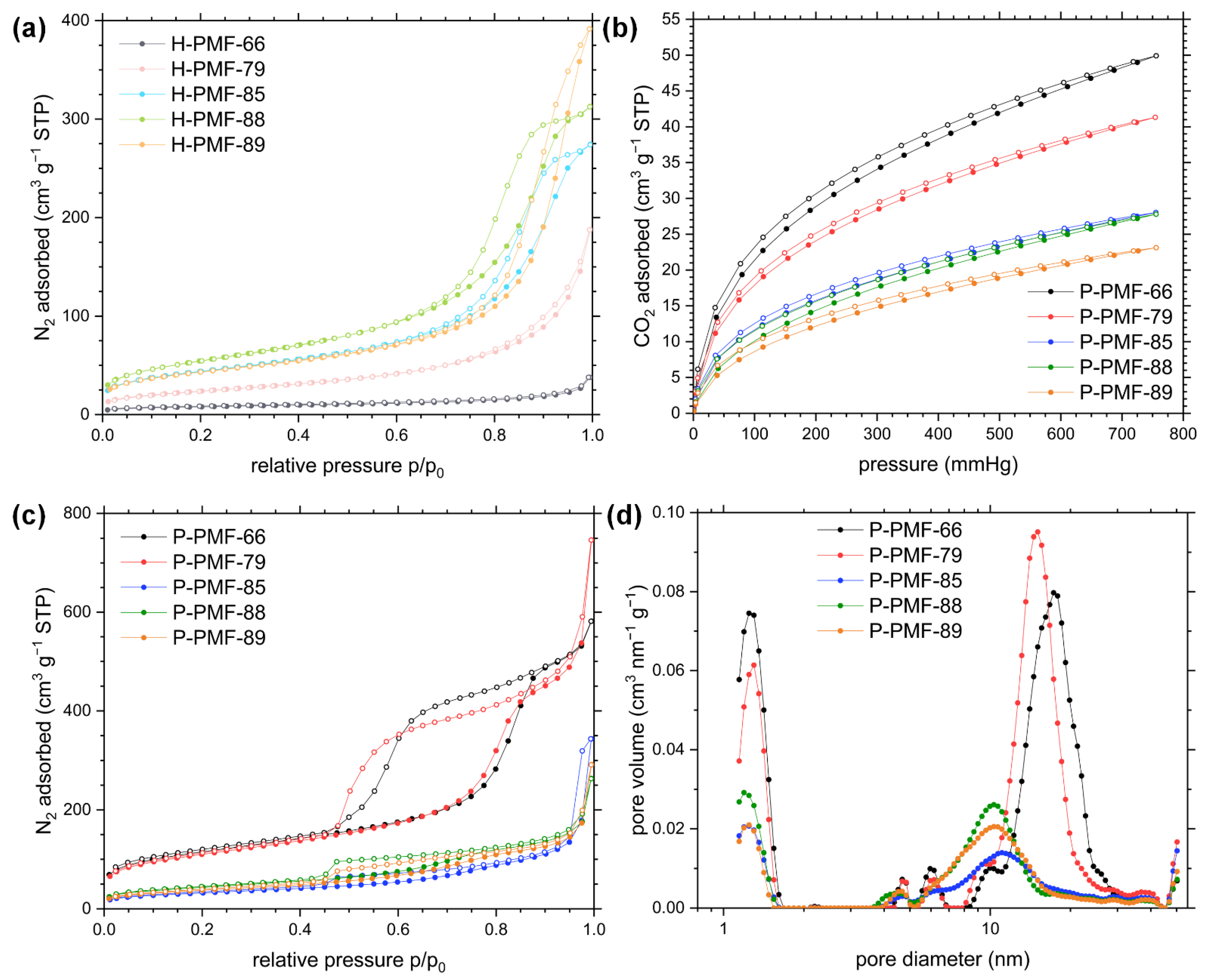
2.1. Synthesis and Characterization of the PMF Particles
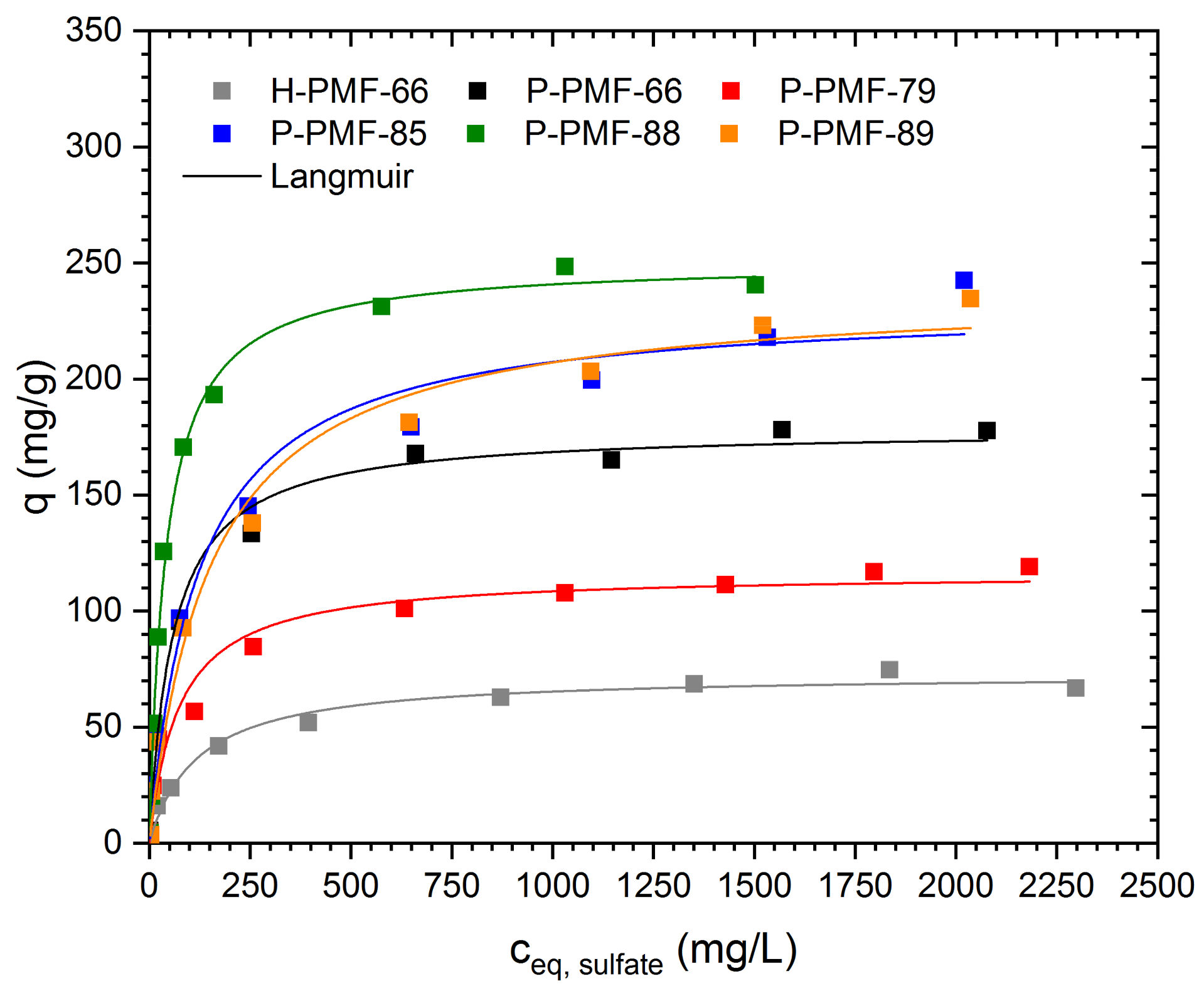
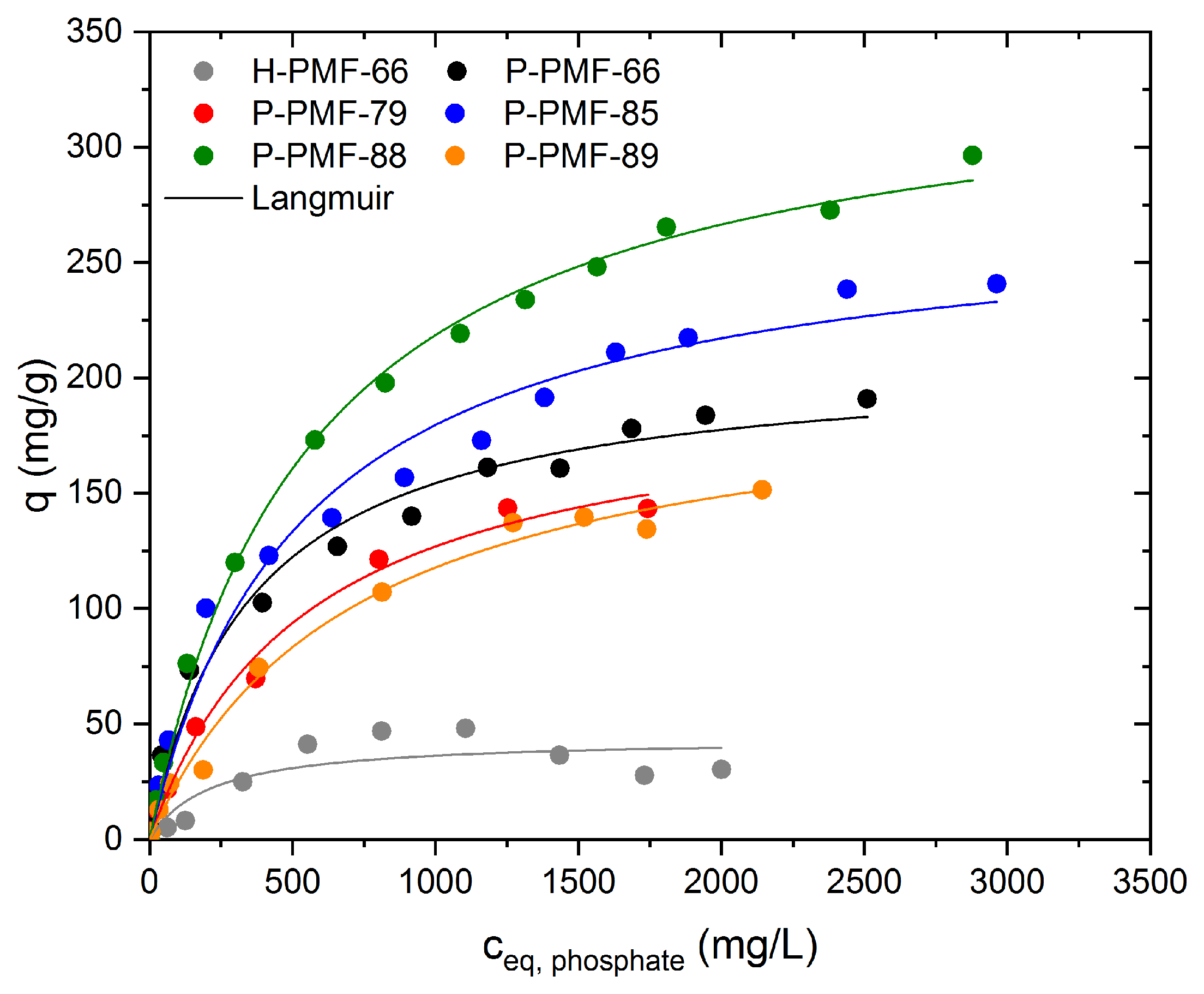
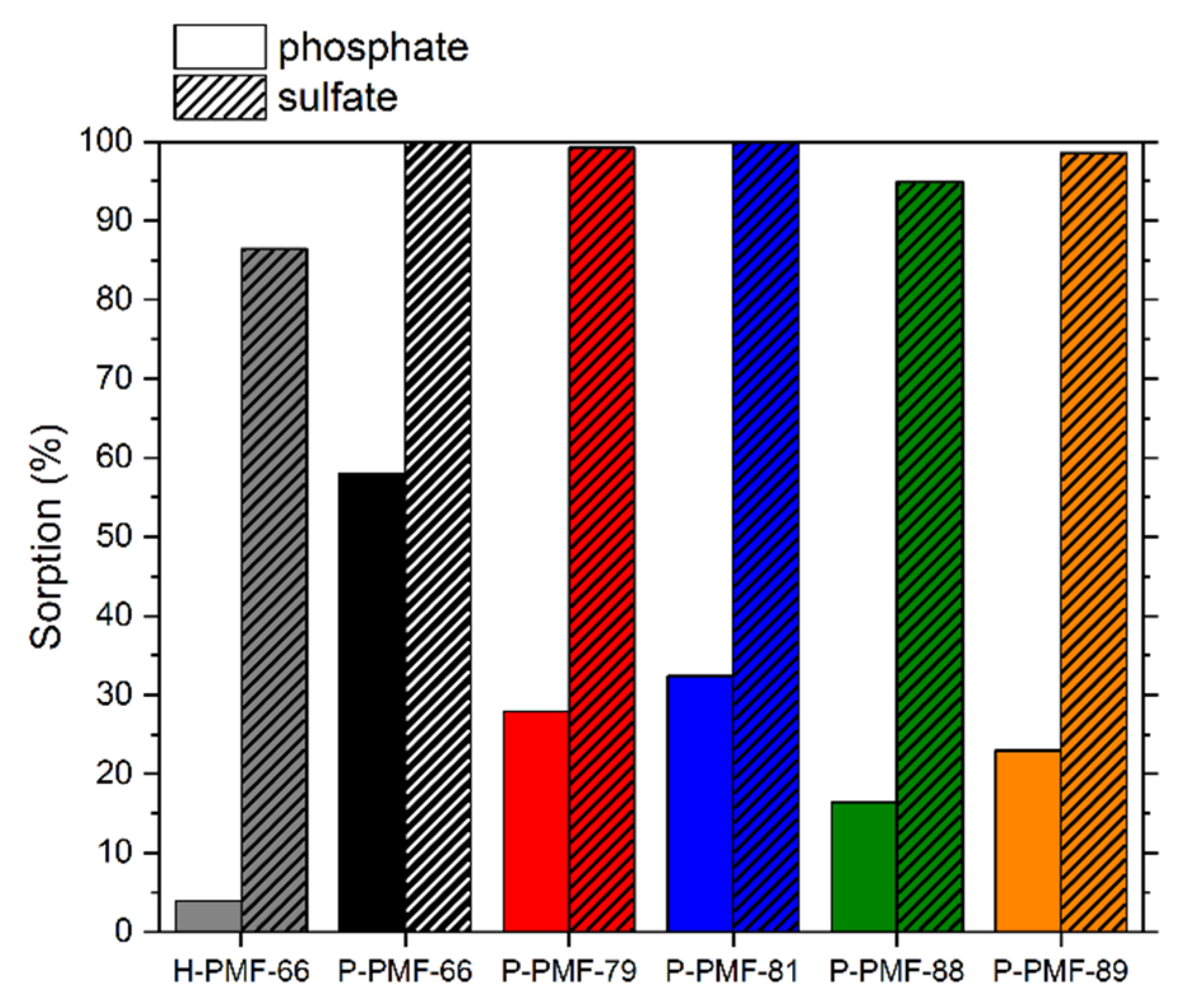
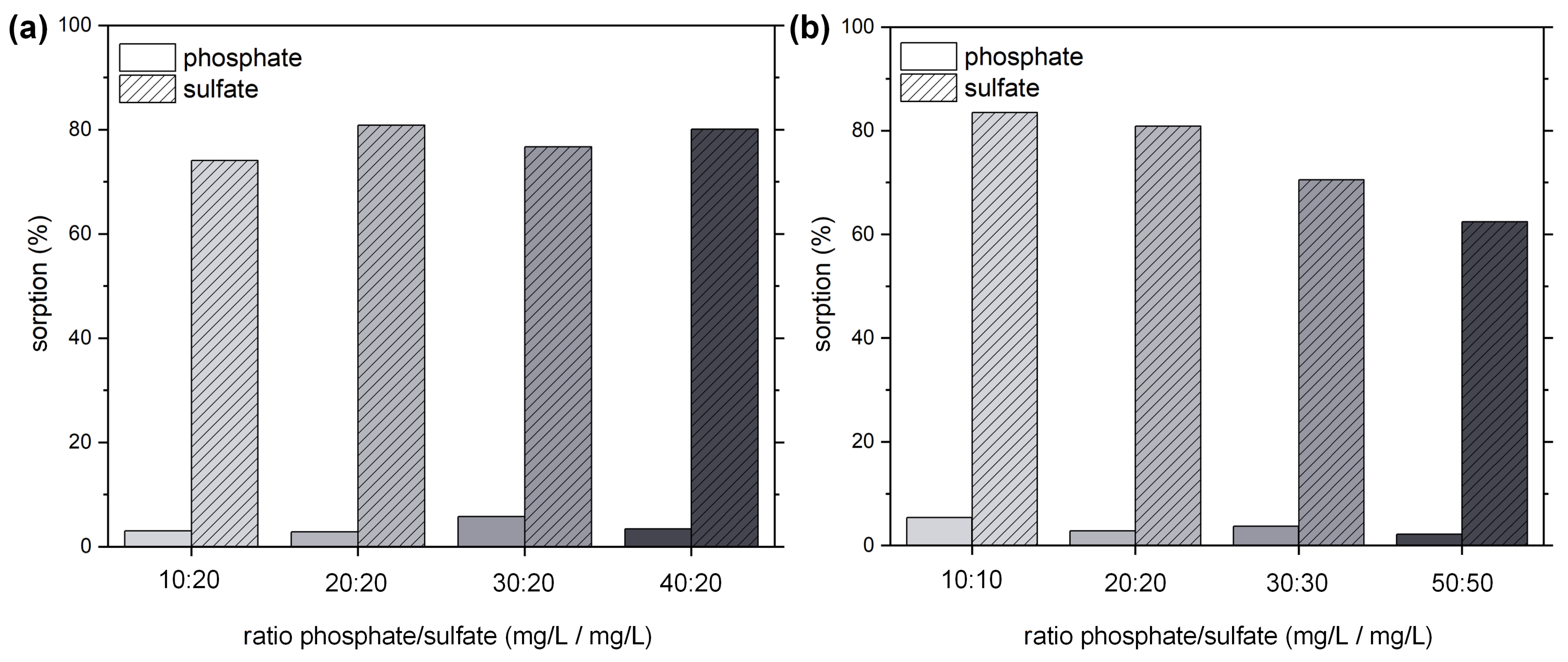
2.2. Sorption Experiments
3. Materials and Methods
3.1. Materials
3.1.1. PMF Particles
3.1.2. Oxyanions
3.1.3. Ultrapure Water
3.1.4. ICP-OES Standard Solutions
3.2. Methods
3.3. Synthesis of the PMF Particles
3.3.1. Synthesis of PMF-66
3.3.2. Synthesis of PMF-79, PMF-85, PMF-88 and PMF-89 Particles
3.4. Water Treatment Experiments
3.4.1. Adsorption Experiments with Sulfuric and Phosphoric Acid
3.4.2. Theoretical Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Fatima, T.; Muntean, A. Sulfate attack in sewer pipes: Derivation of a concrete corrosion model via two-scale convergence. Nonlinear Anal. Real World Appl. 2014, 15, 326–344. [Google Scholar] [CrossRef] [Green Version]
- Katayev, E.A.; Ustynyuk, Y.A.; Sessler, J.L. Receptors for tetrahedral oxyanions. Coord. Chem. Rev. 2006, 250, 3004–3037. [Google Scholar] [CrossRef]
- Elphick, J.R.; Davies, M.; Gilron, G.; Canaria, E.C.; Lo, B.; Bailey, H.C. An aquatic toxicological evaluation of sulfate: The case for considering hardness as a modifying factor in setting water quality guidelines. Environ. Toxicol. Chem. 2010, 30, 247–253. [Google Scholar] [CrossRef]
- Soucek, D.J.; Kennedy, A.J. Effects of hardness, chloride, and acclimation on the acute toxicity of sulfate to freshwater invertebrates. Environ. Toxicol. Chem. 2005, 24, 1204–1210. [Google Scholar] [CrossRef]
- Austigard, Å.D.; Svendsen, K.; Heldal, K.K. Hydrogen sulphide exposure in waste water treatment. J. Occup. Med. Toxicol. 2018, 13, 10. [Google Scholar] [CrossRef] [Green Version]
- Lamers, L.P.M.; Govers, L.L.; Janssen, I.C.J.M.; Geurts, J.J.M.; Van Der Welle, M.E.W.; Van Katwijk, M.M.; Van Der Heide, T.; Roelofs, J.G.M.; Smolders, A.J.P. Sulfide as a soil phytotoxin—a review. Front. Plant Sci. 2013, 4, 268. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, Q. Research Advances in Identifying Sulfate Contamination Sources of Water Environment by Using Stable Isotopes. Int. J. Environ. Res. Public Heal. 2019, 16, 1914. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-Q.; Lang, Y.-C.; Satake, H.; Wu, J.; Li, S.-L. Identification of Anthropogenic and Natural Inputs of Sulfate and Chloride into the Karstic Ground Water of Guiyang, SW China: Combined δ37Cl and δ34S Approach. Environ. Sci. Technol. 2008, 42, 5421–5427. [Google Scholar] [CrossRef]
- Man, K.; Ma, Z.M.; Xu, X.J. Research on the Mechanism of Sulfate Pollution of Groundwater in Jiaozuo Area. Appl. Mech. Mater. 2014, 665, 436–439. [Google Scholar] [CrossRef]
- Miao, Z.; Carroll, K.; Brusseau, M.L. Characterization and quantification of groundwater sulfate sources at a mining site in an arid climate: The Monument Valley site in Arizona, USA. J. Hydrol. 2013, 504, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Mekonnen, M.M.; Hoekstra, A.Y. Global Anthropogenic Phosphorus Loads to Freshwater and Associated Grey Water Footprints and Water Pollution Levels: A High-Resolution Global Study. Water Resour. Res. 2018, 54, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Schindler, D.W.; Carpenter, S.; Chapra, S.C.; Hecky, R.E.; Orihel, D. Reducing Phosphorus to Curb Lake Eutrophication is a Success. Environ. Sci. Technol. 2016, 50, 8923–8929. [Google Scholar] [CrossRef]
- Schindler, D.W.; Vallentyne, J.R. The algal Bowl: Overfertilization of the World’s Freshwaters and Estuaries; University of Alberta Press: Edmonton, AB, Canada, 2008; ISBN 9780888644848. [Google Scholar]
- Sylvan, J.B.; Dortch, Q.; Nelson, D.M.; Brown, A.F.M.; Morrison, W.; Ammerman, J.W. Phosphorus Limits Phytoplankton Growth on the Louisiana Shelf During the Period of Hypoxia Formation. Environ. Sci. Technol. 2006, 40, 7548–7553. [Google Scholar] [CrossRef]
- Seitzinger, S.P.; Harrison, J.; Dumont, E.; Beusen, A.; Bouwman, L. Sources and delivery of carbon, nitrogen, and phosphorus to the coastal zone: An overview of Global Nutrient Export from Watersheds (NEWS) models and their application. Glob. Biogeochem. Cycles 2005, 19, 1–11. [Google Scholar] [CrossRef]
- Farmer, A.M. Phosphate pollution: A global overview of the problem. In Phosphorus: Polluter and Resource of the Future—Removal and Recovery from Wastewater; Schaum, C., Ed.; IWA Publishing: London, UK, 2018; pp. 35–55. ISBN 9781780408361. [Google Scholar]
- Horrigan, L.; Lawrence, R.S.; Walker, P. How sustainable agriculture can address the environmSental and human health harms of industrial agriculture. Environ. Health Perspect. 2002, 110, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Alewell, C.; Ringeval, B.; Ballabio, C.; Robinson, D.A.; Panagos, P.; Borrelli, P. Global phosphorus shortage will be aggravated by soil erosion. Nat. Commun. 2020, 11, 4546. [Google Scholar] [CrossRef]
- Summaries, M.C. Mineral Commodity Summaries; U. S. Government Publishing Office: Washington, DC, USA, 2021; p. 200. [CrossRef]
- Chrispim, M.C.; Scholz, M.; Nolasco, M.A. Phosphorus recovery from municipal wastewater treatment: Critical review of challenges and opportunities for developing countries. J. Environ. Manag. 2019, 248, 109268. [Google Scholar] [CrossRef]
- Petzet, S.; Peplinski, B.; Cornel, P. On wet chemical phosphorus recovery from sewage sludge ash by acidic or alkaline leaching and an optimized combination of both. Water Res. 2012, 46, 3769–3780. [Google Scholar] [CrossRef]
- Remmen, K.; Müller, B.; Köser, J.; Wessling, M.; Wintgens, T. Phosphorus recovery in an acidic environment using layer-by-layer modified membranes. J. Membr. Sci. 2019, 582, 254–263. [Google Scholar] [CrossRef]
- Roy, E. Phosphorus recovery and recycling with ecological engineering: A review. Ecol. Eng. 2017, 98, 213–227. [Google Scholar] [CrossRef]
- Jupp, A.R.; Beijer, S.; Narain, G.C.; Schipper, W.; Slootweg, J.C. Phosphorus recovery and recycling—closing the loop. Chem. Soc. Rev. 2020, 50, 87–101. [Google Scholar] [CrossRef]
- Kong, L.; Hu, X.; Xie, Z.; Ren, X.; Long, J.; Su, M.; Diao, Z.-H.; Chen, D.; Shih, K.; Hou, L. Accelerated phosphorus recovery from aqueous solution onto decorated sewage sludge carbon. Sci. Rep. 2018, 8, 13421. [Google Scholar] [CrossRef]
- Tonini, D.; Saveyn, H.G.M.; Huygens, D. Environmental and health co-benefits for advanced phosphorus recovery. Nat. Sustain. 2019, 2, 1051–1061. [Google Scholar] [CrossRef]
- Atienza-Martínez, M.; Gea, G.; Arauzo, J.; Kersten, S.R.; Kootstra, A.M.J. Phosphorus recovery from sewage sludge char ash. Biomass Bioenergy 2014, 65, 42–50. [Google Scholar] [CrossRef]
- Meng, X.; Huang, Q.; Xu, J.; Gao, H.; Yan, J. A review of phosphorus recovery from different thermal treatment products of sewage sludge. Waste Dispos. Sustain. Energy 2019, 1, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Monea, M.C.; Meyer, C.; Steinmetz, H.; Schönberger, H.; Drenkova-Tuhtan, A. Phosphorus recovery from sewage sludge—phosphorus leaching behavior from aluminum-containing tertiary and anaerobically digested sludge. Water Sci. Technol. 2020, 82, 1509–1522. [Google Scholar] [CrossRef]
- Ohtake, H.; Tsuneda, S. Phosphorus Recovery and Recycling; Springer: Singapore, 2019; ISBN 978-981-10-8030-2. [Google Scholar]
- Du, C.-M.; Gao, X.; Ueda, S.; Kitamura, S.-Y. Separation and recovery of phosphorus from steelmaking slag via a selective leaching–chemical precipitation process. Hydrometallurgy 2019, 189, 105–109. [Google Scholar] [CrossRef]
- Lv, N.-N.; Du, C.-M.; Kong, H.; Yu, Y.-H. Leaching of Phosphorus from Quenched Steelmaking Slags with Different Composition. Metals 2021, 11, 1026. [Google Scholar] [CrossRef]
- Lin, L.; Bao, Y.; Wang, M.; Jiang, W.; Zhou, H. Separation and Recovery of Phosphorus from P-bearing Steelmaking Slag. Journal of Iron and Steel Research International 2014, 21, 496–502. [Google Scholar] [CrossRef]
- Kalmykova, Y.; Fedje, K.K. Phosphorus recovery from municipal solid waste incineration fly ash. Waste Manag. 2013, 33, 1403–1410. [Google Scholar] [CrossRef] [Green Version]
- Schwertmann, U. Solubility and dissolution of iron oxides. Plant Soil 1991, 130, 1–25. [Google Scholar] [CrossRef]
- Han, J.; Kim, M.; Ro, H.-M. Factors modifying the structural configuration of oxyanions and organic acids adsorbed on iron (hydr)oxides in soils. A review. Environ. Chem. Lett. 2020, 18, 631–662. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Guan, K.; Bai, Z.; Liu, F. Facile preparation of acid-resistant magnetite particles for removal of Sb(III) from strong acidic solution. Sci. Technol. Adv. Mater. 2016, 17, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Reis, B.; Gerlach, N.; Steinbach, C.; Carrasco, K.H.; Oelmann, M.; Schwarz, S.; Müller, M.; Schwarz, D. A Complementary and Revised View on the N-Acylation of Chitosan with Hexanoyl Chloride. Mar. Drugs 2021, 19, 385. [Google Scholar] [CrossRef]
- Li, M.; Liu, J.; Xu, Y.; Qian, G. Phosphate adsorption on metal oxides and metal hydroxides: A comparative review. Environ. Rev. 2016, 24, 319–332. [Google Scholar] [CrossRef]
- Ajmal, Z.; Muhmood, A.; Usman, M.; Kizito, S.; Lu, J.; Dong, R.; Wu, S. Phosphate removal from aqueous solution using iron oxides: Adsorption, desorption and regeneration characteristics. J. Colloid Interface Sci. 2018, 528, 145–155. [Google Scholar] [CrossRef]
- Siwek, H.; Bartkowiak, A.; Włodarczyk, M. Adsorption of Phosphates from Aqueous Solutions on Alginate/Goethite Hydrogel Composite. Water 2019, 11, 633. [Google Scholar] [CrossRef] [Green Version]
- Lalley, J.; Han, C.; Li, X.; Dionysiou, D.D.; Nadagouda, M.N. Phosphate adsorption using modified iron oxide-based sorbents in lake water: Kinetics, equilibrium, and column tests. Chem. Eng. J. 2015, 284, 1386–1396. [Google Scholar] [CrossRef]
- Song, Y.; Ma, R.; Jiao, C.; Hao, L.; Wang, C.; Wu, Q.; Wang, Z. Magnetic mesoporous polymelamine-formaldehyde resin as an adsorbent for endocrine disrupting chemicals. Microchim. Acta 2017, 185, 19. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, T.; Schulman, E.; Liu, D.; Meng, J. Hierarchical porous metallized poly-melamine-formaldehyde (PMF) as a low-cost and high-efficiency catalyst for cyclic carbonate synthesis from CO2 and epoxides. J. Mater. Chem. A 2018, 6, 8441–8448. [Google Scholar] [CrossRef]
- Tan, M.X.; Sum, Y.N.; Ying, J.Y.; Zhang, Y. A mesoporous poly-melamine-formaldehyde polymer as a solid sorbent for toxic metal removal. Energy Environ. Sci. 2013, 6, 3254–3259. [Google Scholar] [CrossRef]
- Schwarz, D.; Weber, J. Synthesis of mesoporous poly(melamine-formaldehyde) particles by inverse emulsion polymerization. J. Colloid Interface Sci. 2017, 498, 335–342. [Google Scholar] [CrossRef]
- Schwarz, D.; Weber, J. Organic-solvent free synthesis of mesoporous and narrow-dispersed melamine resin particles for water treatment applications. Polymer 2018, 155, 83–88. [Google Scholar] [CrossRef]
- Ming, G.; Duan, H.; Meng, X.; Sun, G.; Sun, W.; Liu, Y.; Lucia, L. A novel fabrication of monodisperse melamine–formaldehyde resin microspheres to adsorb lead (II). Chem. Eng. J. 2016, 288, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xie, Y.; Zhang, Y.; Tang, S.; Guo, C.; Wu, J.; Lau, R. Anionic and cationic dyes adsorption on porous poly-melamine-formaldehyde polymer. Chem. Eng. Res. Des. 2016, 114, 258–267. [Google Scholar] [CrossRef]
- Zeng, X.; Li, J.; Singh, N. Recycling of Spent Lithium-Ion Battery: A Critical Review. Crit. Rev. Environ. Sci. Technol. 2014, 44, 1129–1165. [Google Scholar] [CrossRef]
- Huang, B.; Pan, Z.; Su, X.; An, L. Recycling of lithium-ion batteries: Recent advances and perspectives. J. Power Sources 2018, 399, 274–286. [Google Scholar] [CrossRef]
- Wang, Y.; An, N.; Wen, L.; Wang, L.; Jiang, X.; Hou, F.; Yin, Y.; Liang, J. Recent progress on the recycling technology of Li-ion batteries. J. Energy Chem. 2020, 55, 391–419. [Google Scholar] [CrossRef]
- Schwarz, D.; Weber, J. Waterborne Colloidal Polymer/Silica Hybrid Dispersions and Their Assembly into Mesoporous Poly(melamine-formaldehyde) Xerogels. Langmuir 2015, 31, 8436–8445. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.; Weber, J. Nanoporous Poly(Melamine Formaldehyde) Networks by Aqueous Dispersion Polycondensation-Synthesis and Adsorption Properties. Macromol. Mater. Eng. 2015, 300, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; John Wiley and Sons, Ltd.: Chichester, UK, 2004; Volume 35, ISBN 978-0-470-09307-8. [Google Scholar]
- Merline, D.J.; Vukusic, S.; Abdala, A. Melamine formaldehyde: Curing studies and reaction mechanism. Polym. J. 2012, 45, 413–419. [Google Scholar] [CrossRef]
- Ullah, S.; Bustam, M.A.; Nadeem, M.; Naz, M.Y.; Tan, W.L.; Shariff, A.M. Synthesis and Thermal Degradation Studies of Melamine Formaldehyde Resins. Sci. World J. 2014, 2014, 940502. [Google Scholar] [CrossRef] [Green Version]
- Devallencourt, C.; Saiter, J.; Fafet, A.; Ubrich, E. Thermogravimetry/Fourier transform infrared coupling investigations to study the thermal stability of melamine formaldehyde resin. Thermochim. Acta 1995, 259, 143–151. [Google Scholar] [CrossRef]
- Bentley, J. Organic film formers. Paint and Surface Coatings; Elsevier: Amsterdam, The Netherlands, 1999; pp. 19–90. ISBN 9781855733480. [Google Scholar]
- Herlinger, H.; Hoffmann, M.; Husemann, E.; Istel, E.; Kern, W.; Krimm, H.; Müller, E.; Rinke, H.; Schmitz-Josten, R.; Schneider, P. (Eds.) Polyadditions-bzw. Polykondensationsprodukten von Carbonyl- und Thiocarbonylverbindungen (X). In Makromolekulare Stoffe; Georg Thieme Verlag: Stuttgart, Germany, 1963; ISBN 9783131972743. [Google Scholar]
- Allen, L.H.; Matijević, E. Stability of colloidal silica. J. Colloid Interface Sci. 1969, 31, 287–296. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Sing, K.S.W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Provisional). Pure Appl. Chem. 1982, 54, 2201–2218. [Google Scholar] [CrossRef]
- Rahman, I.A.; Vejayakumaran, P.; Sipaut, C.S.; Ismail, J.; Chee, C.K. Size-dependent physicochemical and optical properties of silica nanoparticles. Mater. Chem. Phys. 2009, 114, 328–332. [Google Scholar] [CrossRef]
- Kosmulski, M. Surface Charging and Points of Zero Charge; CRC Press: New York, NY, USA, 2009; ISBN 9780429093395. [Google Scholar]
- Ortelli, S.; Costa, A.L.; Matteucci, P.; Miller, M.R.; Blosi, M.; Gardini, D.; Tofail, S.A.M.; Tran, L.; Tonelli, D.; Poland, C.A. Silica modification of titania nanoparticles enhances photocatalytic production of reactive oxygen species without increasing toxicity potential in vitro. RSC Adv. 2018, 8, 40369–40377. [Google Scholar] [CrossRef] [Green Version]
- Casas, J.; Alvarez, F.; Cifuentes, L. Aqueous speciation of sulfuric acid–cupric sulfate solutions. Chem. Eng. Sci. 2000, 55, 6223–6234. [Google Scholar] [CrossRef]
- Campbell, N.A.; Reece, J.B. Biology; Recording for the Blind & Dyslexic: Princeton, NJ, USA, 2005; ISBN 0-8053-7171-0. [Google Scholar]
- Powell, K.J.; Brown, P.L.; Byrne, R.H.; Gajda, T.; Hefter, G.; Sjöberg, S.; Wanner, H. Chemical speciation of environmentally significant heavy metals with inorganic ligands. Part 1: The Hg2+–Cl–, OH–, CO32–, SO42–, and PO43– aqueous systems (IUPAC Technical Report). Pure Appl. Chem. 2005, 77, 739–800. [Google Scholar] [CrossRef] [Green Version]
- Helfferich, F.G. Ion Exchange; Dover Publications: New York, NY, USA, 1995; ISBN 0-486-68784-8. [Google Scholar]
- Tan, L.C.; Calix, E.M.; Rene, E.R.; Nancharaiah, Y.V.; Van Hullebusch, E.D.; Lens, P.N.L. Amberlite IRA-900 Ion Exchange Resin for the Sorption of Selenate and Sulfate: Equilibrium, Kinetic, and Regeneration Studies. J. Environ. Eng. 2018, 144, 04018110-1–04018110-12. [Google Scholar] [CrossRef]
- Haghsheno, R.; Mohebbi, A.; Hashemipour, H.; Sarrafi, A. Study of kinetic and fixed bed operation of removal of sulfate anions from an industrial wastewater by an anion exchange resin. J. Hazard. Mater. 2009, 166, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, S.; Li, Y.; Arai, Y. Novel Application of Hybrid Anion Exchange Resin for Phosphate Desorption Kinetics in Soils: Minimizing Re-Adsorption of Desorbed Ions. Soil Syst. 2020, 4, 36. [Google Scholar] [CrossRef]
- Nur, T.; Johir, M.; Loganathan, P.; Nguyen, T.; Vigneswaran, S.; Kandasamy, J. Phosphate removal from water using an iron oxide impregnated strong base anion exchange resin. J. Ind. Eng. Chem. 2013, 20, 1301–1307. [Google Scholar] [CrossRef] [Green Version]
- Sowmya, A.; Meenakshi, S. Removal of nitrate and phosphate anions from aqueous solutions using strong base anion exchange resin. Desalination Water Treat. 2013, 51, 7145–7156. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Langmuir, I. The constitution and fundamental properties of solids and liquids. part i. solids. J. Am. Chem. Soc. 1916, 38, 2221–2295. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.M. The Equation of the Characteristic Curve of Activated Charcoal. Proc. USSR Acad. Sci. 1947, 55, 327–329. [Google Scholar]
- Foo, K.Y.; Hameed, B.H. Insights into the modeling of adsorption isotherm systems. Chem. Eng. J. 2010, 156, 2–10. [Google Scholar] [CrossRef]
- Kamga, F.T. Modeling adsorption mechanism of paraquat onto Ayous (Triplochiton scleroxylon) wood sawdust. Appl. Water Sci. 2018, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X. Correction to the calculation of Polanyi potential from Dubinnin-Rudushkevich equation. J. Hazard. Mater. 2019, 384, 121101. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | C (wt.%) | H (wt.%) | N (wt.%) | Residue (wt.%) | n/n (C/N) |
|---|---|---|---|---|---|
| H-PMF-66 | 14.6 | 2.4 | 19.5 | 63.5 | 0.87 |
| P-PMF-66 | 33.5 | 4.9 | 49.4 | 12.1 | 0.79 |
| H-PMF-79 | 11.9 | 2.2 | 16.8 | 69.1 | 0.82 |
| P-PMF-79 | 35.6 | 4.8 | 52.4 | 7.1 | 0.79 |
| H-PMF-85 | 7.3 | 1.6 | 9.9 | 81.2 | 0.85 |
| P-PMF-85 | 35.6 | 4.8 | 53.5 | 6.0 | 0.78 |
| H-PMF-88 | 5.7 | 1.4 | 7.7 | 85.3 | 0.87 |
| P-PMF-88 | 35.5 | 4.8 | 53.7 | 6.1 | 0.77 |
| H-PMF-89 | 5.4 | 1.2 | 7.1 | 86.3 | 0.89 |
| P-PMF-89 | 35.3 | 4.7 | 53.7 | 6.2 | 0.77 |
| Sample Code | SBET (m2 g−1) a | PV (cm3 g−1) b | MPV (cm3 g−1) c | CO2 Uptake (mmol g−1) d |
|---|---|---|---|---|
| H-PMF-66 | 28 | n.a. | n.a. | n.a. |
| P-PMF-66 | 409 | 0.78 | 0.15 | 2.23 |
| H-PMF-79 | 87 | 0.19 | n.a. | n.a. |
| P-PMF-79 | 393 | 0.76 | 0.15 | 1.84 |
| H-PMF-85 | 159 | 0.39 | 0.06 | n.a. |
| P-PMF-85 | 116 | 0.23 | 0.04 | 1.25 |
| H-PMF-88 | 198 | 0.45 | 0.07 | n.a. |
| P-PMF-88 | 156 | 0.24 | 0.06 | 1.24 |
| H-PMF-89 | 155 | 0.50 | 0.06 | n.a. |
| P-PMF-89 | 136 | 0.23 | 0.05 | 1.03 |
| Ludox® HS-40 e | 185 | 0.71 | 0.07 | n.a. |
| Sample | Model | Qm mg/g | K L/mg | mol2/J2 | Eads,DR kJ/mol | R2 (COD) |
|---|---|---|---|---|---|---|
| H-PMF-66 | Langmuir | 73.0 ± 2.4 | 8.46 ± 1.39 | -- | -- | 0.987 |
| Dubinin–Radushkevich | 64.1 ± 3.5 | -- | 1.56 × 10−7 ± 4.7 × 10−8 | 1.79 ± 0.27 | 0.934 | |
| P-PMF-66 | Langmuir | 178.4 ± 4.2 | 17.15 ± 2.47 | -- | -- | 0.992 |
| Dubinin–Radushkevich | 162.2 ± 7.3 | -- | 6.12 × 10−8 ± 1.56 × 10−8 | 2.86 ± 0.36 | 0.957 | |
| P-PMF-79 | Langmuir | 116.4 ± 4.8 | 13.67 ± 3.44 | -- | -- | 0.968 |
| Dubinin–Radushkevich | 103.22 ± 5.7 | -- | 5.58 × 10−8 ± 1.71 × 10−8 | 2.99 ± 0.45 | 0.917 | |
| P-PMF-85 | Langmuir | 232.4 ± 10.8 | 8.3 ± 1.96 | -- | -- | 0.979 |
| Dubinin–Radushkevich | 205.0 ± 12.4 | -- | 1.96 × 10−7 ± 6.83 × 10−8 | 1.59 ± 0.27 | 0.934 | |
| P-PMF-88 | Langmuir | 250.7 ± 4.5 | 24.25 ± 1.84 | -- | -- | 0.995 |
| Dubinin–Radushkevich | 230.3 ± 6.1 | -- | 5.59 × 10−8 ± 5.28 × 10−9 | 2.99 ± 0.14 | 0.985 | |
| P-PMF-89 | Langmuir | 238.4 ± 8.8 | 6.61 ± 1.16 | -- | -- | 0.987 |
| Dubinin–Radushkevich | 206.5 ± 12.0 | -- | 2.59 × 10−7 ± 8.36 × 10−8 | 1.38 ± 0.22 | 0.940 |
| Sample | Model | Qm mg/g | K L/mg | mol2/J2 | Eads,DR kJ/mol | R2 (COD) |
|---|---|---|---|---|---|---|
| H-PMF-66 | Langmuir | 43.9 ± 7.6 | 4.72 ± 3.51 | -- | -- | 0.748 |
| Dubinin–Radushkevich | 39.0 ± 3.8 | -- | 7.13 × 10−7 ± 3.58 × 10−7 | 0.83 ± 0.21 | 0.825 | |
| P-PMF-66 | Langmuir | 208.9 ± 9.5 | 2.83 ± 0.50 | -- | -- | 0.984 |
| Dubinin–Radushkevich | 164.2 ± 8.9 | -- | 6.47 × 10−7 ± 2.36 × 10−7 | 0.88 ± 0.16 | 0.907 | |
| P-PMF-79 | Langmuir | 196.4 ± 11.2 | 1.83 ± 0.28 | -- | -- | 0.993 |
| Dubinin–Radushkevich | 138.0 ± 9.7 | -- | 1.16 × 10−7 ± 3.42 × 10−7 | 0.66 ± 0.10 | 0.942 | |
| P-PMF-85 | Langmuir | 274.6 ± 14. 4 | 1.89 ± 0.33 | -- | -- | 0.978 |
| Dubinin–Radushkevich | 204.5 ± 12.2 | -- | 1.19 × 10−6 ± 4.42 × 10−7 | 0.65 ± 0.12 | 0.867 | |
| P-PMF-88 | Langmuir | 341.4 ± 7.3 | 1.78 ± 0.12 | -- | -- | 0.997 |
| Dubinin–Radushkevich | 256.4 ± 12.4 | -- | 1.60 × 10−6 ± 4.64 × 10−7 | 0.56 ± 0.08 | 0.935 | |
| P-PMF-89 | Langmuir | 201.3 ± 12.4 | 1.41 ± 0.23 | -- | -- | 0.990 |
| Dubinin–Radushkevich | 142.6 ± 6.4 | -- | 1.96 × 10−6 ± 4.28 × 10−7 | 0.50 ± 0.05 | 0.964 |
| Sample | m (M) | m (F) | m (HS-40) | Ratio (M-F-HS-40) a | pH (HS-40) b | pH c | pH d |
|---|---|---|---|---|---|---|---|
| PMF-66 | 9.1 g | 12.95 g | 42 g | 66% | 9.8 | 2.14 | 3.52 |
| PMF-79 | 9.1 g | 12.95 g | 84 g | 79% | 9.8 | 2.44 | 3.89 |
| PMF-85 | 9.1 g | 12.95 g | 126 g | 85% | 9.8 | 3.27 | 4.32 |
| PMF-88 | 9.1 g | 12.95 g | 168 g | 88% | 9.8 | 4.07 | 5.01 |
| PMF-89 | 9.1 g | 12.95 g | 182 g | 89% | 9.8 | 4.15 | 5.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borchert, K.B.L.; Steinbach, C.; Reis, B.; Gerlach, N.; Zimmermann, P.; Schwarz, S.; Schwarz, D. Mesoporous Poly(melamine-co-formaldehyde) Particles for Efficient and Selective Phosphate and Sulfate Removal. Molecules 2021, 26, 6615. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216615
Borchert KBL, Steinbach C, Reis B, Gerlach N, Zimmermann P, Schwarz S, Schwarz D. Mesoporous Poly(melamine-co-formaldehyde) Particles for Efficient and Selective Phosphate and Sulfate Removal. Molecules. 2021; 26(21):6615. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216615
Chicago/Turabian StyleBorchert, Konstantin B. L., Christine Steinbach, Berthold Reis, Niklas Gerlach, Philipp Zimmermann, Simona Schwarz, and Dana Schwarz. 2021. "Mesoporous Poly(melamine-co-formaldehyde) Particles for Efficient and Selective Phosphate and Sulfate Removal" Molecules 26, no. 21: 6615. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216615