
2-APCAs, the Novel Microtubule Targeting Agents Active against Distinct Cancer Cell Lines

 ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. 2-APCAs Inhibit the Viability of the Multiple Cancer Cell Lines
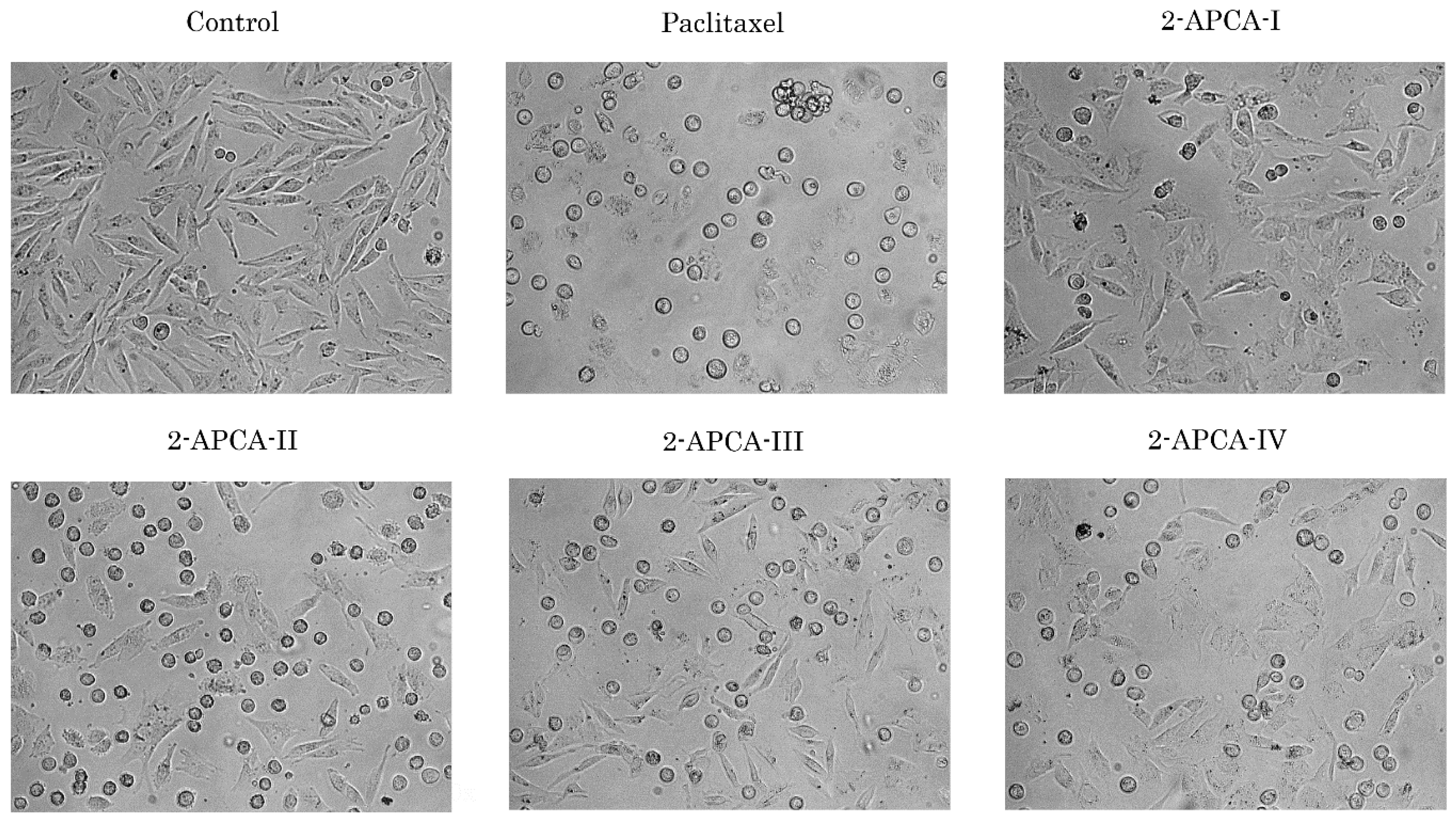
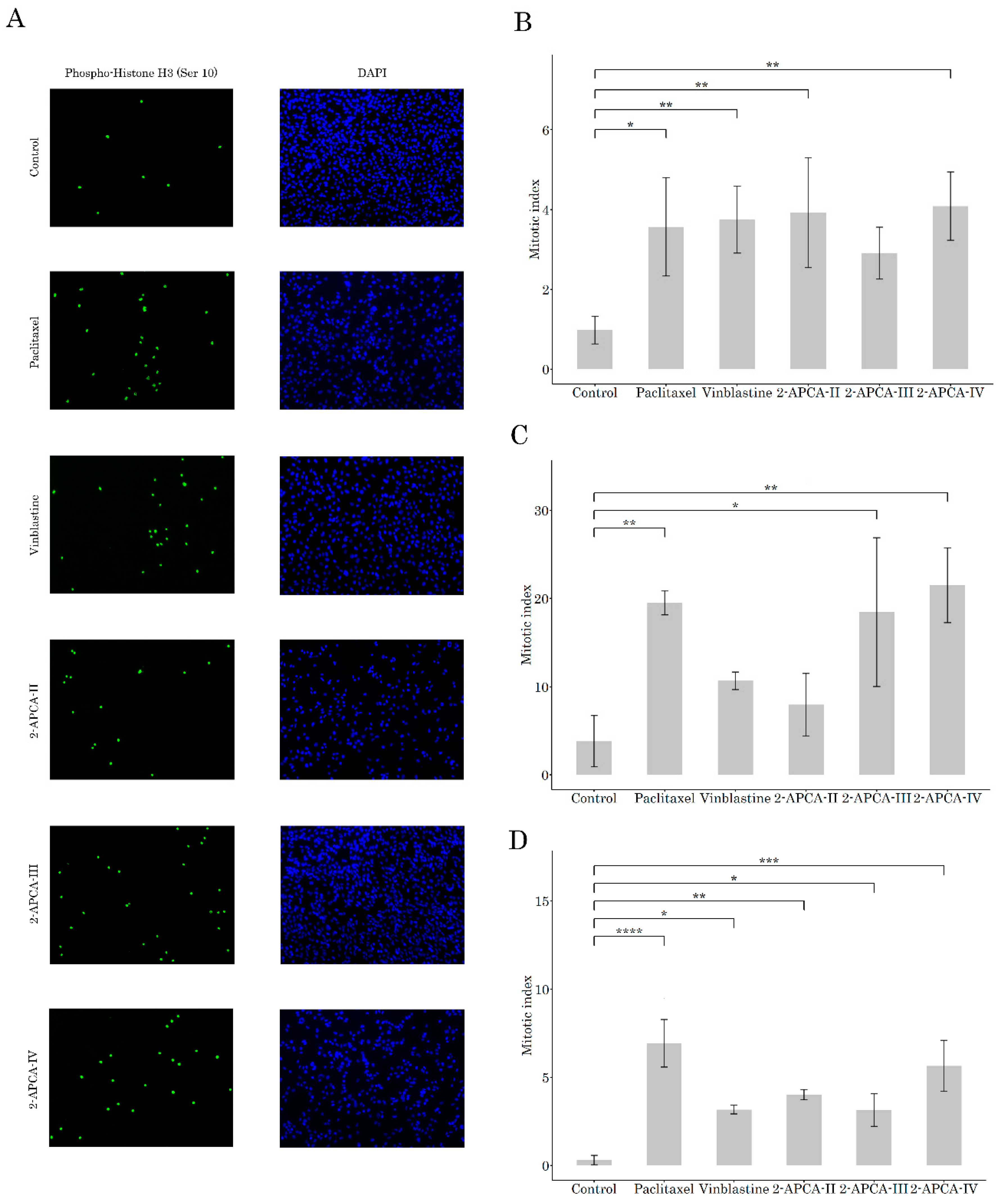
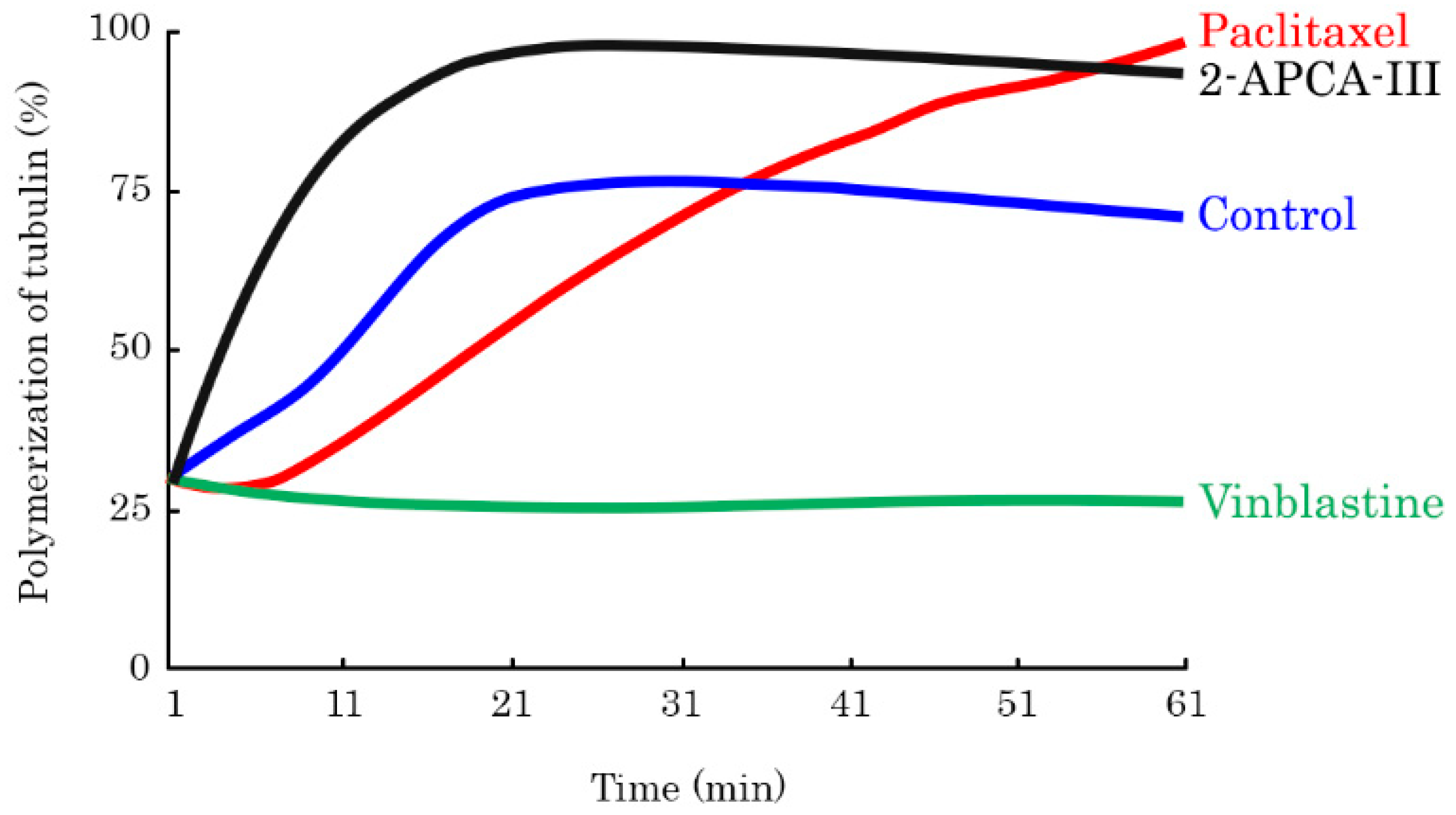
2.2. 2-APCAs Induce Mitotic Arrest of Cancer Cells and Enhance Tubulin Polymerization
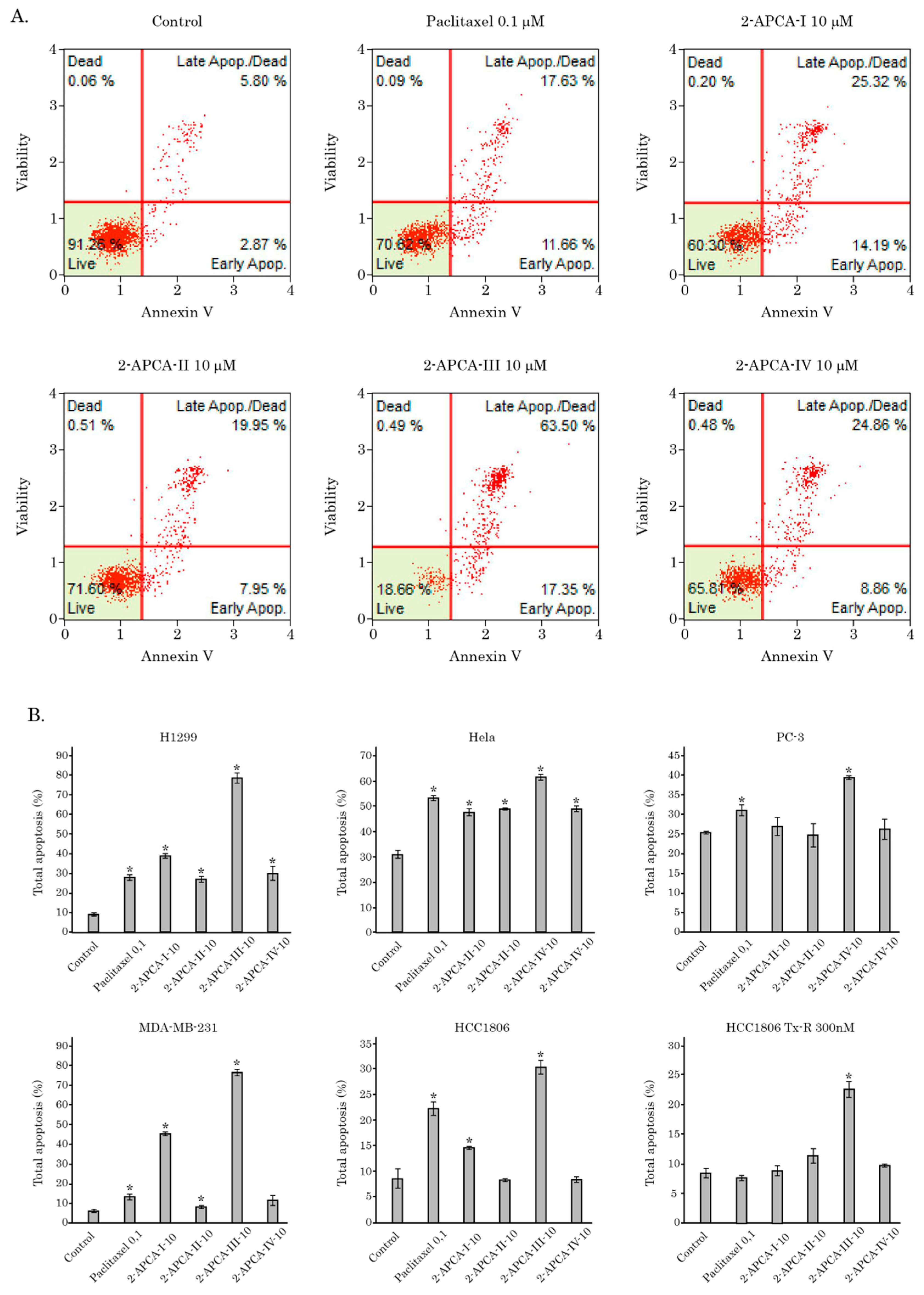
2.3. The 2-APCAs Induce Apoptosis of Breast, Lung, and Prostate Cancer Cells
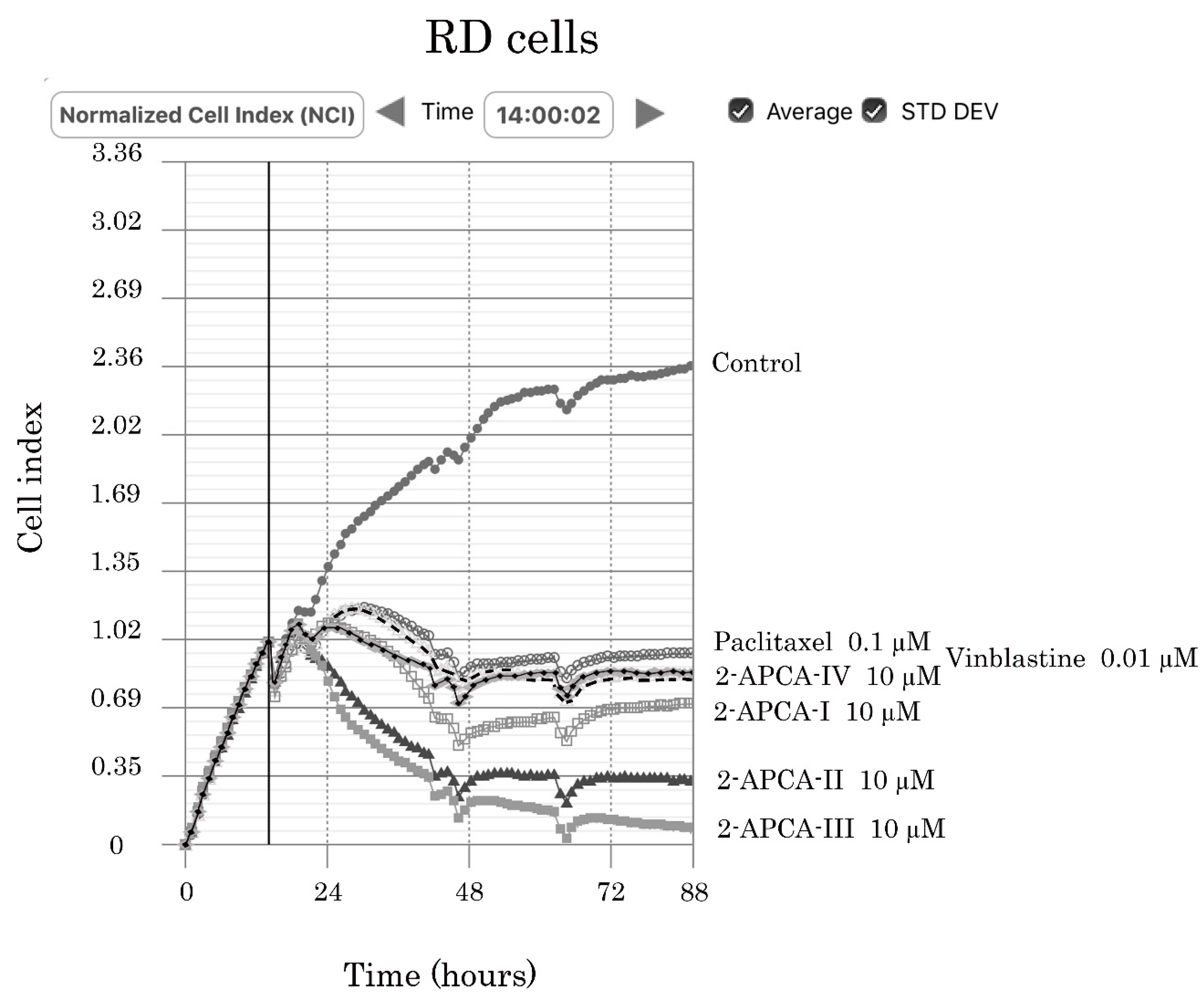
2.4. The 2-APCAs Are Effective against Osteosarcomas (OS), STS, and GIST
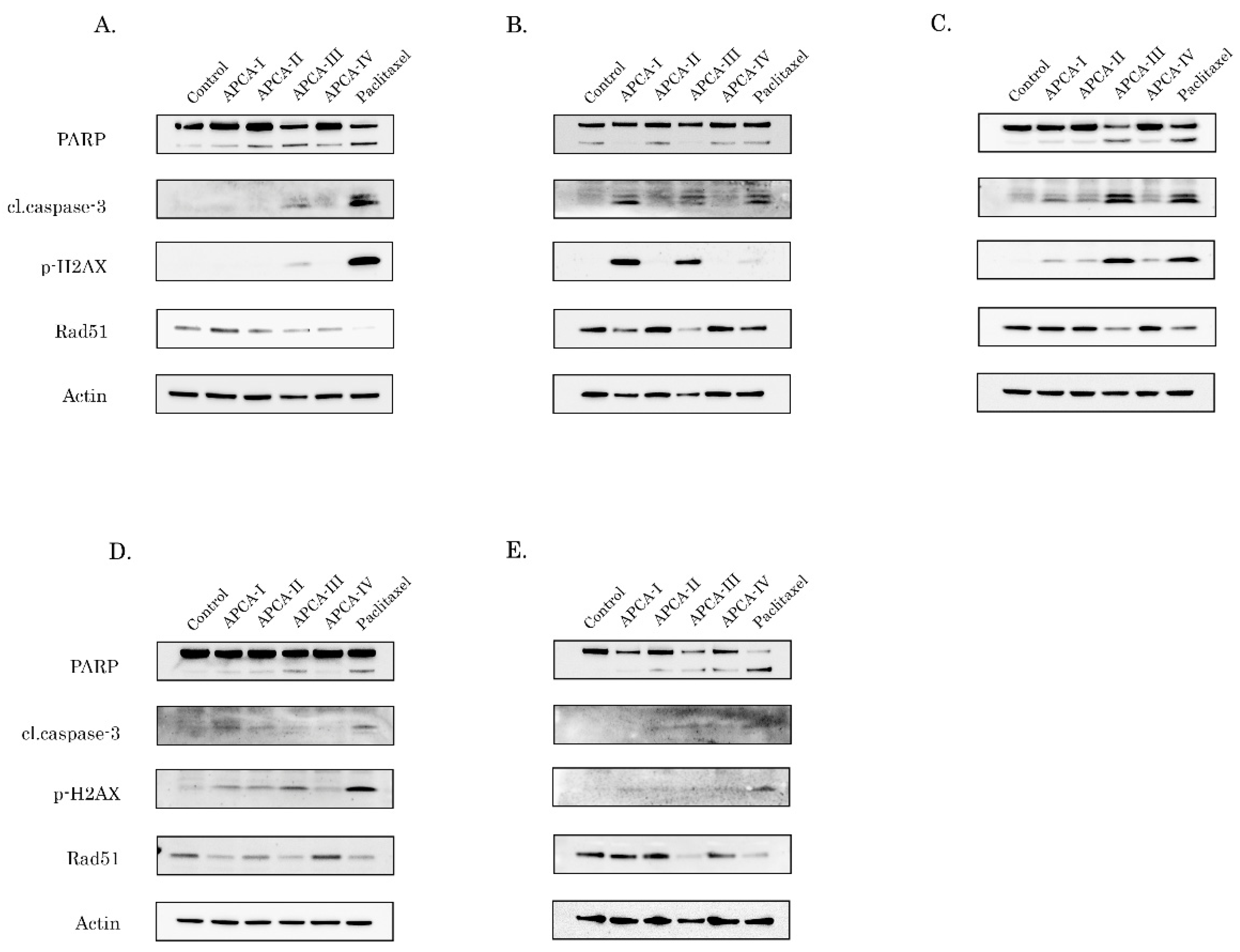
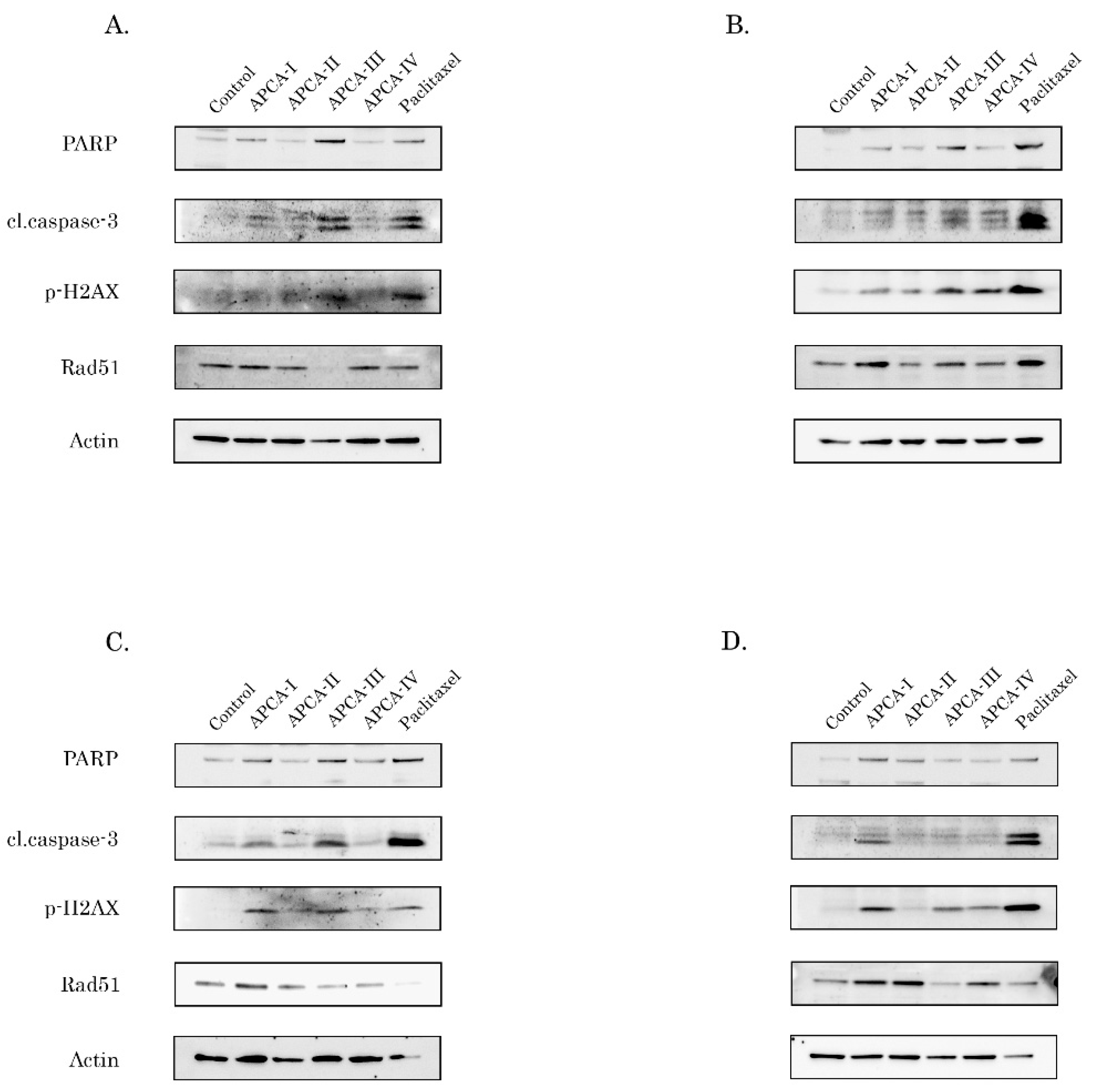
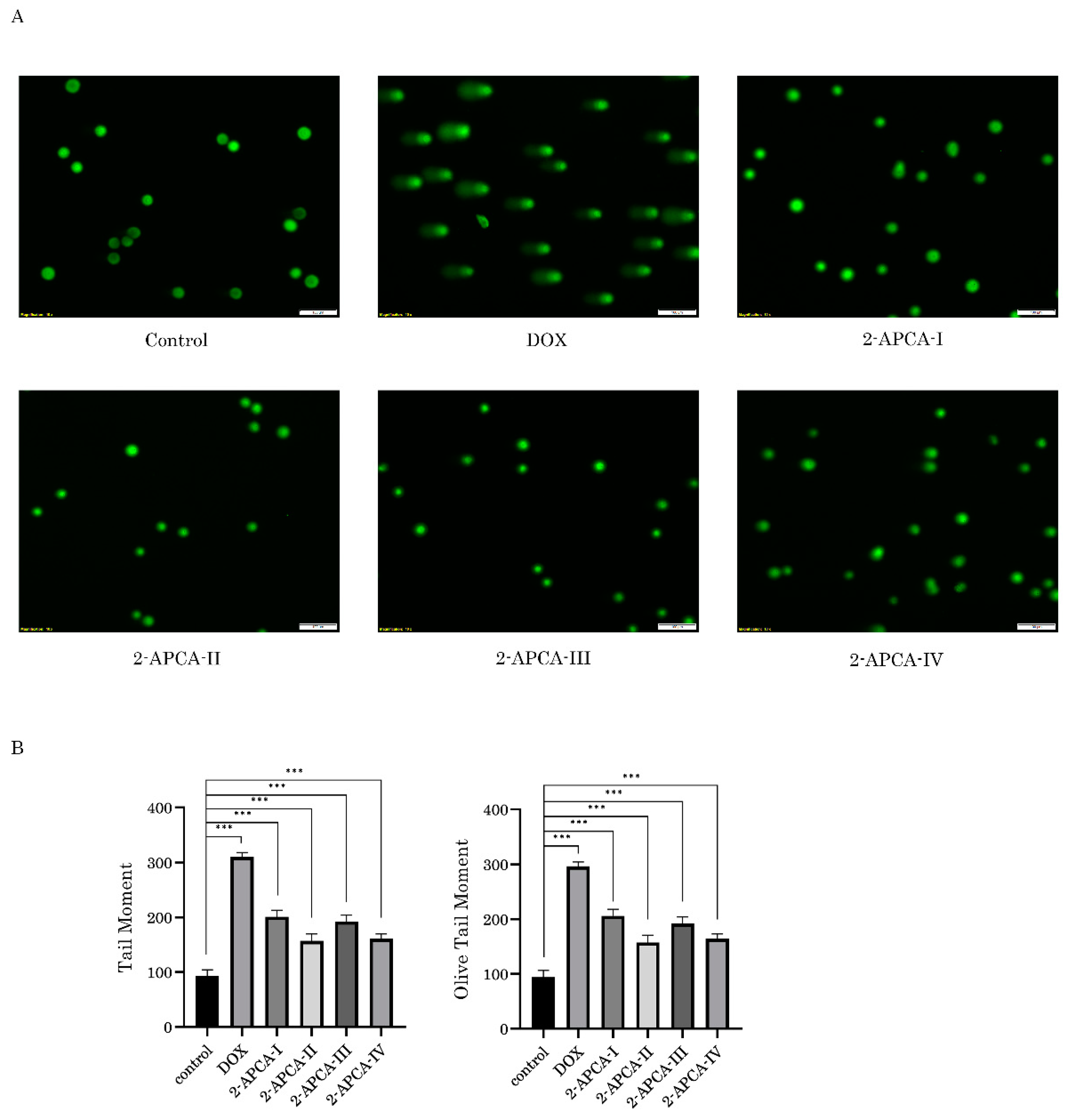
2.5. 2-APCAs Induce an Increased Expression of γ-H2AX without DNA Damage
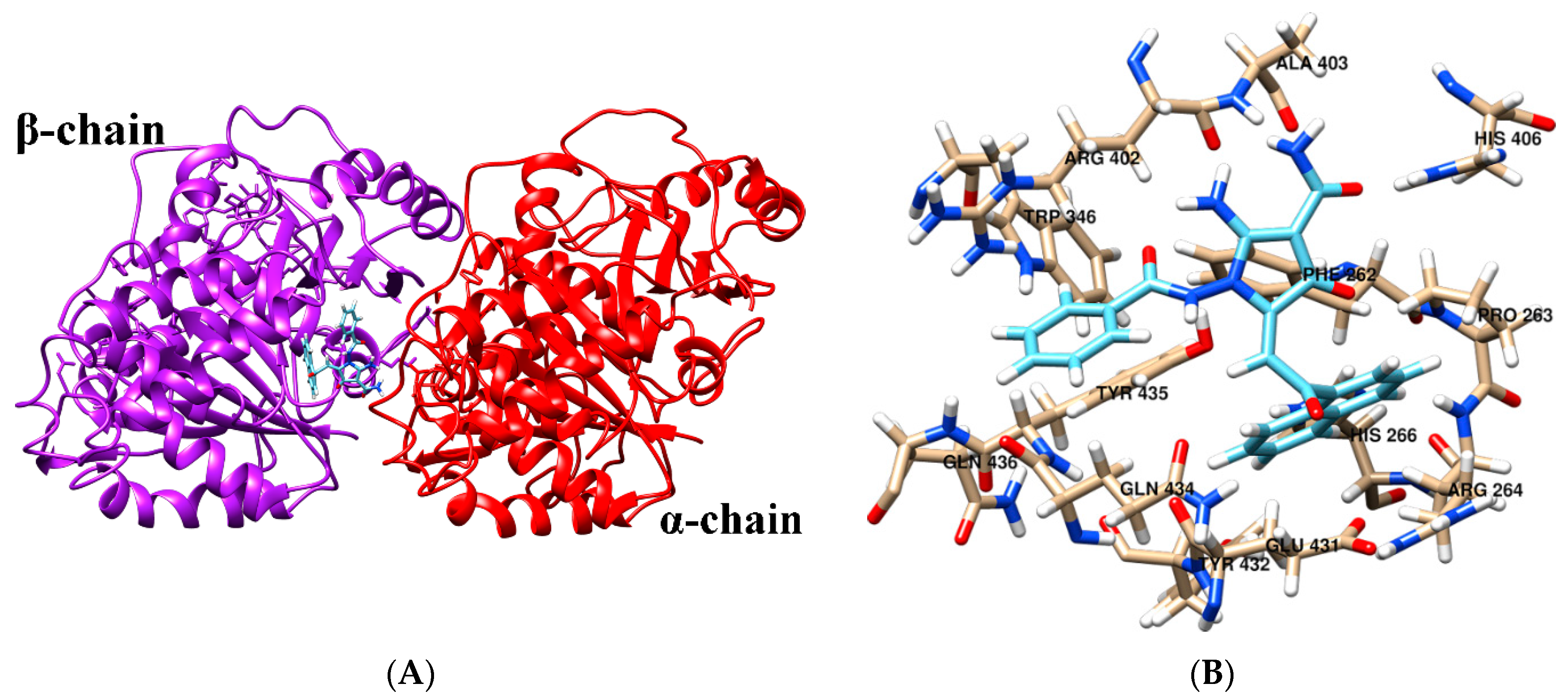
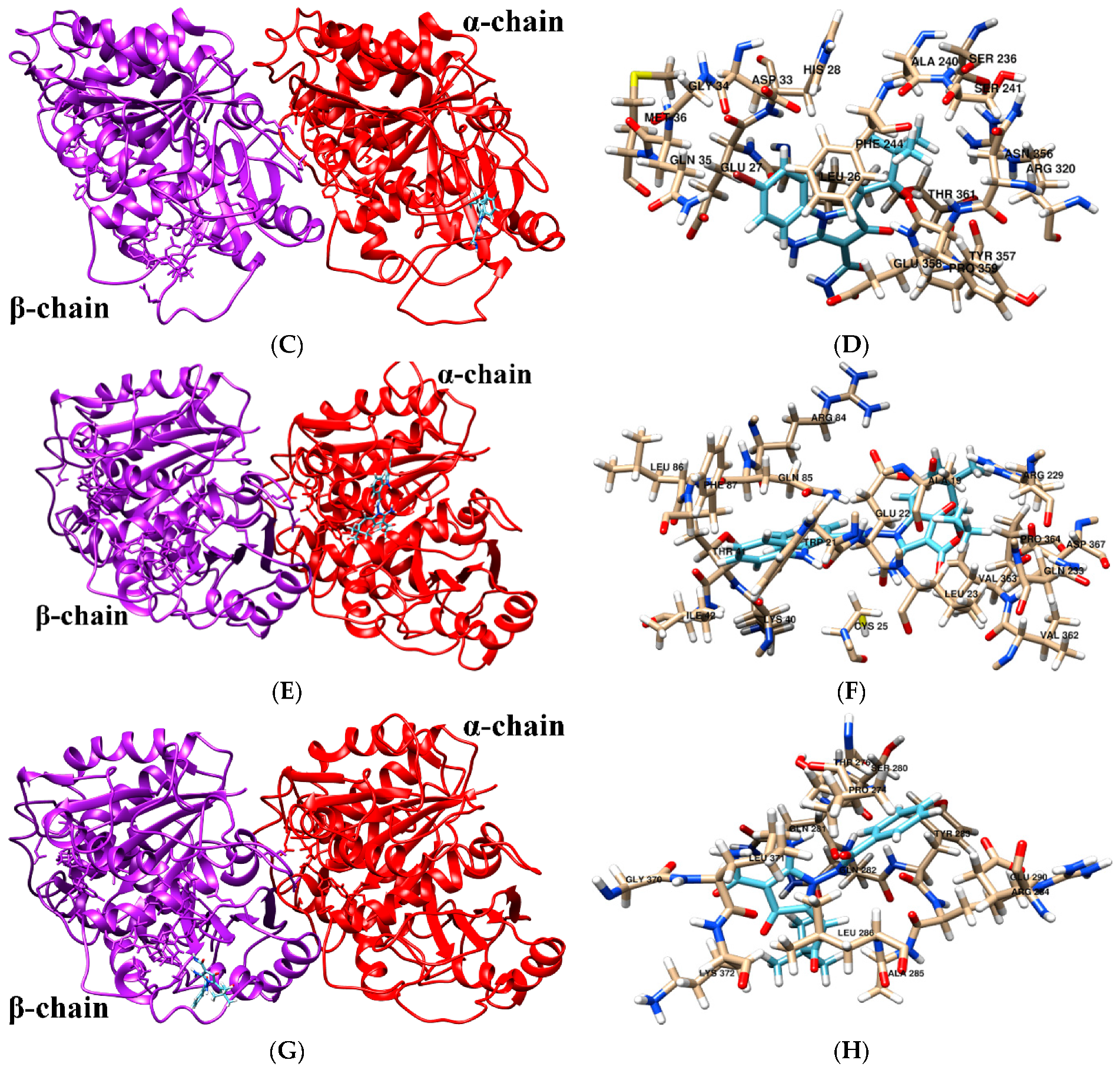
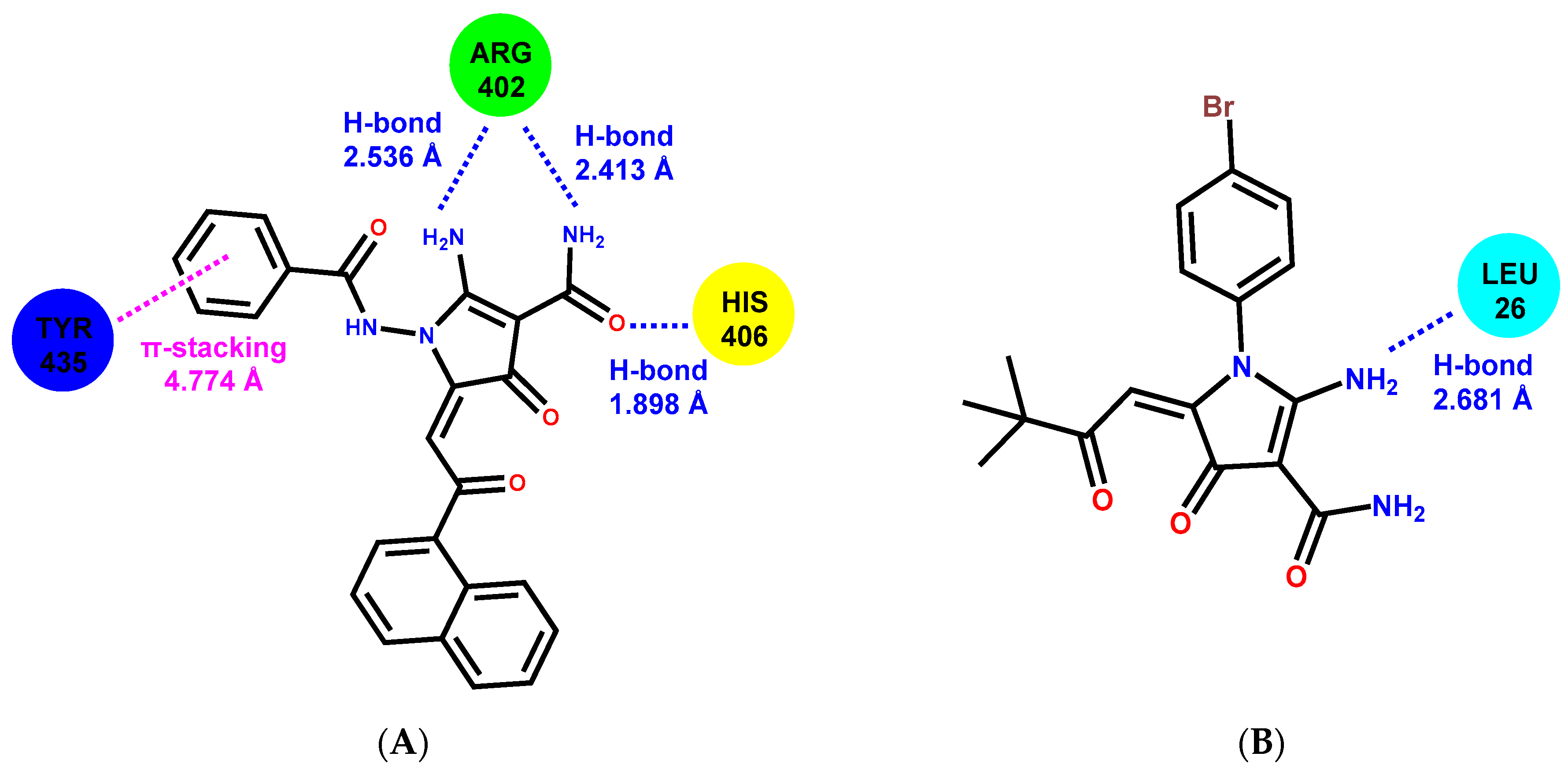
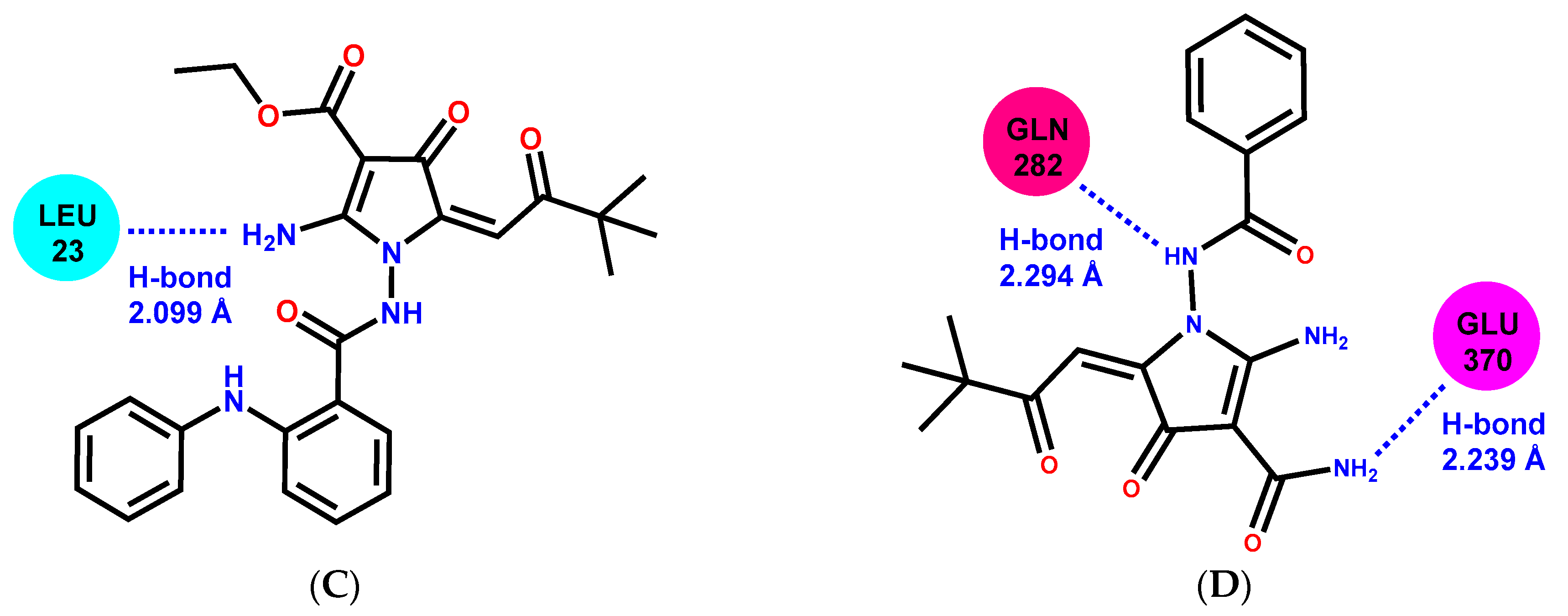
2.6. Molecular Docking
3. Discussion
4. Materials and Methods
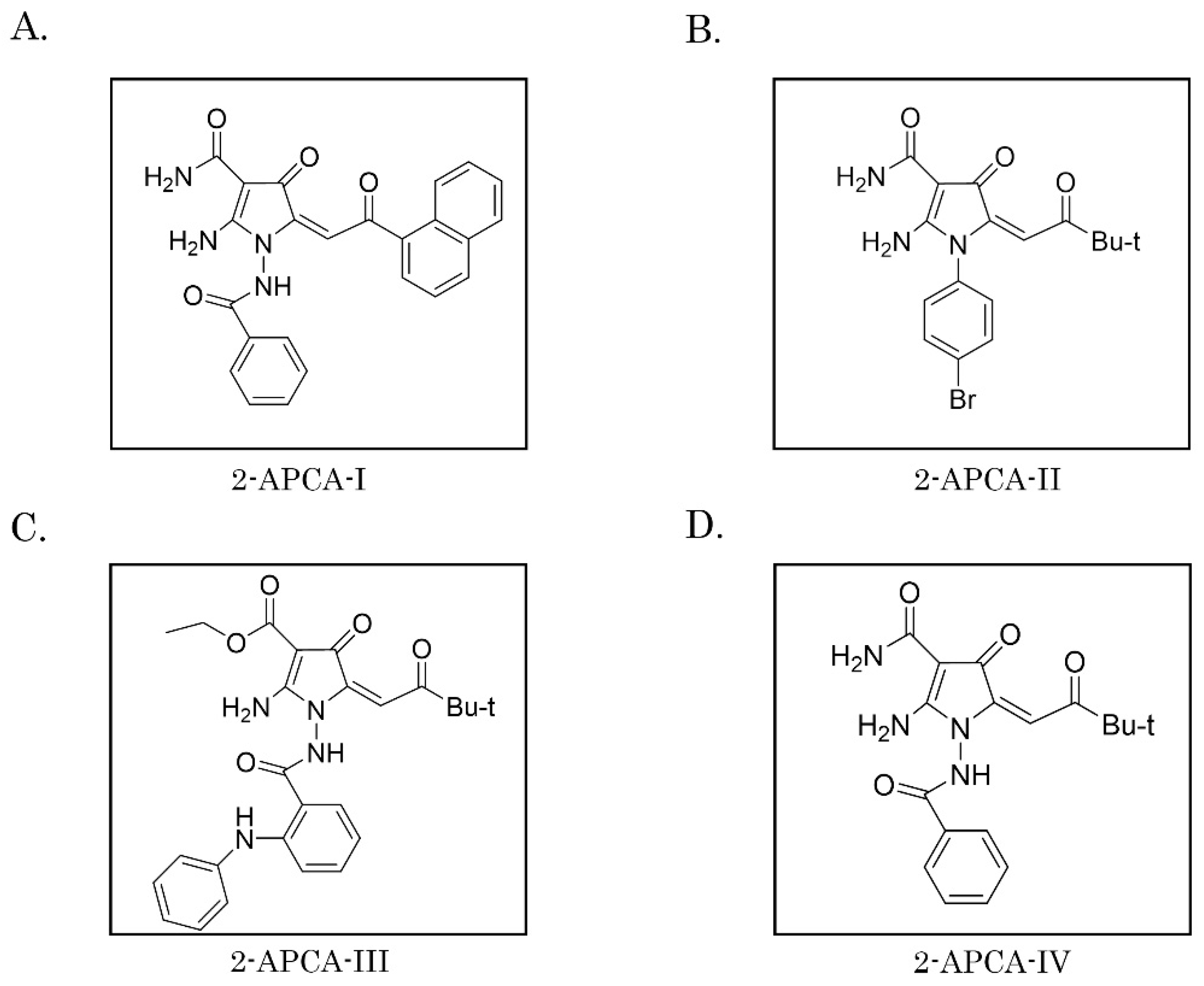
4.1. Chemical Compounds
4.2. Cell Lines and Culture Conditions
4.3. Antibodies
4.4. Cellular Survival MTS-Based Assay
4.5. Tubulin Polymerization Assay
4.6. Single-Cell Electrophoresis (Comet Assay)
4.7. Western Blotting
4.8. Immunofluorescence Staining
4.9. Real-Time Monitoring of Cell Proliferation
4.10. Molecular Docking
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Prota, A.E.; Bargsten, K.; Diaz, J.F.; Marsh, M.; Cuevas, C.; Liniger, M.; Neuhaus, C.; Andreu, J.M.; Altmann, K.-H.; O Steinmetz, M. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 13817–13821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Wang, Y.; Wang, T.; Jiang, J.; Botting, C.H.; Liu, H.; Chen, Q.; Yang, J.; Naismith, J.H.; Zhu, X.; et al. Pironetin reacts covalently with cysteine-316 of α-tubulin to destabilize microtubule. Nat. Commun. 2016, 7, 12103. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Setter, J.; Waight, A.B.; Bargsten, K.; Murga, J.; Diaz, J.F.; Steinmetz, M.O. Pironetin Binds Covalently to αCys316 and Perturbs a Major Loop and Helix of α-Tubulin to Inhibit Microtubule Formation. J. Mol. Biol. 2016, 428, 2981–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorléans, A.; Gigant, B.; Ravelli, R.B.G.; Mailliet, P.; Mikol, V.; Knossow, M. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc. Natl. Acad. Sci. USA 2009, 106, 13775–13779. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Benz, F.W.; Wu, Y.; Wang, Q.; Chen, Y.; Chen, X.; Li, H.; Zhang, Y.; Zhang, R.; Yang, J. Structural Insights into the Pharmacophore of Vinca Domain Inhibitors of Microtubules. Mol. Pharmacol. 2015, 89, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Steinmetz, M.; Prota, A.E. Microtubule-Targeting Agents: Strategies to Hijack the Cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef]
- Fanale, D.; Bronte, G.; Passiglia, F.; Calo, V.; Castiglia, M.; Di Piazza, F.; Barraco, N.; Cangemi, A.; Catarella, M.T.; Insalaco, L.; et al. Stabilizing versus destabilizing the microtubules: A double-edge sword for an effective cancer treatment option? Anal. Cell. Pathol. 2015, 2015, 690916. [Google Scholar] [CrossRef] [Green Version]
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res. 1999, 59, 653–660. [Google Scholar]
- West, L.M.; Northcote, P.T.; Battershill, C.N. ChemInform Abstract: Peloruside A: A Potent Cytotoxic Macrolide Isolated from the New Zealand Marine Sponge Mycale sp. J. Org. Chem. 2010, 31, 445–449. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.; Miller, J.H.; Díaz, J.F.; Steinmetz, M.O. Structural Basis of Microtubule Stabilization by Laulimalide and Peloruside A. Angew. Chem. Int. Ed. 2014, 53, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-G.; Horwitz, S.B. Differential mitotic responses to microtubule-stabilizing and -destabilizing drugs. Cancer Res. 2002, 62, 1935–1938. [Google Scholar] [PubMed]
- Canta, A.; Chiorazzi, A.; Cavaletti, G. Tubulin: A target for antineoplastic drugs into the cancer cells but also in the peripheral nervous system. Curr. Med. Chem. 2009, 16, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Carlson, K.; Ocean, A.J. Peripheral Neuropathy with Microtubule-Targeting Agents: Occurrence and Management Approach. Clin. Breast Cancer 2011, 11, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Krishna, R.; Mayer, L.D. Multidrug resistance (MDR) in cancer: Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci. 2000, 11, 265–283. [Google Scholar] [CrossRef]
- Bradley, G.; Ling, V. P-glycoprotein, multidrug resistance and tumor progression. Cancer Metastasis Rev. 1994, 13, 223–233. [Google Scholar] [CrossRef]
- Mechetner, E.; Kyshtoobayeva, A.; Zonis, S.; Kim, H.; Stroup, R.; Garcia, R.; Parker, R.J.; Fruehauf, J.P. Levels of multidrug resistance (MDR1) P-glycoprotein expression by human breast cancer correlate with in vitro resistance to taxol and doxorubicin. Clin. Cancer Res. 1998, 4, 389–398. [Google Scholar]
- Kavallaris, M.; Kuo, D.Y.; A Burkhart, C.; Regl, D.L.; Norris, M.D.; Haber, M.; Horwitz, S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J. Clin. Investig. 1997, 100, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef]
- Coulup, S.; Georg, G. Revisiting microtubule targeting agents: α-Tubulin and the pironetin-binding site as unexplored targets for cancer therapeutics. Bioorg. Med. Chem. Lett. 2019, 29, 1865–1873. [Google Scholar] [CrossRef]
- Zykova, S.; Boĭchuk, S.V.; Galembikova, A.R.; Ramazanov, B.R.; Mustafin, I.G.; Igidov, N.M.; Odegova, T.F. [3-Hydroxy-1,5-diaryl-4-pivaloyl-2,5-dihydro-2-pyrrolones induce the mitotic exit failure and cell death in tumor cells in vitro]. Tsitologiya 2014, 56, 439–442. [Google Scholar]
- Boichuk, S.; Galembikova, А.; Zykova, S.; Ramazanov, B.; Khusnutdinov, R.; Dunaev, P.; Khaibullina, S.; Lombardi, V. Ethyl-2-amino-pyrrole-3-carboxylates are novel potent anticancer agents that affect tubulin polymerization, induce G2/M cell-cycle arrest, and effectively inhibit soft tissue cancer cell growth in vitro. Anti Cancer Drugs 2016, 27, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Micheeva, E.; Novikova, M.; Khromova, N.; Kopnin, P. Ethyl-2-amino-pyrrole-3-carboxylates are active against imatinib-resistant gastrointestinal stromal tumors in vitro and in vivo. Anti Cancer Drugs 2019, 30, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Zykova, S.; Kizimova, I.A.; Syutkina, A.I.; Toksarova, Y.S.; Igidov, N.M.; Ibishov, D.F.; Boichuk, S.V.; Dunaev, P.D.; Galembikova, A.R.; Korochkina, R.R. Synthesis and Cytostatic Activity of (E)-Ethyl-2-Amino-5-(3,3-Dimethyl-4-Oxobutyliden)-4-Oxo-1- (2-Phenylaminobenzamido)-4,5-Dihydro-1Hpyrrol-3-Carboxylate. Pharm. Chem. J. 2020, 53, 895–898. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA Double-stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, W.-Z.; Li, B.; Huang, B.; Wang, Y.; Liu, X.-D.; Guan, H.; Zhang, S.-M.; Tang, Y.; Rang, W.-Q.; Zhou, P.-K. γH2AX foci formation in the absence of DNA damage: Mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013, 587, 3437–3443. [Google Scholar] [CrossRef] [Green Version]
- Noble, R.L.; Beer, C.T.; Cutts, J.H. Role of chance observations in chemotherapy: Vinca rosea *. Ann. N. Y. Acad. Sci. 1958, 76, 882–894. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Demagalhaes-Silverman, M.; Hammert, L.; Lembersky, B.; Lister, J.; Rybka, W.; Ball, E. High-dose chemotherapy and autologous stem cell support followed by post-transplant doxorubicin and taxol as initial therapy for metastatic breast cancer: Hematopoietic tolerance and efficacy. Bone Marrow Transplant. 1998, 21, 1207–1211. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, S.; Sekine, I.; Saijo, N. Paclitaxel (taxol): A review of its antitumor activity and toxicity in clinical studies. Gan kagaku ryoho. Cancer Chemother. 1998, 25, 605–615. [Google Scholar]
- Camirand, A.; Fadhil, I.; Luco, A.-L.; Ochietti, B.; Kremer, R. Enhancement of taxol, doxorubicin and zoledronate anti-proliferation action on triple-negative breast cancer cells by a PTHrP blocking monoclonal antibody. Am. J. Cancer Res. 2013, 3, 500–508. [Google Scholar] [PubMed]
- Hardin, C.; Shum, E.; Singh, A.P.; Perez-Soler, R.; Cheng, H. Emerging treatment using tubulin inhibitors in advanced non-small cell lung cancer. Expert Opin. Pharmacother. 2017, 18, 701–716. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, M.F.; Gerber, D.E. Chemotherapy for Advanced Non-small Cell Lung Cancer. Cancer Treat. Res. 2016, 170, 119–149. [Google Scholar] [PubMed]
- Balar, A.V. The impact of taxanes on the management of genitourinary cancers. Anti-Cancer Drugs 2014, 25, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, J.M.; de Wit, R. Taxane mechanisms of action: Potential implications for treatment sequencing in metastatic castration-resistant prostate cancer. Eur. Urol. 2014, 65, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Chen, S.; Wang, Y.; Watahiki, A.; Bohrer, L.; Sun, Z.; Wang, Y.; Huang, H. Inhibition of the Androgen Receptor as a Novel Mechanism of Taxol Chemotherapy in Prostate Cancer. Cancer Res. 2009, 69, 8386–8394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmonson, J.H.; Ryan, L.M.; Blum, R.H.; Brooks, J.S.; Shiraki, M.; Frytak, S.; Parkinson, D.R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J. Clin. Oncol. 1993, 11, 1269–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, A.; Tursz, T.; Mouridsen, H.; Verweij, J.; Steward, W.; Somers, R.; Buesa, J.; Casali, P.; Spooner, D.; Rankin, E. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: A randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J. Clin. Oncol. 1995, 13, 1537–1545. [Google Scholar] [CrossRef]
- Benjamin, R.S.; Lee, J.J. One step forward, 2 steps back. Lancet Oncol. 2014, 15, 366–367. [Google Scholar] [CrossRef]
- Verweij, J.; Lee, S.M.; Ruka, W.; Buesa, J.; Coleman, R.; Van Hoessel, R.; Seynaeve, C.; Di Paola, E.D.; Van Glabbeke, M.; Tonelli, D.; et al. Randomized Phase II Study of Docetaxel Versus Doxorubicin in First- and Second-Line Chemotherapy for Locally Advanced or Metastatic Soft Tissue Sarcomas in Adults: A Study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J. Clin. Oncol. 2000, 18, 2081–2086. [Google Scholar] [CrossRef]
- Edmonson, J.H.; Ebbert, L.P.; Nascimento, A.G.; Jung, S.-H.; McGaw, H.; Gerstner, J.B. Phase II Study of Docetaxel in Advanced Soft Tissue Sarcomas. Am. J. Clin. Oncol. 1996, 19, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, Q.G.C.M.; Verweij, J.; Catimel, G.; Clavel, M.; Kerbrat, P.; Van Oosterom, A.; Kerger, J.; Tursz, T.; Van Glabbeke, M.; Van Pottelsberghe, C.; et al. Phase II study with Docetaxel (Taxotere®) in advanced soft tissue sarcomas of the adult. Ann. Oncol. 1994, 5, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Hensley, M.L.; Maki, R.; Venkatraman, E.; Geller, G.; Lovegren, M.; Aghajanian, C.; Sabbatini, P.; Tong, W.; Barakat, R.; Spriggs, D.R. Gemcitabine and Docetaxel in Patients With Unresectable Leiomyosarcoma: Results of a Phase II Trial. J. Clin. Oncol. 2002, 20, 2824–2831. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.G.; Wathen, J.K.; Patel, S.R.; Priebat, D.A.; Okuno, S.H.; Samuels, B.; Fanucchi, M.; Harmon, D.C.; Schuetze, S.M.; Reinke, D.; et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: Results of Sarcoma Alliance for Research through Collaboration study 002 [corrected]. J. Clin. Oncol. 2007, 25, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Pautier, P.; Floquet, A.; Penel, N.; Piperno-Neumann, S.; Isambert, N.; Rey, A.; Bompas, E.; Cioffi, A.; Delcambre, C.; Cupissol, D.; et al. Randomized Multicenter and Stratified Phase II Study of Gemcitabine Alone Versus Gemcitabine and Docetaxel in Patients with Metastatic or Relapsed Leiomyosarcomas: A Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) French Sarcoma Group Study (TAXOGEM study). Oncologist 2012, 17, 1213–1220. [Google Scholar] [CrossRef] [Green Version]
- Seddon, B.; Whelan, J.; Strauss, S.; Leahy, M.G.; Woll, P.J.; Cowie, F.; Rothermundt, C.A.; Wood, Z.; Forsyth, S.; Khan, I.; et al. GeDDiS: A prospective randomized controlled phase III trial of gemcitabine and docetaxel compared with doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft tissue sarcomas [abstract]. J. Clin. Oncol. 2015, 33, 10500. [Google Scholar] [CrossRef]
- Sarkissian, S.; Fruehauf, J.P. Pazopanib and taxane combinations for metastatic or recurrent soft tissue sarcoma: A retrospective case series. J. Clin. Oncol. 2016, 34, e22528. [Google Scholar] [CrossRef]
- Horwitz, S.B.; Cohen, D.; Rao, S.; Ringel, I.; Shen, H.J.; Yang, C.P. Taxol: Mechanisms of action and resistance. J. Natl. Cancer Inst. Monogr. 1993, 15, 55–61. [Google Scholar] [CrossRef]
- Distefano, M.; Scambia, G.; Ferlini, C.; Gallo, D.; De Vincenzo, R.; Filippini, P.; Riva, A.; Bombardelli, E.; Mancuso, S. Antitumor activity of paclitaxel (taxol) analogues on MDR-positive human cancer cells. Anti Cancer Drug Des. 1998, 13, 489–499. [Google Scholar]
- Poruchynsky, M.S.; Komlodi-Pasztor, E.; Trostel, S.; Wilkerson, J.; Regairaz, M.; Pommier, Y.; Zhang, X.; Maity, T.K.; Robey, R.W.; Burotto, M.; et al. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 1571–1576. [Google Scholar] [CrossRef] [Green Version]
- Chernikova, S.B.; Nguyen, R.B.; Truong, J.T.; Mello, S.S.; Stafford, J.H.; Hay, M.P.; Olson, A.; Solow-Cordero, D.; Wood, D.; Henry, S.; et al. Dynamin impacts homology-directed repair and breast cancer response to chemotherapy. J. Clin. Investig. 2018, 128, 5307–5321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassing, I.; Saayman, X.; Rampazzo, L.; Ralf, C.; Bassett, A.; Esashi, F. The RAD51 recombinase protects mitotic chromatin in human cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, T.; Sonobe, H.; Toyonaga, S.-I.; Yamasaki, I.; Shuin, T.; Takano, A.; Araki, K.; Akimaru, K.; Yuri, K. Conventional and Molecular Cytogenetic Characterization of a New Human Cell Line, GIST-T1, Established from Gastrointestinal Stromal Tumor. Lab. Investig. 2002, 82, 663–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boichuk, S.; Galembikova, А.; Dunaev, P.; Valeeva, E.; Shagimardanova, E.; Gusev, O.; Khaiboullina, S.F. A Novel Receptor Tyrosine Kinase Switch Promotes Gastrointestinal Stromal Tumor Drug Resistance. Molecules 2017, 22, 2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boichuk, S.; Galembikova, А.; Sitenkov, A.; Khusnutdinov, R.; Dunaev, P.; Valeeva, E.; Usolova, N. Establishment and characterization of a triple negative basal-like breast cancer cell line with multi-drug resistance. Oncol. Lett. 2017, 14, 5039–5045. [Google Scholar] [CrossRef] [Green Version]
- Khusnutdinov, R.; Galembikova, A.; Boichuk, S. Establishment of the clone of gastrointestinal stromal tumor cells with the signs of multidrug resistance and assessment if its properties. Сoвременные Технoлoгии Медицине 2016, 8, 36–38. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sarcoma Cell Lines | 2-APCA-I (µmol) | 2-APCA-II (µmol) | 2-APCA-III (µmol) | 2-APCA-IV (µmol) | Paclitaxel | Vinblastine |
|---|---|---|---|---|---|---|
| BJ tert | 11.3 ± 2 | 38.3 ± 3.3 | 6.2 ± 0.5 | 54.3 ± 2.8 | 57.2 ± 3.7 nmol | 45.3 ± 5.9 nmol |
| HT1080 | 8.2 ± 0.2 | 30.6 ± 3.5 | 13.8 ± 2.6 | 60 ± 5.5 | 57.2 ± 6.7 pmol | 18.9 ± 2 pmol |
| RD | 4.2 ± 0.5 | 5.2 ± 0.4 | 2.5 ± 0.2 | 7.9 ± 0.7 | 9.8 ± 0.7 nmol | 1.1 ± 0.1 nmol |
| SK-LMS-1 | 7.1 ± 0.2 | 12.5 ± 0.6 | 11.3 ± 0.9 | 15.4 ± 2.4 | 23.9 ± 3.3 nmol | 20 ± 2.4 nmol |
| А673 | 28.9 ± 1.3 | 27.1 ± 3.8 | 10.2 ± 1.1 | 64.3 ± 6.4 | 18.8 ± 4.4 pmol | 740.4 ± 174.9 fmol |
| U2OS | 12 ± 0.13 | 28.8 ± 1.1 | 10.6 ± 0.7 | 63.1 ± 8.5 | 15.8 ± 2.8 nmol | 124.2 ± 22.9 nmol |
| Epithelial Cancer Cell Lines | 2-APCA-I (µmol) | 2-APCA-II (µmol) | 2-APCA-III (µmol) | 2-APCA-IV (µmol) | Paclitaxel | Vinblastine |
|---|---|---|---|---|---|---|
| HCC1806 | 13.5 ± 2.3 | 23.7 ± 1.1 | 12.4 ± 1 | 29.2 ± 5.7 | 31.6 ± 6 pmol | 322.3 ± 36.5 pmol |
| MDA-MB-231 | 20.3 ± 4 | 29.5 ± 4.8 | 7.2 ± 0.3 | 19.7 ± 1.3 | 35 ± 10.9 pmol | 852.6 ± 271 pmol |
| Н1299 | 29.9 ± 5.7 | 29.5 ± 4.8 | 5 ± 0.1 | 5 ± 0.1 | 1.7 ± 0.5 pmol | 6.7 ± 1.9 pmol |
| РС-3 | 15.8 ± 3.5 | 43.1 ± 0.9 | 10.7 ± 1.1 | 32.2 ± 4.5 | 42 ± 9.6 pmol | 40.4 ± 10.8 fmol |
| Hela | 2.8 ± 0.4 | 33 ± 2 | 14.4 ± 2 | 33.9 ± 3.2 | 352.4 ± 51 pmol | 1.7 ± 0.4 pmol |
| Cell Lines | 2-APCA-I (µmol) | 2-APCA-II (µmol) | 2-APCA-III (µmol) | 2-APCA-IV (µmol) | Paclitaxel | Vinblastine |
|---|---|---|---|---|---|---|
| GIST T1 | 1.3 ± 0.2 | 8.3 ± 0.3 | 2.6 ± 0.1 | 8.4 ± 0.5 | 51.7 ± 9.5 pmol | 109.1 ± 18.9 pmol |
| GIST T1-IM-R | 9.3 ± 0.4 | 9.7 ± 0.3 | 2 ± 0.1 | 13.8 ± 0.9 | 7.8 ± 1 pmol | 481.3 ± 77.4 pmol |
| GIST T1-Tx-R | 6.7 ± 0.2 | 9.9 ± 0.2 | 4.6 ± 0.3 | 12.4 ± 1.2 | 431.6 ± 69.8 nmol | 41.8 ± 0.6 nmol |
| GIST 430 | 8.9 ± 0.5 | 9 ± 0.5 | 8.9 ± 0.3 | 9 ± 0.5 | 30.4 ± 7.6 µmol | 33.4 ± 6.5 µmol |
| HCC1806 | 13.5 ± 2.3 | 23.7 ± 1.1 | 12.4 ± 1 | 29.2± 5.7 | 31.6 ± 6 pmol | 322.3 ± 36.5 pmol |
| HCC1806 Tx-R | 21.8 ± 1.9 | 13.5 ± 0.7 | 10.2 ± 1.2 | 22.3 ± 2.1 | 949 ± 78.8 nmol | 415.8 ± 81.7 nmol |
| 2-APCA | Amino Acid Composition of the Binding Site |
|---|---|
| 2-APCA-I | A: ARG-402, ALA-403, HIS-406 B: PHE-262, PRO-263, ARG-264, HIS-266, TRP-346, GLU-431, TYR-432, GLN-434, TYR-435, GLN-436 |
| 2-APCA-II | LEU-26, GLU-27, HIS-28, ASP-33, GLY-34, GLN-35, MET-36, SER-236, ALA-240, SER-241, PHE-244, ARG-320, ASN-356, TYR-357, GLU-358, PRO-359, THR-361 |
| 2-APCA-III | ALA-19, TRP-21, GLU-22, LEU-23, CYS-25, LYS-40, THR-41, ILE-42, ARG-84, GLN-85, LEU-86, PHE-87, ARG-229, GLN-233, VAL-362, VAL-363, PRO-364, ASP-367 |
| 2-APCA-IV | PRO-274, THR-276, SER-280, GLN-281, GLN-282, TYR-283, ARG-284, ALA-285, LEU-286, GLU-290, GLY-370, LEU-371, LYS-372 |
| Gold Fitness | ChemScore dG (kJ·mol−1) | |
|---|---|---|
| 2-АРСА-I | 53.39 | −26.32 |
| 2-АРСА-II | 52.54 | −26.37 |
| 2-АРСА-III | 65.43 | −27.71 |
| 2-АРСА-IV | 57.91 | −25.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boichuk, S.; Galembikova, A.; Bikinieva, F.; Dunaev, P.; Aukhadieva, A.; Syuzov, K.; Zykova, S.; Igidov, N.; Ksenofontov, A.; Bocharov, P. 2-APCAs, the Novel Microtubule Targeting Agents Active against Distinct Cancer Cell Lines. Molecules 2021, 26, 616. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26030616
Boichuk S, Galembikova A, Bikinieva F, Dunaev P, Aukhadieva A, Syuzov K, Zykova S, Igidov N, Ksenofontov A, Bocharov P. 2-APCAs, the Novel Microtubule Targeting Agents Active against Distinct Cancer Cell Lines. Molecules. 2021; 26(3):616. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26030616
Chicago/Turabian StyleBoichuk, Sergei, Aigul Galembikova, Firuza Bikinieva, Pavel Dunaev, Aida Aukhadieva, Kirill Syuzov, Svetlana Zykova, Nazim Igidov, Alexander Ksenofontov, and Pavel Bocharov. 2021. "2-APCAs, the Novel Microtubule Targeting Agents Active against Distinct Cancer Cell Lines" Molecules 26, no. 3: 616. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26030616