
PSC-db: A Structured and Searchable 3D-Database for Plant Secondary Compounds

 ,
, 
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. System Architecture
- i.
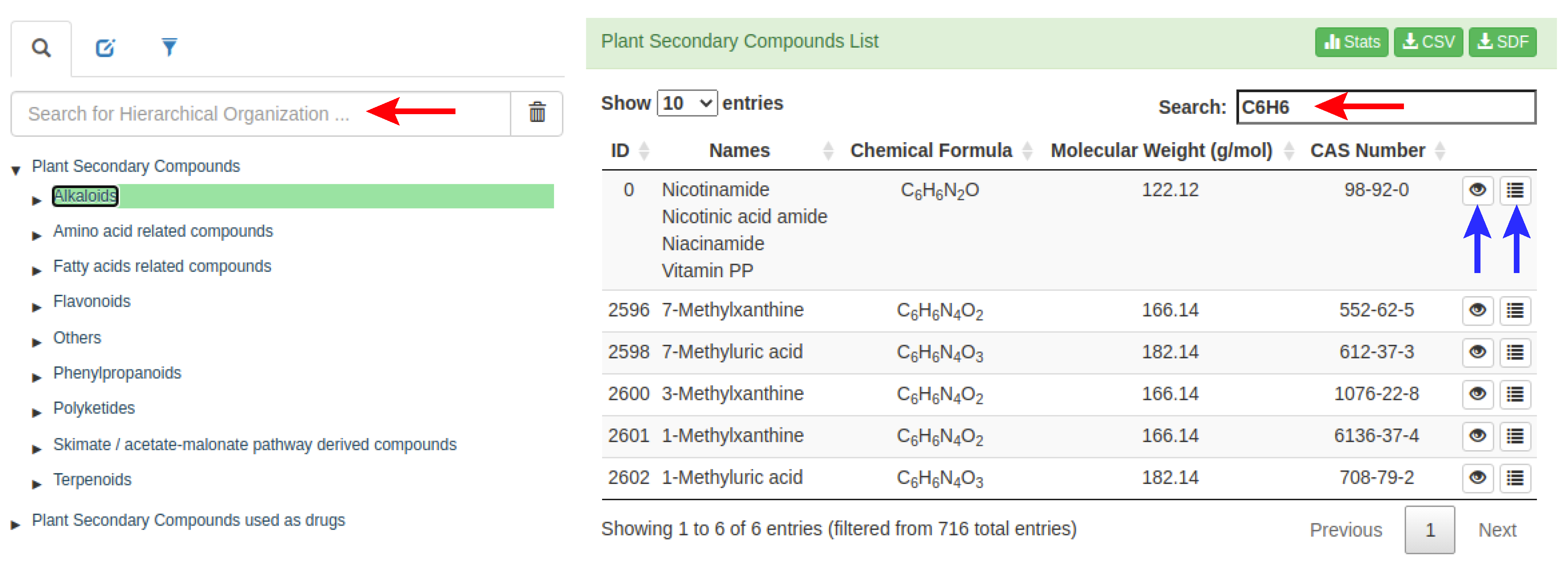
- HierarchicalView: performs a hierarchical representation of the compounds organized by subfamilies.
- ii.
- TableView: shows the list of compounds as data tables. It is possible to filter the results in the data table writing some patterns into a text input. Also, a dynamic CSV or SDF file can be downloaded containing only the compounds listed (filtered) in the data table.
- iii.
- DetailsView: allows access to detailed information of the compounds, such as external links, hierarchical organization, and its properties (physicochemical, lipophilicity, water-solubility, pharmacokinetics, and drug-likeness), source organism and biological activity.
- iv.
- StatsView: generates statistics of the compounds and determines the correlation value (r2).
- v.
- 2D/3DView: shows the 2D and 3D visualization of each chemical compound with its corresponding Radar ADME/Tox graph.
4.2. Database Model
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wink, M. Evolution of secondary metabolites from an ecological and molecular phylogenetic perspective. Phytochemistry 2003, 64, 3–19. [Google Scholar] [CrossRef]
- Demain, A.L.; Fang, A. The natural functions of secondary metabolites. In History of Modern Biotechnology I; Springer: Berlin/Heidelberg, Germany, 2000; pp. 1–39. [Google Scholar]
- Wolfram, S.; Wang, Y.; Thielecke, F. Anti-obesity effects of green tea: From bedside to bench. Mol. Nutr. Food Res. 2006, 50, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Tipoe, G.L.; Leung, T.-M.; Hung, M.-W.; Fung, M.-L. Green tea polyphenols as an anti-oxidant and anti-inflammatory agent for cardiovascular protection. Cardiovasc. Hematol. Disord. Targets 2007, 7, 135–144. [Google Scholar] [CrossRef]
- Patil, K.R.; Goyal, S.N.; Sharma, C.; Patil, C.R.; Ojha, S. Phytocannabinoids for cancer therapeutics: Recent updates and future prospects. Curr. Med. Chem. 2015, 22, 3472–3501. [Google Scholar] [CrossRef]
- Hill, A.J.; Williams, C.M.; Whalley, B.J.; Stephens, G.J. Phytocannabinoids as novel therapeutic agents in CNS disorders. Pharmacol. Ther. 2012, 133, 79–97. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, R.G.; Hallak, J.E.C.; Leite, J.P.; Zuardi, A.W.; Crippa, J.A.S. Phytocannabinoids and epilepsy. J. Clin. Pharm. Ther. 2015, 40, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Catalan, U.; Barrubes, L.; Valls, R.M.; Sola, R.; Rubio, L. In vitro metabolomic approaches to investigating the potential biological effects of phenolic compounds: An update. Genomics. Proteom. Bioinform. 2017, 15, 236–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corpas, R.; Griñán-Ferré, C.; Rodríguez-Farré, E.; Pallàs, M.; Sanfeliu, C. Resveratrol induces brain resilience against Alzheimer neurodegeneration through proteostasis enhancement. Mol. Neurobiol. 2019, 56, 1502–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorokina, M.; Steinbeck, C. Review on natural products databases: Where to find data in 2020. J. Cheminform. 2020, 12, 1–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, P.; Erehman, J.; Gohlke, B.-O.; Wilhelm, T.; Preissner, R.; Dunkel, M. Super Natural II—A database of natural products. Nucleic Acids Res. 2015, 43, D935–D939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.-Z.; Xu, X. Use of natural products as chemical library for drug discovery and network pharmacology. PLoS ONE 2013, 8, e62839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Santen, J.A.; Jacob, G.; Singh, A.L.; Aniebok, V.; Balunas, M.J.; Bunsko, D.; Neto, F.C.; Castaño-Espriu, L.; Chang, C.; Clark, T.N.; et al. The natural products atlas: An open access knowledge base for microbial natural products discovery. ACS Cent. Sci. 2019, 5, 1824–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhang, P.; He, W.; Qin, C.; Chen, S.; Tao, L.; Wang, Y.; Tan, Y.; Gao, D.; Wang, B.; et al. NPASS: Natural product activity and species source database for natural product research, discovery and tool development. Nucleic Acids Res. 2018, 46, D1217–D1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein--ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomiki, T.; Saito, T.; Ueki, M.; Konno, H.; Asaoka, T.; Suzuki, R.; Uramoto, M.; Kakeya, H.; Osada, H. RIKEN natural products encyclopedia (RIKEN NPEdia), a chemical database of RIKEN natural products depository (RIKEN NPDepo). J. Comput. Aided Chem. 2006, 7, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Maeda, M.H.; Kondo, K. Three-dimensional structure database of natural metabolites (3DMET): A novel database of curated 3D structures. J. Chem. Inf. Model. 2013, 53, 527–533. [Google Scholar] [CrossRef]
- Shen, J.; Xu, X.; Cheng, F.; Liu, H.; Luo, X.; Shen, J.; Chen, K.; Zhao, W.; Shen, X.; Jiang, H. Virtual screening on natural products for discovering active compounds and target information. Curr. Med. Chem. 2003, 10, 2327–2342. [Google Scholar] [CrossRef]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [Green Version]
- Hou, T.J.; Zhang, W.; Xia, K.; Qiao, X.B.; Xu, X.J. ADME evaluation in drug discovery. 5. Correlation of Caco-2 permeation with simple molecular properties. J. Chem. Inf. Comput. Sci. 2004, 44, 1585–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, T.; Wang, J.; Zhang, W.; Xu, X. ADME evaluation in drug discovery. 7. Prediction of oral absorption by correlation and classification. J. Chem. Inf. Model. 2007, 47, 208–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durán-Iturbide, N.A.; Díaz-Eufracio, B.I.; Medina-Franco, J.L. In silico ADME/Tox profiling of natural products: A focus on BIOFACQUIM. ACS Omega 2020, 5, 16076–16084. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, D.B.; Singh, S.; Shukla, R.; Ramteke, P.W.; Misra, K. Exploring medicinal plant legacy for drug discovery in post-genomic era. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 2019, 89, 1141–1151. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Holliday, J.D.; Salim, N.; Whittle, M.; Willett, P. Analysis and display of the size dependence of chemical similarity coefficients. J. Chem. Inf. Comput. Sci. 2003, 43, 819–828. [Google Scholar] [CrossRef]
- Rego, N.; Koes, D. 3Dmol. js: Molecular visualization with WebGL. Bioinformatics 2015, 31, 1322–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimazu, S.; Miklya, I. Pharmacological studies with endogenous enhancer substances: β-phenylethylamine, tryptamine, and their synthetic derivatives. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2004, 28, 421–427. [Google Scholar] [CrossRef]
- Wölfel, R.; Graefe, K.-H. Evidence for various tryptamines and related compounds acting as substrates of the platelet 5-hydroxytryptamine transporter. Naunyn. Schmiedebergs. Arch. Pharmacol. 1992, 345, 129–136. [Google Scholar] [CrossRef]
- Callaway, J.C.; McKenna, D.J.; Grob, C.S.; Brito, G.S.; Raymon, L.P.; Poland, R.E.; Andrade, E.N.; Andrade, E.O.; Mash, D.C. Pharmacokinetics of Hoasca alkaloids in healthy humans. J. Ethnopharmacol. 1999, 65, 243–256. [Google Scholar] [CrossRef]
- Wan, F.; Zhu, Y.; Hu, H.; Dai, A.; Cai, X.; Chen, L.; Gong, H.; Xia, T.; Yang, D.; Wang, M.-W.; et al. DeepCPI: A deep learning-based framework for large-scale in silico drug screening. Genomics. Proteom. Bioinform. 2019, 17, 478–495. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Shimura, N.; Otabe, Y.; Hirai-Morita, A.; Nakamura, Y.; Ono, N.; Ul-Amin, M.A.; Kanaya, S. KNApSAcK-3D: A Three-Dimensional Structure Database of Plant Metabolites. Plant Cell Physiol. 2013, 54, e4. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka Michałand Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; Davies, M.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Westbrook, J.D.; Shao, C.; Feng, Z.; Zhuravleva, M.; Velankar, S.; Young, J. The chemical component dictionary: Complete descriptions of constituent molecules in experimentally determined 3D macromolecules in the Protein Data Bank. Bioinformatics 2015, 31, 1274–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, E.M.; Jorgensen, W.L. Prediction of properties from simulations: Free energies of solvation in hexadecane, octanol, and water. J. Am. Chem. Soc. 2000, 122, 2878–2888. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdés-Jiménez, A.; Peña-Varas, C.; Borrego-Muñoz, P.; Arrue, L.; Alegría-Arcos, M.; Nour-Eldin, H.; Dreyer, I.; Nuñez-Vivanco, G.; Ramírez, D. PSC-db: A Structured and Searchable 3D-Database for Plant Secondary Compounds. Molecules 2021, 26, 1124. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26041124
Valdés-Jiménez A, Peña-Varas C, Borrego-Muñoz P, Arrue L, Alegría-Arcos M, Nour-Eldin H, Dreyer I, Nuñez-Vivanco G, Ramírez D. PSC-db: A Structured and Searchable 3D-Database for Plant Secondary Compounds. Molecules. 2021; 26(4):1124. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26041124
Chicago/Turabian StyleValdés-Jiménez, Alejandro, Carlos Peña-Varas, Paola Borrego-Muñoz, Lily Arrue, Melissa Alegría-Arcos, Hussam Nour-Eldin, Ingo Dreyer, Gabriel Nuñez-Vivanco, and David Ramírez. 2021. "PSC-db: A Structured and Searchable 3D-Database for Plant Secondary Compounds" Molecules 26, no. 4: 1124. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26041124