
Vibrational Spectroscopic Analyses and Imaging of the Early Middle Ages Hemp Bast Fibres Recovered from Lake Sediments

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
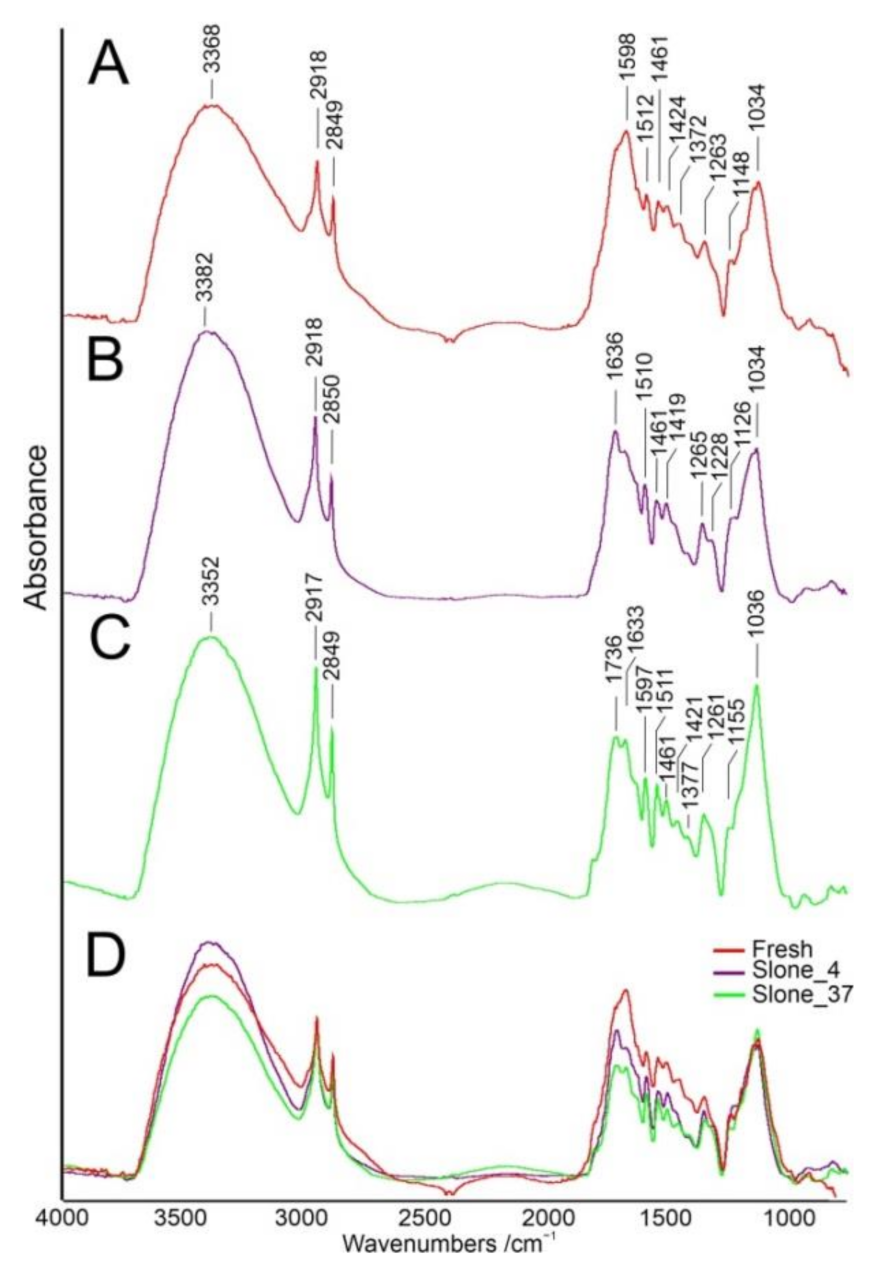
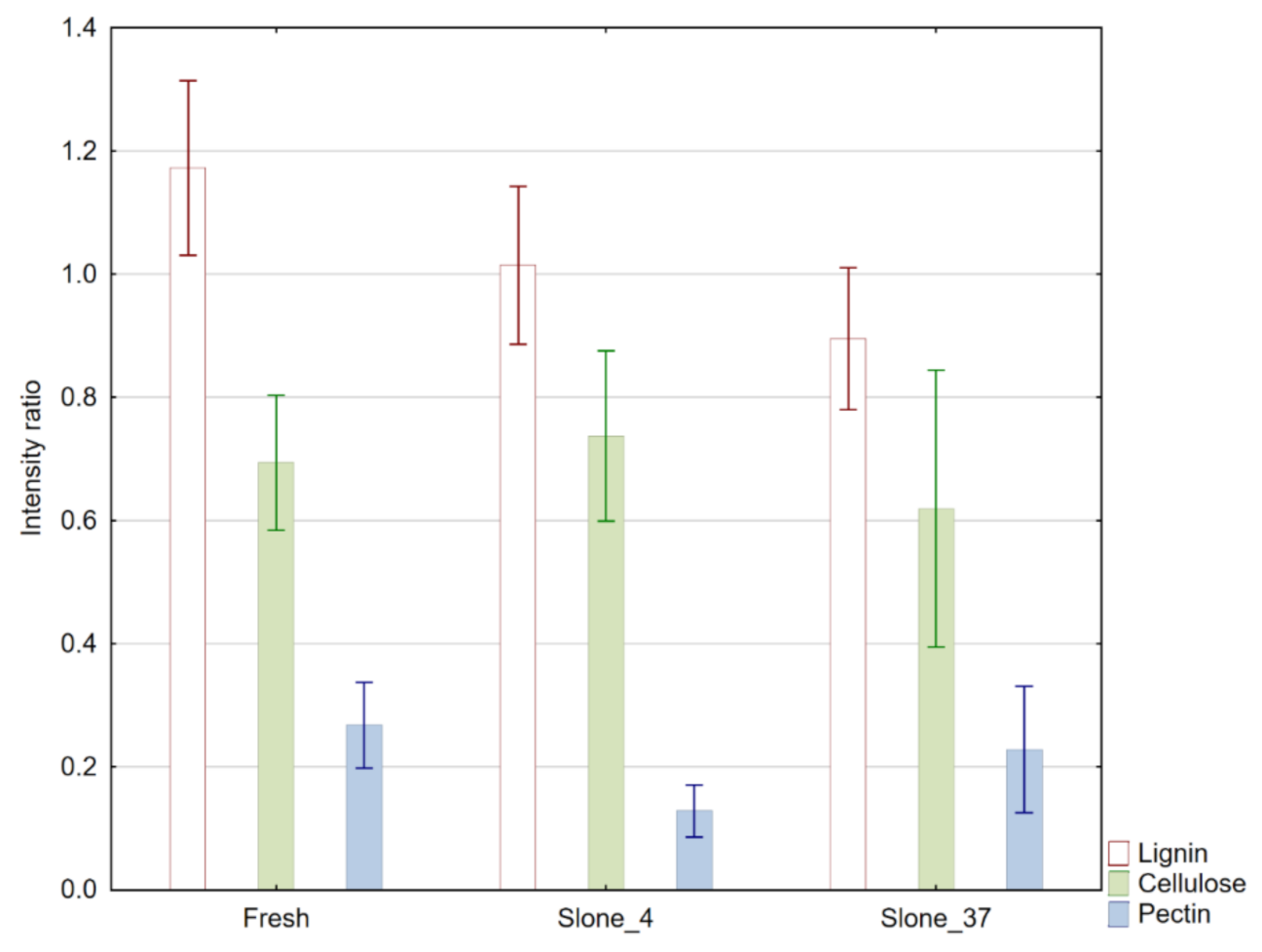
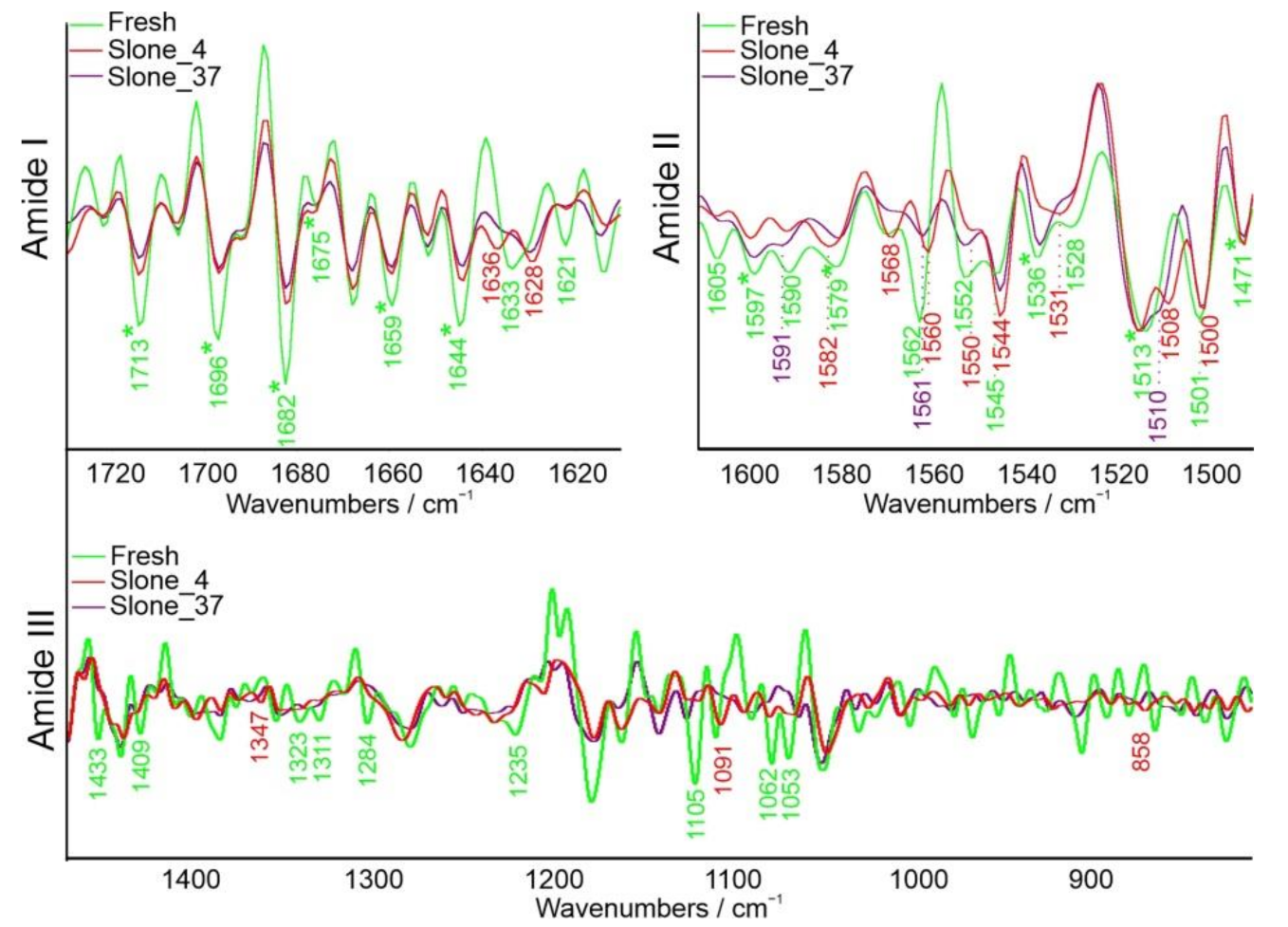
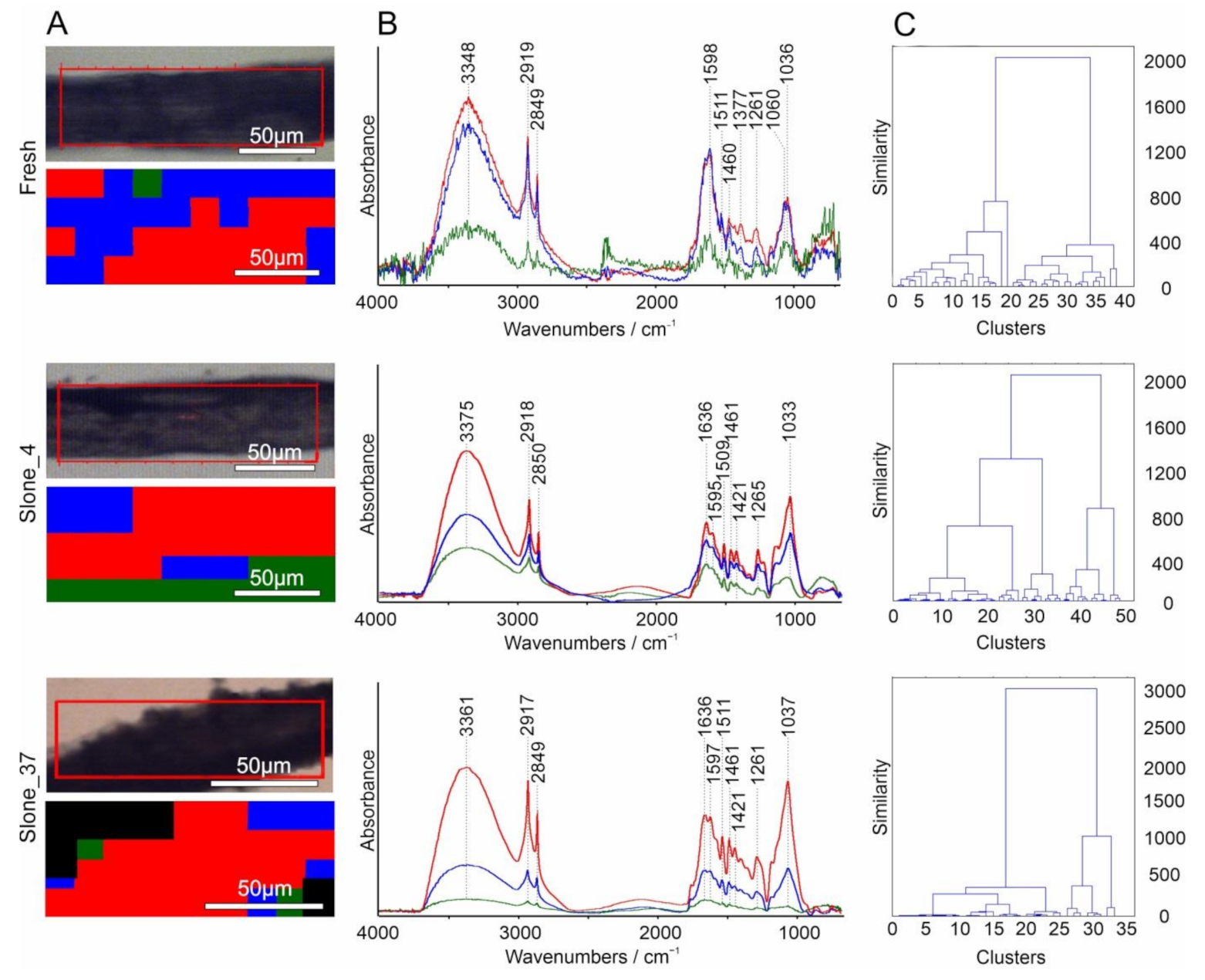
2.1. FT-IR Spectroscopy
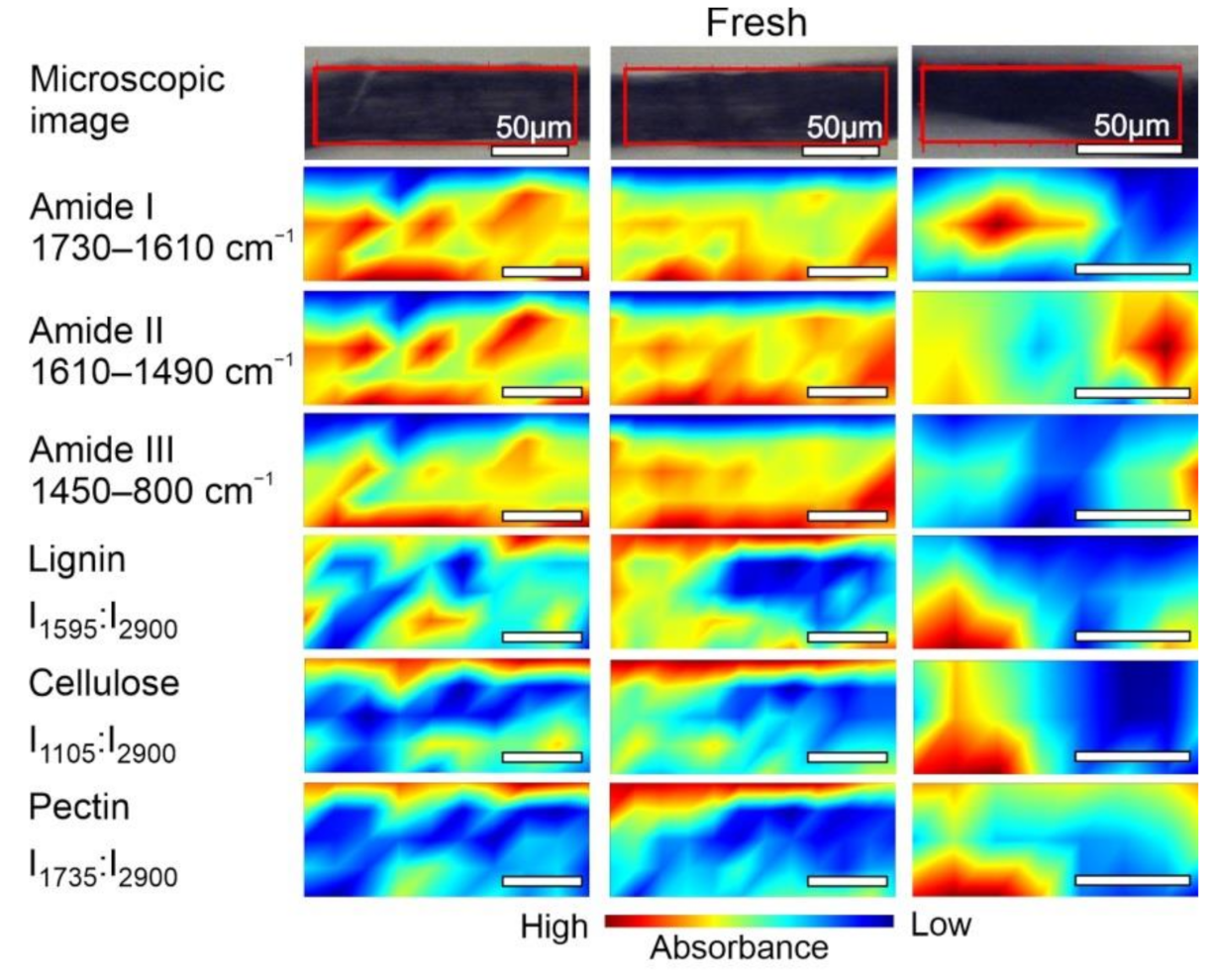
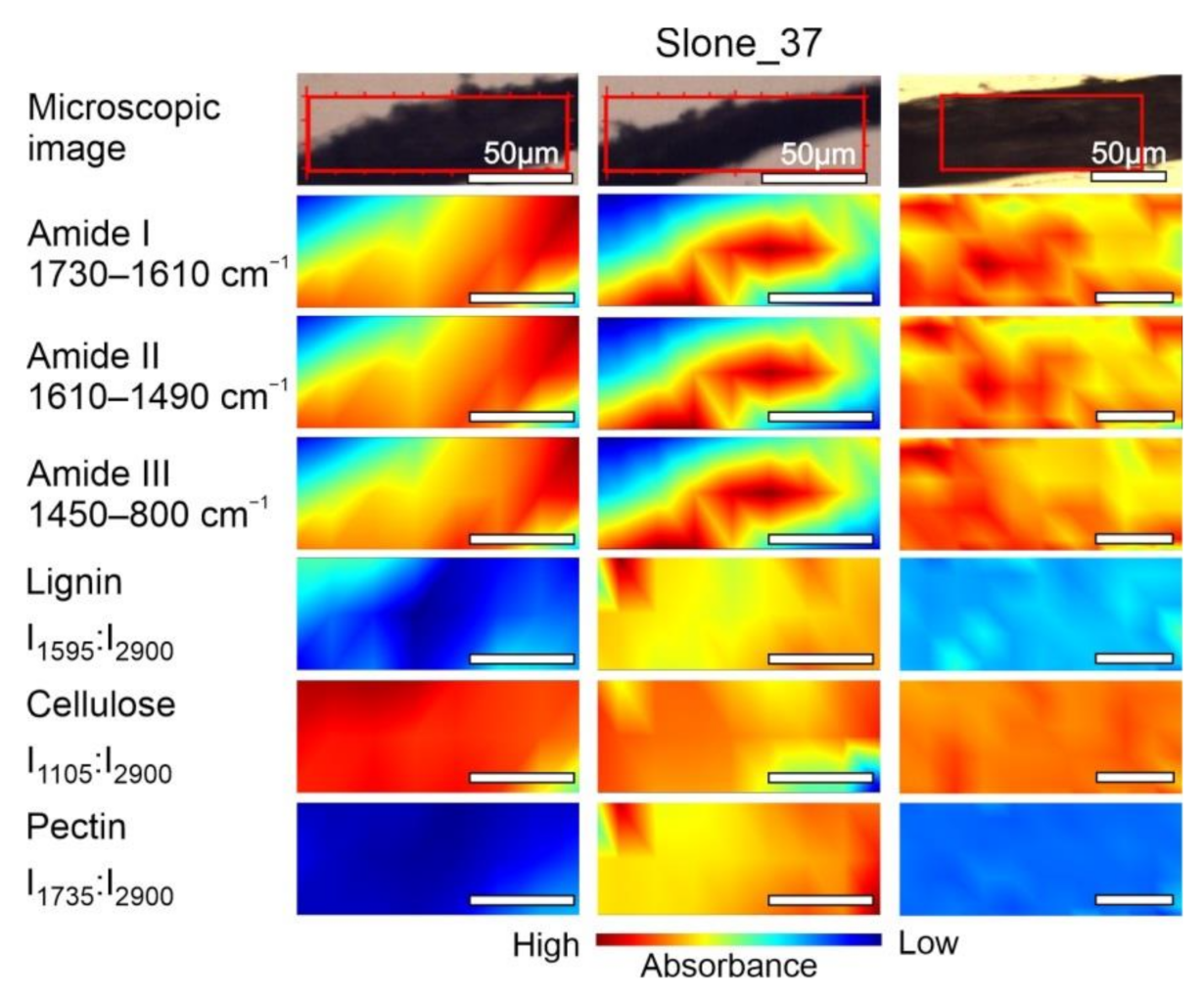
2.2. FT-IR Spectroscopic Imaging
2.3. Hierarchical Cluster Analysis (HCA)
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Preparation
4.2. FT-IR Microspectroscopy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Band/Ratio | Absorbance Intensity/a.u. ±SD |
|---|---|---|
| Fresh | 2900 cm−1 | 0.171 ± 0.046 |
| 1595 cm−1 | 0.189 ± 0.047 | |
| 1105 cm−1 | 0.104 ± 0.033 | |
| 1735 cm−1 | 0.046 ± 0.018 | |
| R1=I1595/I2900 | 1.136 ± 0.224 | |
| R2=I1105/I2900 | 0.628 ± 0.177 | |
| R3=I1735/I2900 | 0.277 ± 0.118 | |
| Slone_4 | 2900 cm−1 | 0.197 ± 0.055 |
| 1595 cm−1 | 0.192 ± 0.069 | |
| 1105 cm−1 | 0.145 ± 0.044 | |
| 1735 cm−1 | 0.019 ± 0.018 | |
| R1=I1595/I2900 | 0.967 ± 0.158 | |
| R2=I1105/I2900 | 0.752 ± 0.231 | |
| R3=I1735/I2900 | 0.094 ± 0.057 | |
| Slone_37 | 2900 cm−1 | 0.220 ± 0.112 |
| 1595 cm−1 | 0.185 ± 0.081 | |
| 1105 cm−1 | 0.138 ± 0.082 | |
| 1735 cm−1 | 0.045 ± 0.020 | |
| R1=I1595/I2900 | 0.904 ± 0.206 | |
| R2=I1105/I2900 | 0.631 ± 0.250 | |
| R3=I1735/I2900 | 0.235 ± 0.131 |
References
- Kulesza, P.; Suchora, M.; Pidek, I.A.; Dobrowolski, R.; Alexandrowicz, W.P. The Holocene palaeoenvironmental changes reflected in the multi-proxy studies of Lake Słone sediments (SE Poland). Palaeogeogr. Palaeoclimatol. Palaeoecol. 2012, 363–364, 79–98. [Google Scholar] [CrossRef]
- Kulesza, P.; Suchora, M.; Pidek, I.A.; Alexandrowicz, W.P. Chronology and directions of Late Glacial paleoenvironmental changes: A multi-proxy study on sediments of Lake Słone (SE Poland). Quat. Int. 2011, 238, 89–106. [Google Scholar] [CrossRef]
- Dai, D.; Fan, M. Characteristic and Performance of Elementary Hemp Fibre. Mater. Sci. Appl. 2010, 1, 336–342. [Google Scholar] [CrossRef] [Green Version]
- Le Troedec, M.; Sedan, D.; Peyratout, C.; Bonnet, J.P.; Smith, A.; Guinebretiere, R.; Gloaguen, V.; Krausz, P. Influence of various chemical treatments on the composition and structure of hemp fibres. Compos. Part A Appl. Sci. Manuf. 2008, 39, 514–522. [Google Scholar] [CrossRef]
- Franck, R.R. Bast and Other Plant Fibres; Woodhead Publishing Limited: Cambridge, UK, 2005; ISBN 978-1-85573-684-9. [Google Scholar]
- Guerriero, G.; Behr, M.; Legay, S.; Mangeot-Peter, L.; Zorzan, S.; Ghoniem, M.; Hausman, J.-F. Transcriptomic profiling of hemp bast fibres at different developmental stages. Sci. Rep. 2017, 7, 4961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Sain, M.; Oksman, K. Study of Structural Morphology of Hemp Fiber from the Micro to the Nanoscale. Appl. Compos. Mater. 2007, 14, 89–103. [Google Scholar] [CrossRef]
- Dussán, K.J.; Silva, D.D.V.; Moraes, E.J.C.; Arruda, P.V.; Felipe, M.G.A. Dilute-acid hydrolysis of cellulose to glucose from sugarcane bagasse. Chem. Eng. Trans. 2014, 38, 433–438. [Google Scholar] [CrossRef]
- Gurunathan, T.; Mohanty, S.; Nayak, S.K. A review of the recent developments in biocomposites based on natural fibres and their application perspectives. Compos. Part A Appl. Sci. Manuf. 2015, 77, 1–25. [Google Scholar] [CrossRef]
- Mansor, A.M.; Lim, J.S.; Ani, F.N.; Hashim, H.; Ho, W.S. Characteristics of cellulose, hemicellulose and lignin of MD2 pineapple biomass. Chem. Eng. Trans. 2019, 79–84. [Google Scholar] [CrossRef]
- Mohnen, D. Pectin structure and biosynthesis. Curr. Opin. Plant Biol. 2008, 11, 266–277. [Google Scholar] [CrossRef]
- Ralph, J.; Lapierre, C.; Boerjan, W. Lignin structure and its engineering. Curr. Opin. Biotechnol. 2019, 56, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Manaia, J.P.; Manaia, A.T.; Rodriges, L. Industrial Hemp Fibers: An Overview. Fibers 2019, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Edwards, H.G.M.; Farwell, D.W.; Webster, D. FT Raman microscopy of untreated natural plant fibres. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1997, 53, 2383–2392. [Google Scholar] [CrossRef]
- Talari, A.C.S.; Martinez, M.A.G.; Movasaghi, Z.; Rehman, S.; Rehman, I.U. Advances in Fourier transform infrared (FTIR) spectroscopy of biological tissues. Appl. Spectrosc. Rev. 2017, 52, 456–506. [Google Scholar] [CrossRef]
- Kazarian, S.G.; Chan, K.L.A. ATR-FTIR spectroscopic imaging: Recent advances and applications to biological systems. Analyst 2013, 138, 1940–1951. [Google Scholar] [CrossRef]
- Kimber, J.A.; Kazarian, S.G. Spectroscopic imaging of biomaterials and biological systems with FTIR microscopy or with quantum cascade lasers. Anal. Bioanal. Chem. 2017, 409, 5813–5820. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, U.P. 1064 nm FT-Raman spectroscopy for investigations of plant cell walls and other biomass materials. Front. Plant Sci. 2014, 5, 490. [Google Scholar] [CrossRef] [PubMed]
- Durak, T.; Depciuch, J. Effect of plant sample preparation and measuring methods on ATR-FTIR spectra results. Environ. Exp. Bot. 2020, 169, 103915. [Google Scholar] [CrossRef]
- Fan, M.; Dai, D.; Huang, B. Fourier Transform Infrared Spectroscopy for Natural Fibres. In Fourier Transform—Materials Analysis; InTech: Rijeka, Croatia, 2012; pp. 45–68. [Google Scholar]
- Baker, M.J.; Trevisan, J.; Bassan, P.; Bhargava, R.; Butler, H.J.; Dorling, K.M.; Fielden, P.R.; Fogarty, S.W.; Fullwood, N.J.; Heys, K.A.; et al. Using Fourier transform IR spectroscopy to analyze biological materials. Nat. Protoc. 2014, 9, 1771–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazarian, S.G.; Chan, K.L.A. Applications of ATR-FTIR spectroscopic imaging to biomedical samples. Biochim. Biophys. Acta Biomembr. 2006, 1758, 858–867. [Google Scholar] [CrossRef] [Green Version]
- Zimniewska, M.; Rozańska, W.; Gryszczynska, A.; Romanowska, B.; Kicinska-Jakubowska, A. Antioxidant potential of hemp and flax fibers depending on their chemical composition. Molecules 2018, 23, 1993. [Google Scholar] [CrossRef] [Green Version]
- Garside, P.; Wyeth, P. Identification of Cellulosic Fibres by FTIR Spectroscopy—Thread and Single Fibre Analysis by Attenuated Total Reflectance. Stud. Conserv. 2003, 48, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Dorado, J.; Almendros, G.; Field, J.A.; Sierra-Alvarez, R. Infrared spectroscopy analysis of hemp (Cannabis sativa) after selective delignification by Bjerkandera sp. at different nitrogen levels. Enzyme Microb. Technol. 2001, 28, 550–559. [Google Scholar] [CrossRef]
- Ying, W.; Shi, Z.; Yang, H.; Xu, G.; Zheng, Z.; Yang, J. Effect of alkaline lignin modification on cellulase–lignin interactions and enzymatic saccharification yield. Biotechnol. Biofuels 2018, 11, 214. [Google Scholar] [CrossRef]
- Célino, A.; Gonçalves, O.; Jacquemin, F.; Fréour, S. Qualitative and quantitative assessment of water sorption in natural fibres using ATR-FTIR spectroscopy. Carbohydr. Polym. 2014, 101, 163–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synytsya, A.; Čopíková, J.; Matějka, P.; Machovič, V. Fourier transform Raman and infrared spectroscopy of pectins. Carbohydr. Polym. 2003, 54, 97–106. [Google Scholar] [CrossRef]
- Tamm, L.K.; Tatulian, S.A. Infrared spectroscopy of proteins and peptides in lipid bilayers. Q. Rev. Biophys. 1997, 30, 365–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fries, M.; Ihrig, J.; Brocklehurst, K.; Shevchik, V.E.; Pickersgill, R.W. Molecular basis of the activity of the phytopathogen pectin methylesterase. EMBO J. 2007, 26, 3879–3887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.; Singh, B.R. A Distinct Utility of the Amide III Infrared Band for Secondary Structure Estimation of Aqueous Protein Solutions Using Partial Least Squares Methods †. Biochemistry 2004, 43, 2541–2549. [Google Scholar] [CrossRef]
- Richard, J.-A.; Kelly, I.; Marion, D.; Auger, M.; Pézolet, M. Structure of β-Purothionin in Membranes: A Two-Dimensional Infrared Correlation Spectroscopy Study †. Biochemistry 2005, 44, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yang, X.; Ji, Z.; Zhu, L.; Ma, N.; Chen, D.; Jia, X.; Tang, J.; Cao, Y. DFT-Calculated IR Spectrum Amide I, II, and III Band Contributions of N -Methylacetamide Fine Components. ACS Omega 2020, 5, 8572–8578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiercigroch, E.; Szafraniec, E.; Czamara, K.; Pacia, M.Z.; Majzner, K.; Kochan, K.; Kaczor, A.; Baranska, M.; Malek, K. Raman and infrared spectroscopy of carbohydrates: A review. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 185, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhong, Z.; Feng, L. Advances in the performance and application of hemp fiber. Int. J. Simul. Syst. Sci. Technol. 2016, 17, 1–18. [Google Scholar] [CrossRef]
- Miras, Y.; Beauger, A.; Lavrieux, M.; Berthon, V.; Serieyssol, K.; Andrieu-Ponel, V.; Ledger, P.M. Tracking long-term human impacts on landscape, vegetal biodiversity and water quality in the Lake Aydat catchment (Auvergne, France) using pollen, non-pollen palynomorphs and diatom assemblages. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2015, 424, 76–90. [Google Scholar] [CrossRef]
- Schofield, J.E.; Waller, M.P. A pollen analytical record for hemp retting from Dungeness Foreland, UK. J. Archaeol. Sci. 2005, 32, 715–726. [Google Scholar] [CrossRef]
- Liu, M.; Fernando, D.; Meyer, A.S.; Madsen, B.; Daniel, G.; Thygesen, A. Characterization and biological depectinization of hemp fibers originating from different stem sections. Ind. Crops Prod. 2015, 76, 880–891. [Google Scholar] [CrossRef]
- Magnusson, K.; Svennerstedt, B. Influence of Temperature on the Water Retting Process of Hemp ( Cannabis sativa L.) Cultivated Under Swedish Climate Conditions. J. Ind. Hemp 2007, 12, 3–17. [Google Scholar] [CrossRef]
- Tamburini, E.; Leon, A.G.; Perito, B.; Mastromei, G. Characterization of bacterial pectinolytic strains involved in the water retting process. Environ. Microbiol. 2003, 5, 730–736. [Google Scholar] [CrossRef]
- Abdollahzadeh, R.; Pazhang, M.; Najavand, S.; Fallahzadeh-Mamaghani, V.; Amani-Ghadim, A.R. Screening of pectinase-producing bacteria from farmlands and optimization of enzyme production from selected strain by RSM. Folia Microbiol. (Praha). 2020, 65, 705–719. [Google Scholar] [CrossRef]
- Garg, G.; Singh, A.; Kaur, A.; Singh, R.; Kaur, J.; Mahajan, R. Microbial pectinases: An ecofriendly tool of nature for industries. 3 Biotech 2016, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickersgill, R.W.; Jenkins, J.A. The Structures and Active Sites of Pectinases. In Advances in Pectin and Pectinase Research; Springer: Dordrecht, The Netherlands, 2003; pp. 267–275. [Google Scholar]
- Jayani, R.S.; Saxena, S.; Gupta, R. Microbial pectinolytic enzymes: A review. Process Biochem. 2005, 40, 2931–2944. [Google Scholar] [CrossRef]
- Adapa, V.; Ramya, L.N.; Pulicherla, K.K.; Rao, K.R.S.S. Cold Active Pectinases: Advancing the Food Industry to the Next Generation. Appl. Biochem. Biotechnol. 2014, 172, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Wani, N.; Shezad, F.; Ahanger, F.A.; Malik, S.M.; Sahay, S.; Jain, K. Cold-Active Pectinases and their Commercial Applications A Review. Int. J. Sci. Res. Biol. Sci. 2018, 5, 125–133. [Google Scholar] [CrossRef]
- Janusz, G.; Pawlik, A.; Sulej, J.; Świderska-Burek, U.; Jarosz-Wilkołazka, A.; Paszczyński, A. Lignin degradation: Microorganisms, enzymes involved, genomes analysis and evolution. FEMS Microbiol. Rev. 2017, 41, 941–962. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Khalina, A.; Lee, S.H.; Liu, M. A Comprehensive Review on Bast Fibre Retting Process for Optimal Performance in Fibre-Reinforced Polymer Composites. Adv. Mater. Sci. Eng. 2020, 2020, 1–27. [Google Scholar] [CrossRef]
- Dimarogona, M.; Topakas, E.; Christakopoulos, P. Cellulose degradation by oxydative enzymes. Comput. Struct. Biotechnol. J. 2012, 2, e201209015. [Google Scholar] [CrossRef] [Green Version]
- Margariti, C. The application of FTIR microspectroscopy in a non-invasive and non-destructive way to the study and conservation of mineralised excavated textiles. Herit. Sci. 2019, 7, 63. [Google Scholar] [CrossRef]
- McCann, M.C.; Hammouri, M.; Wilson, R.; Belton, P.; Roberts, K. Fourier Transform Infrared Microspectroscopy Is a New Way to Look at Plant Cell Walls. Plant Physiol. 1992, 100, 1940–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieppo, L.; Saarakkala, S.; Närhi, T.; Helminen, H.J.; Jurvelin, J.S.; Rieppo, J. Application of second derivative spectroscopy for increasing molecular specificity of fourier transform infrared spectroscopic imaging of articular cartilage. Osteoarthr. Cartil. 2012, 20, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soukup, A. Selected Simple Methods of Plant Cell Wall Histochemistry and Staining for Light Microscopy. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; pp. 25–40. ISBN 9781627036429. [Google Scholar]
- Englyst, H.; Wiggins, H.S.; Cummings, J.H. Determination of the non-starch polysaccharides in plant foods by gas-liquid chromatography of constituent sugars as alditol acetates. Analyst 1982, 107, 307. [Google Scholar] [CrossRef] [PubMed]
- Hubbe, M.; Chandra, R.; Dogu, D.; van Velzen, S.T.J. Analytical staining of cellulosic materials: A review. BioResources 2019, 14, 7387–7464. [Google Scholar] [CrossRef]
- Pattathil, S.; Avci, U.; Baldwin, D.; Swennes, A.G.; McGill, J.A.; Popper, Z.; Bootten, T.; Albert, A.; Davis, R.H.; Chennareddy, C.; et al. A Comprehensive Toolkit of Plant Cell Wall Glycan-Directed Monoclonal Antibodies. Plant Physiol. 2010, 153, 514–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Park, Y.B.; Cosgrove, D.J.; Hong, M. Cellulose-Pectin Spatial Contacts Are Inherent to Never-Dried Arabidopsis Primary Cell Walls: Evidence from Solid-State Nuclear Magnetic Resonance. Plant Physiol. 2015, 168, 871–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovur, S.K.; Schenzel, K.C.; Grimm, E.; Diepenbrock, W. Characterization of refined hemp fibers using NIR FT Raman micro spectroscopy and environmental scanning electron microscopy. BioResources 2008, 3, 1081–1091. [Google Scholar] [CrossRef]
- Szymańska-Chargot, M.; Chylińska, M.; Pieczywek, P.M.; Rösch, P.; Schmitt, M.; Popp, J.; Zdunek, A. Raman imaging of changes in the polysaccharides distribution in the cell wall during apple fruit development and senescence. Planta 2016, 243, 935–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gierlinger, N.; Schwanninger, M. The potential of Raman microscopy and Raman imaging in plant research. Spectroscopy 2007, 21, 69–89. [Google Scholar] [CrossRef] [Green Version]







| FT-IR Bands (Wavenumbers/cm−1) | Approximate Assignment of Vibrational Mode | Remarks [23,24,25] | ||
|---|---|---|---|---|
| Fresh | Slone_4 | Slone_37 | ||
| 3368 | 3382 | 3352 | ν(OH) free [23] | cellulose, hemicellulose, lignin, pectin |
| 2918 | 2918 | 2917 | ν(CH) [24] | general organic content |
| 2849 | 2850 | 2849 | νs(CH2) [24] | waxes |
| - | - | 1736 | ν C=O ester band [23,24] | pectin, waxes |
| - | 1636 | 1633 | ν(C=O) in plane, O-H [23] | amide I adsorbed water |
| 1598 | - | 1597 | ν(C=C) aromatic in-plane [24] | lignin |
| 1512 | 1510 | 1511 | Aromatic skeletal vibrations [26] | amide II |
| 1461 | 1461 | 1461 | δ(CH2), δ(COH) [14] τO-H [23] | lignin, cellulose, hemicellulose, pectins, waxes and fats |
| 1424 | 1419 | 1421 | δ(C-H) [24] νCOO [23] τ(HCH) τ(OCH) in-plane [3] | carboxylic acid, pectins, cellulose |
| 1372 | - | 1377 | δ(C-H) [20,23] δC-CH3 symmetrical [23] | lignin, cellulose |
| 1263 | 1265 | 1261 | δ(CH2) twisting | amide III |
| 1148 | - | 1155 | νas(CC) ring breathing [25] τC-O-C [23] | cellulose, hemicellulose, pectin |
| - | 1126 | - | νas(COC) glycosidic [14] νC-O [23,27] | cellulose, hemicellulose, pectin |
| 1034 | 1034 | 1036 | ν(CO), 1° alcohol [14] | cellulose, hemicellulose, pectin |
| 894 | - | - | νs(C-O-C) in plane [23] Characteristic of β-links in cellulose [26] | cellulose, hemicellulose, pectin |
| 814 | 714 | - | δ(C–OH)ring [28] | cellulose |
| Depth (cm) | Laboratory Code | 14C Date [14C BP] | Calibrated Age—1σ Range (cal BCE/CE) 68.3% Probability | Calibrated Age—2σ Range (cal BCE/CE) 95.4% Probability | Dated Material |
|---|---|---|---|---|---|
| 65 | Poz-123833 | 1345 ± 30 BP | 650 CE (55.4%) 680 CE 747 CE (12.9%) 758 CE | 643 CE (68.3%) 705 CE 738 CE (27.1%) 774 CE | Terrestrial Plant Macrofossil —Cannabis sativa L. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalisz, G.; Gieroba, B.; Chrobak, O.; Suchora, M.; Starosta, A.L.; Sroka-Bartnicka, A. Vibrational Spectroscopic Analyses and Imaging of the Early Middle Ages Hemp Bast Fibres Recovered from Lake Sediments. Molecules 2021, 26, 1314. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051314
Kalisz G, Gieroba B, Chrobak O, Suchora M, Starosta AL, Sroka-Bartnicka A. Vibrational Spectroscopic Analyses and Imaging of the Early Middle Ages Hemp Bast Fibres Recovered from Lake Sediments. Molecules. 2021; 26(5):1314. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051314
Chicago/Turabian StyleKalisz, Grzegorz, Barbara Gieroba, Olga Chrobak, Magdalena Suchora, Agata L. Starosta, and Anna Sroka-Bartnicka. 2021. "Vibrational Spectroscopic Analyses and Imaging of the Early Middle Ages Hemp Bast Fibres Recovered from Lake Sediments" Molecules 26, no. 5: 1314. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051314