
Design, Synthesis and Pharmacological Evaluation of Three Novel Dehydroabietyl Piperazine Dithiocarbamate Ruthenium (II) Polypyridyl Complexes as Potential Antitumor Agents: DNA Damage, Cell Cycle Arrest and Apoptosis Induction

Abstract
:1. Introduction
2. Results and Discussion
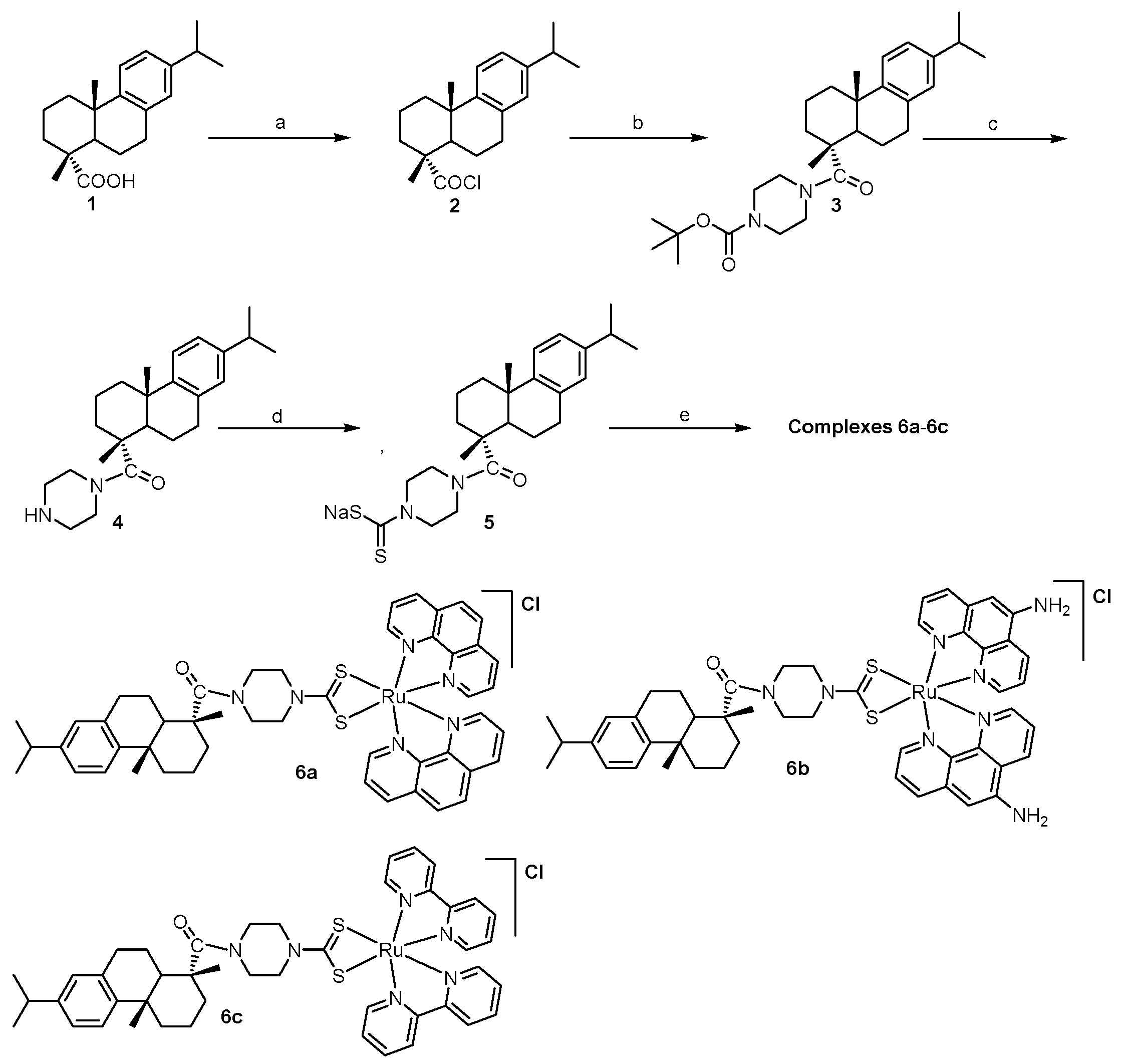
2.1. Chemistry
2.2. Antiproliferative Activity
2.2.1. In Vitro Antiproliferative Activity
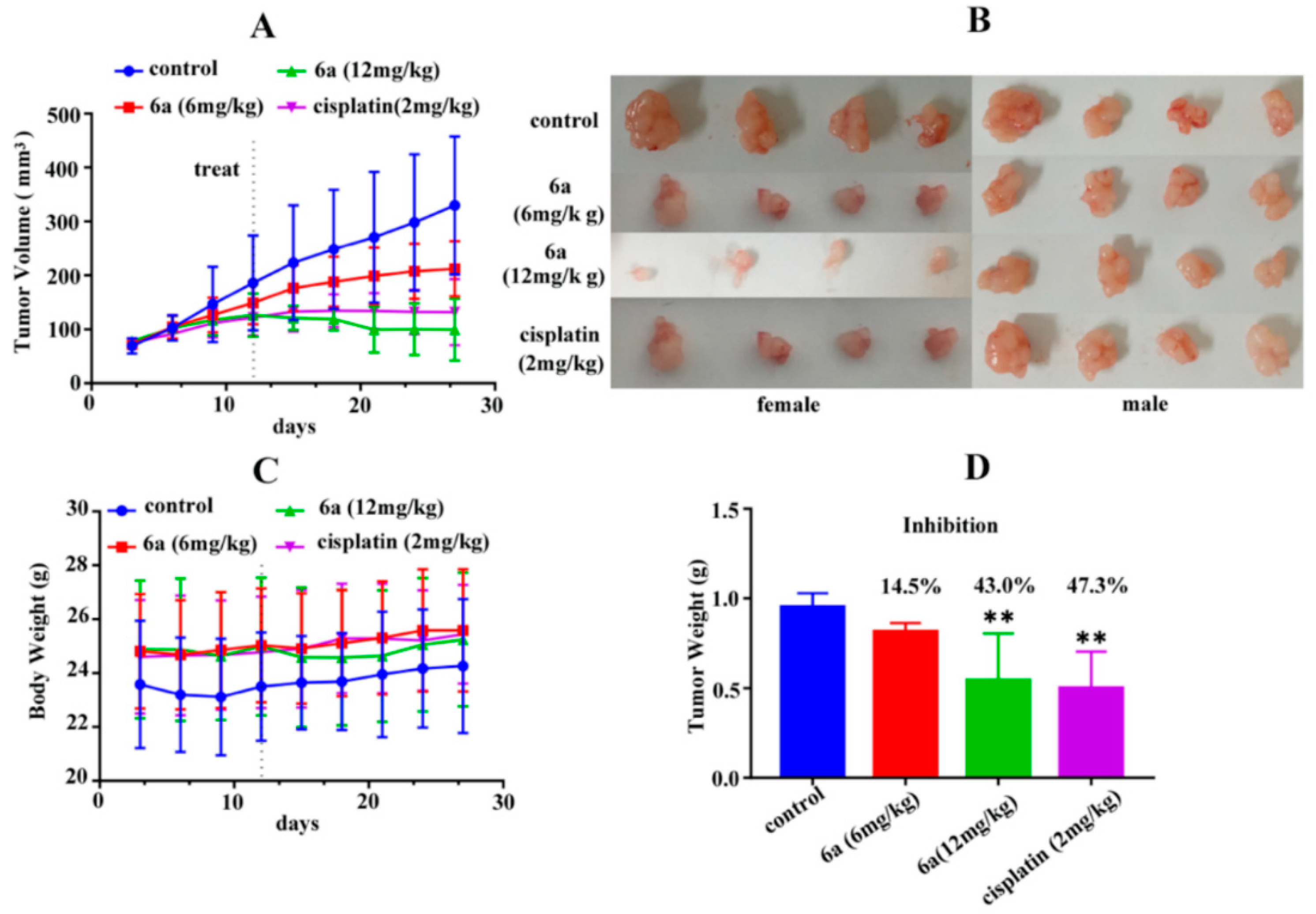
2.2.2. In Vivo Antiproliferative Activity
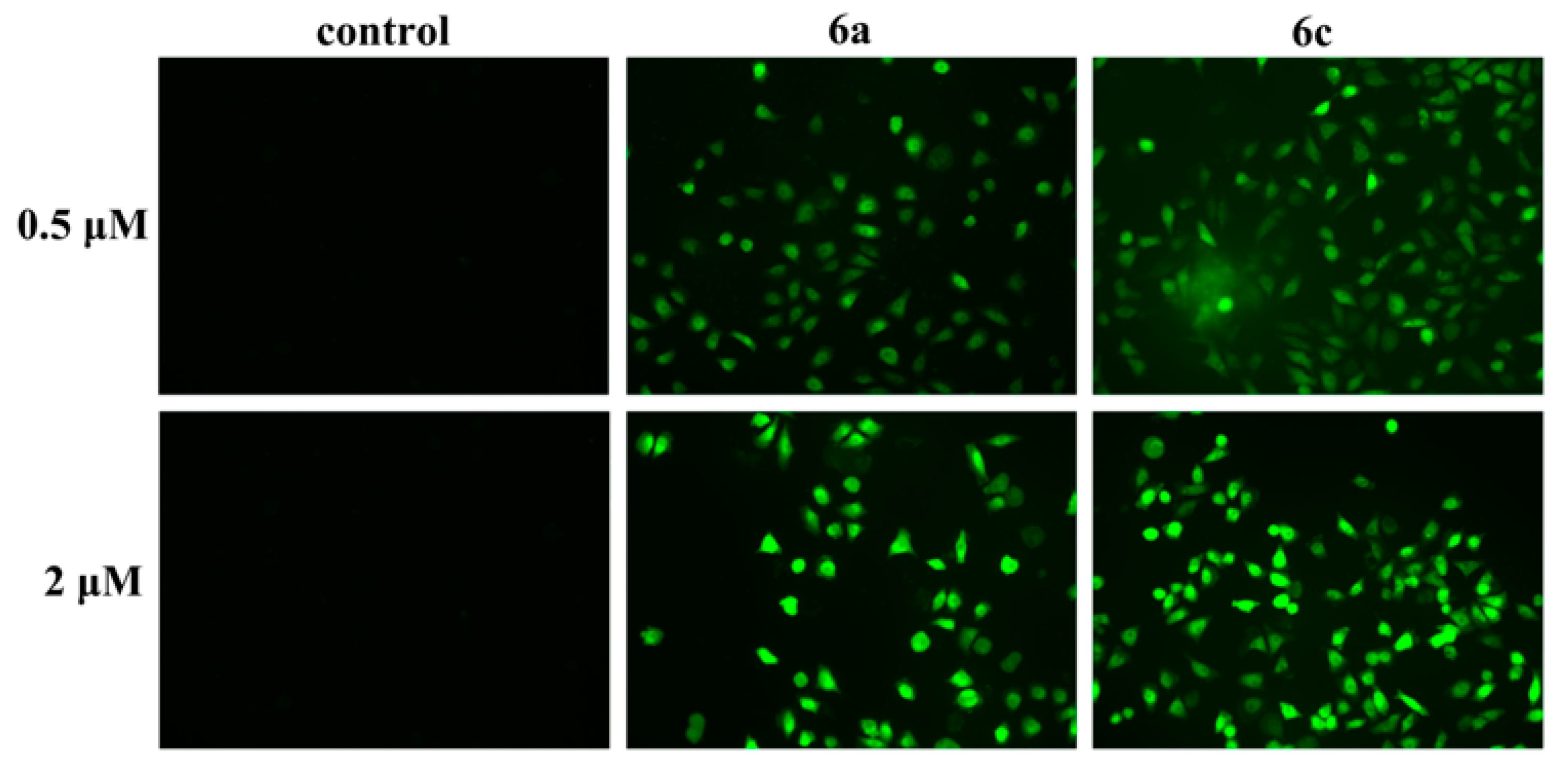
2.2.3. Concentration in T-24 Cells of 6a and 6c
2.3. Antitumor Mechanism
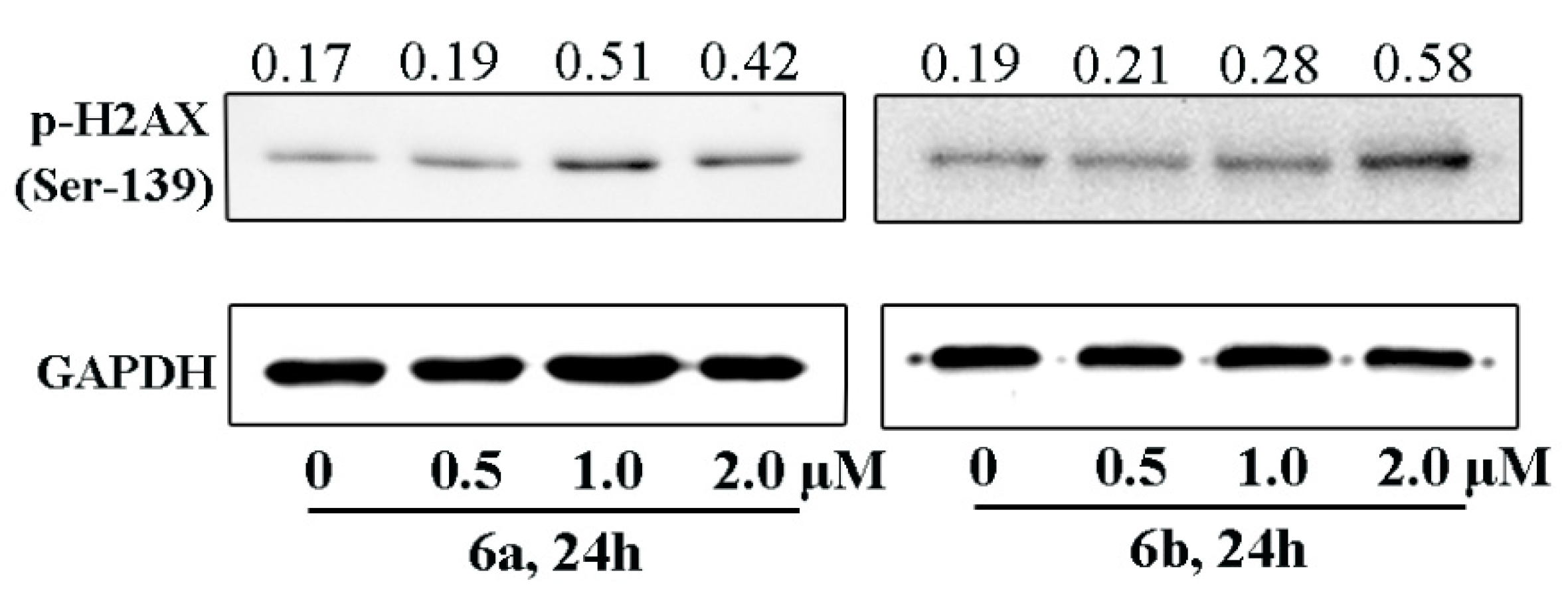
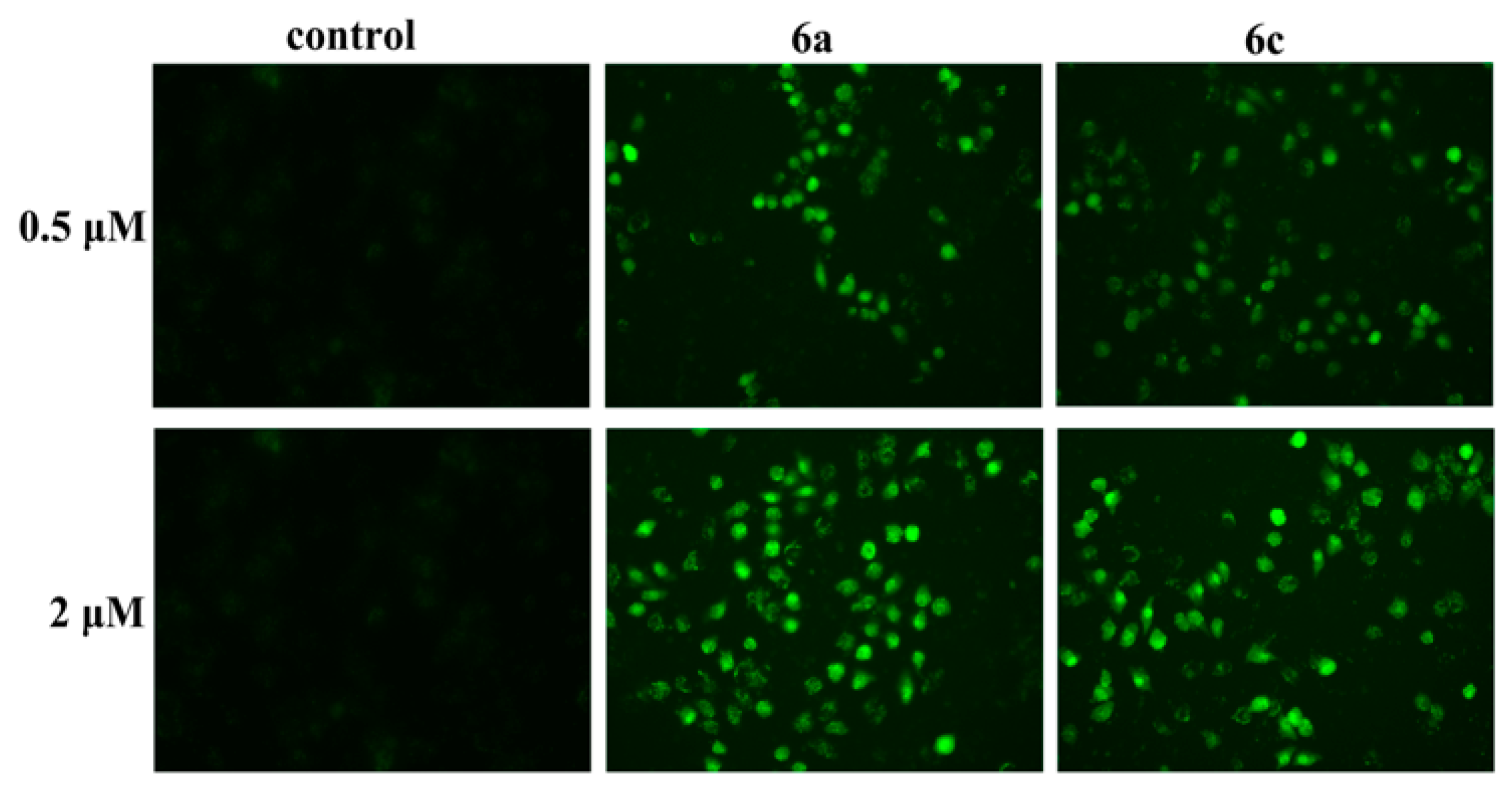
2.3.1. DNA Intercalation and Damage
2.3.2. Cell Cycle Arrest Analysis
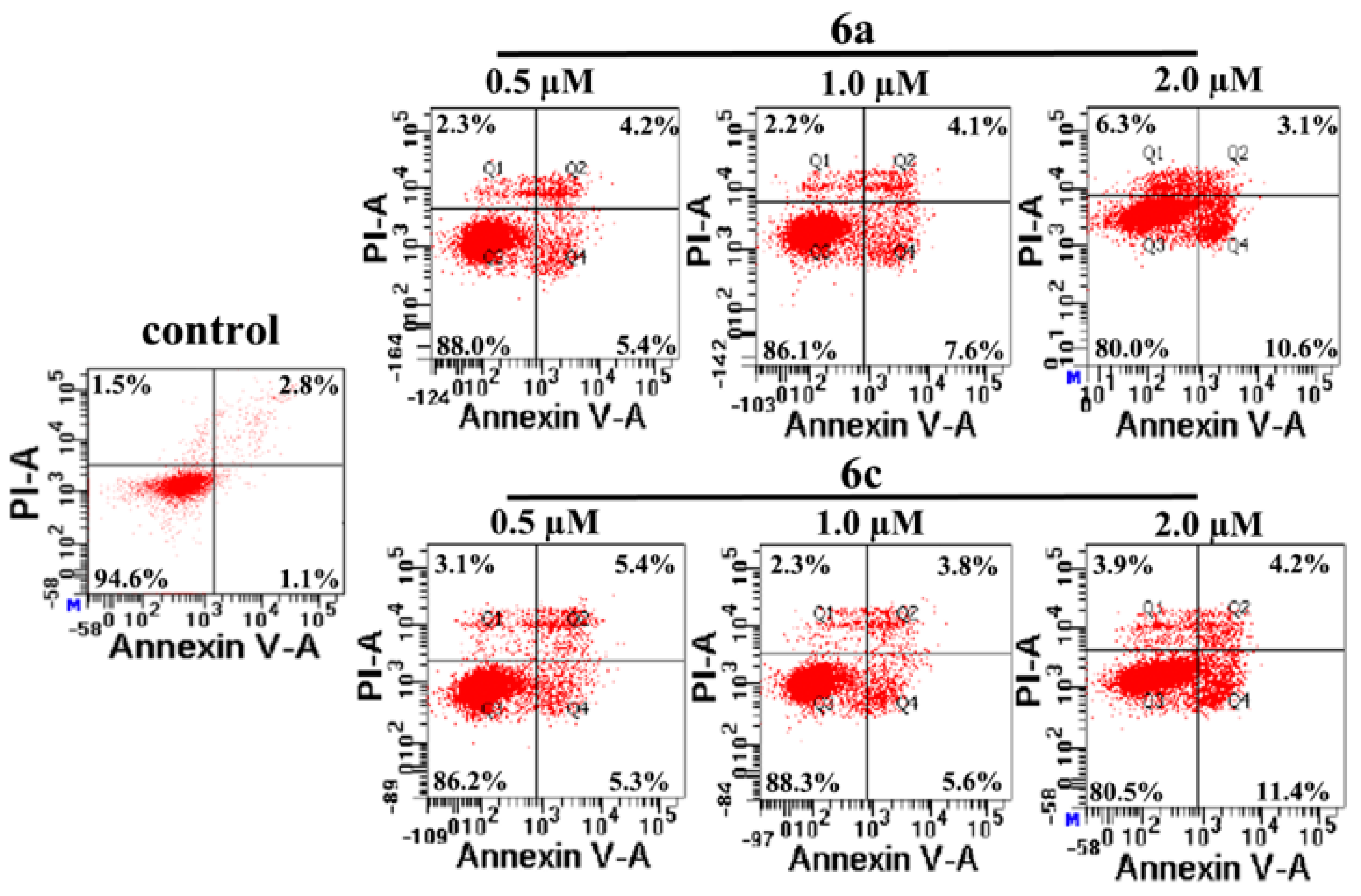
2.3.3. Apoptosis: Mode of Cell Death Induced by Compounds 6a and 6c
3. Materials and Methods
3.1. Materials and Instruments
3.1.1. Chemistry: General Synthesis Procedure for Compounds 4–7
3.1.2. Biological Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef]
- Skladanowski, A.; Bozko, P.; Sabisz, M. DNA structure and integrity checkpoints during the cell cycle and their role in drug targeting and sensitivity of tumor cells to anticancer treatment. Chem. Rev. 2009, 109, 2951–2973. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Bloodgood, B.L.; Sharma, N.; Browne, H.A.; Trepman, A.Z.; Greenberg, M.E. The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature 2013, 503, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt (II) agents, nanoparticle delivery, and Pt (IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelland, L. The resurgence of platinum- based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Canc. Canc. Treat Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.L.; Gupta, P.; Chen, Y.L.; Wang, E.J.; Ji, L.N.; Chao, H.; Chen, Z.S. The development of anticancer ruthenium(II) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Wang, X.Y.; Jin, S.X.; Muhammad, N.; Guo, Z.J. Stimuli-responsive therapeutic metallodrugs. Chem. Rev. 2019, 119, 1138–1192. [Google Scholar] [CrossRef]
- Clarke, M.J.; Zhu, F.; Frasca, D.R. Non-platinum chemotherapeutic metallopharmaceuticals. Chem. Rev. 1999, 99, 2511–2533. [Google Scholar] [CrossRef]
- Allardyce, C.S.; Dyson, P.J. Ruthenium in medicine: Current clinical uses and future prospects. Platin. Met. Rev. 2001, 45, 62–69. [Google Scholar]
- Levina, A.; Mitra, A.; Lay, P.A. Recent developments in ruthenium anticancer drugs. Metallomics 2009, 1, 458–470. [Google Scholar] [CrossRef]
- Pacor, S.; Zorzet, S.; Cocchietto, M.; Bacac, M.; Vadori, M.; Turrin, C.; Gava, B.; Castellarin, A.; Sava, G. Intratumoral NAMI-A treatment triggers metastasis reduction, which correlates to CD44 regulation and tumor infiltrating lymphocyte recruitment. J. Pharmacol. Exp. Ther. 2004, 310, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Hartinger, C.G.; Seifried, S.Z.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside—Preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef]
- Erkkila, K.E.; Odom, D.T.; Barton, J.K. Recognition and reaction of metallointercalators with DNA. Chem. Rev. 1999, 99, 2777–2796. [Google Scholar] [CrossRef]
- Smith, J.A.; Keene, F.R.; Li, F.; Collins, J.G. Non-covalent DNA binding of metal complexes. In Comprehensive Inorganic Chemistry II.; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier: Oxford, UK, 2013; Volume 3, pp. 709–750. [Google Scholar]
- Ji, L.N.; Zou, X.H.; Liu, J.G. Shape and enantioselective interaction of Ru (II)/Co (III) polypyridyl complexes with DNA. Coord. Chem. Rev. 2001, 216–217, 513–536. [Google Scholar] [CrossRef]
- Keene, F.R.; Smith, J.A.; Collins, J.G. Metal complexes as structure-selective binding agents for nucleic acid. Coord. Chem. Rev. 2009, 253, 2021–2035. [Google Scholar] [CrossRef]
- Huang, H.Y.; Zhang, P.Y.; Yu, B.L.; Chen, Y.; Wang, J.Q.; Ji, L.N.; Chao, H. Targeting nucleus DNA with a cyclometalated dipyridophenazineruthenium (II) complex. J. Med. Chem. 2014, 57, 8971–8983. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Guo, Z.J. Targeting and delivery of platinum-based anticancer drugs. Chem. Soc. Rev. 2013, 42, 202–224. [Google Scholar]
- Shibata, K.; Ito, Y.; Hongo, A.; Yasoshima, A.; Endo, T.; Ohashi, M. Bacterial activity of a new antiulcer agent, ecabet sodium, against Helicobacter pylori under acidic conditions. Antimicrob. Agents Chemother. 1995, 39, 1295–1299. [Google Scholar] [CrossRef] [Green Version]
- Gigante, B.; Silva, A.M.; Marcelo-Curto, M.J.; Feio, S.S.; Roseiro, J.; Reis, L.V. Structural effects on the bioactivity of dehydroabietic acid derivatives. Planta. Med. 2002, 68, 680–684. [Google Scholar] [CrossRef]
- Wasowski, C.; Marder, M. Central nervous system activities of two diterpenes isolated from Aloysia virgate. Phytomedicine 2011, 18, 393–401. [Google Scholar] [CrossRef]
- Lee, C.L.; Chiang, L.C.; Cheng, L.H.; Liaw, C.C.; Abd El-Razek, M.H.; Chang, F.R.; Wu, Y.C. Influenza A (H1N1) antiviral and cytotoxic agents from Ferula assa-foetida. J. Nat. Prod. 2009, 72, 1568–1572. [Google Scholar] [CrossRef]
- Zapata, B.; Rojas, M.; Betancur-Galvis, L.; Mesa-Arango, A.C.; Pérez-Guaita, D.; González, M.A. Cytotoxic, immunomodula- tory, antimycotic, and antiviral activities of semisynthetic 14-hydroxyabietane derivatives and triptoquinone C-4 epimers. MedChemComm 2013, 4, 1239–1246. [Google Scholar] [CrossRef]
- González, M.A.; Pérez-Guaita, D.; Correa-Royero, J.; Zapata, B.; Agudelo, L.; Mesa-Arango, A.; Betancur-Galvis, L. Synthesis and biological evaluation of dehydroabietic acid derivatives. Eur. J. Med. Chem. 2010, 45, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Qu, H.E.; Huang, X.C.; Pan, Y.M.; Liang, D.; Chen, Z.F.; Wang, H.S.; Zhang, Y. Synthesis and biological evaluation of novel dehydroabietic acid derivatives conjugated with acyl-thiourea peptide moiety as antitumor agents. Int. J. Mol. Sci. 2015, 16, 14571–14593. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.C.; Wang, M.; Wang, H.S.; Chen, Z.F.; Zhang, Y.; Pan, Y.M. Synthesis and antitumor activities of novel dipeptide derivativesderived from dehydroabietic acid. Bioorg. Med. Chem. Lett. 2014, 24, 1511–1518. [Google Scholar] [CrossRef]
- Huang, X.C.; Wang, M.; Pan, Y.M.; Yao, G.Y.; Wang, H.S.; Tian, X.Y.; Qin, J.K.; Zhang, Y. Synthesis and antitumor activities of novel thioureaa-aminophosphonates from dehydroabietic acid. Eur. J. Med. Chem. 2013, 69, 508–520. [Google Scholar] [CrossRef]
- Huang, X.C.; Wang, M.; Pan, Y.M.; Tian, X.Y.; Wang, H.S.; Zhang, Y. Synthesis and antitumor activities of novel aminophosphonatesdehydroabietic acid derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 5283–5289. [Google Scholar] [CrossRef]
- Nagy, E.M.; Ronconi, L.; Nardon, C.; Fregona, D. Noble metal-dithiocarbamates precious allies in the fight against cancer, Mini-Rev. Med. Chem. 2012, 12, 1216–1229. [Google Scholar]
- Braña, M.F.; Ramos, A. Naphthalimides as anticancer agents: Synthesis and biological activity. Curr. Med. Chem. Anticancer Agents 2001, 1, 237–255. [Google Scholar] [CrossRef]
- Zou, S.S.; Li, G.Y.; Rees, T.W.; Jin, C.Z.; Huang, J.J.; Chen, Y.; Ji, L.N.; Chao, H. Interfering with DNA high-order structures using chiral ruthenium (II) Complexes. Chem. Eur. J. 2018, 24, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Hattab, S.; Chouba, L.; Kheder, M.B.; Mahouachi, T.; Boussetta, H. Cadmium- and copper- induced DNA damage in Pisumsativum roots and leaves as determined by the Comet assay. Plant Biosyst. 2009, 143, S6–S11. [Google Scholar] [CrossRef]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–322. [Google Scholar] [CrossRef]
- Ludivine, F.; Bastien, B.; Moussa, A.; Meng, X.J.; Collin, J.P.; Claude, S.; Gaiddonb, C.; Pfeffer, M. Library of second-generation cycloruthenated compounds and evaluation of their biological properties as potential anticancer drugs: Passing the nanomolar barrier. Dalton Trans. 2011, 40, 8869–8878. [Google Scholar]
- Wei, J.H.; Chen, Z.F.; Qin, J.L.; Liu, Y.C.; Li, Z.Q.; Khan, T.M.; Wang, M.; Jiang, Y.H.; Shen, W.Y.; Liang, H. Water-soluble oxoglaucine-Y(III), Dy (III) complexes: In vitro and in vivo anticancer activities by triggering DNA damage, leading to S phase arrest and apoptosis. Dalton Trans. 2015, 44, 11408–11419. [Google Scholar] [CrossRef] [PubMed]
- Kuang, W.B.; Huang, R.Z.; Qin, J.L.; Lu, X.; Qin, Q.P.; Zou, B.Q.; Chen, Z.F.; Liang, H.; Zhang, Y. Design, synthesis and pharmacological evaluation of new 3-(1Hbenzimidazol-2-yl) quinolin-2(1H)-one derivatives as potential antitumor agents. Eur. J. Med. Chem. 2018, 157, 139–150. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MGC-803 | T-24 | HepG2 | CNE-2 | MDA-MB-231 | MCF-7 | |
|---|---|---|---|---|---|---|
| 5 | >50 | >50 | >50 | >50 | >50 | >50 |
| A | >50 | >50 | >50 | >50 | >50 | >50 |
| B | >50 | >50 | >50 | >50 | >50 | >50 |
| C | >50 | >50 | >50 | >50 | >50 | >50 |
| 6a | 1.2 ± 0.9 | 1.2 ± 0.7 | 2.0 ± 0.3 | 3.2 ± 0.1 | 3.4 ± 0.7 | 2.1 ± 0.6 |
| 6b | >50 | >50 | >50 | >50 | >50 | >50 |
| 6c | 1.6 ± 0.5 | 1.0 ± 0.2 | 2.2 ± 0.4 | 4.2 ± 0.7 | 1.3 ± 0.5 | 3.6 ± 0.3 |
| Cisplatin | 10.1 ± 0.2 | 9.5 ± 0.6 | 7.1 ± 0.7 | >50 | 14.7 ± 1.9 | 21.5 ± 0.6 |
| A549 | A549-DDP | |
|---|---|---|
| 5 | >50 | >50 |
| A | >50 | >50 |
| B | >50 | >50 |
| C | >50 | >50 |
| 6a | 5.2 ± 0.6 | 1.3 ± 0.3 |
| 6b | >50 | >50 |
| 6c | 5.5 ± 0.9 | 1.8 ± 0.4 |
| Cisplatin | 6.4 ± 1.0 | 30.5 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Wei, J.; Jiang, H.; Zhang, Y.; Jiang, C.; Ma, X. Design, Synthesis and Pharmacological Evaluation of Three Novel Dehydroabietyl Piperazine Dithiocarbamate Ruthenium (II) Polypyridyl Complexes as Potential Antitumor Agents: DNA Damage, Cell Cycle Arrest and Apoptosis Induction. Molecules 2021, 26, 1453. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051453
Wang H, Wei J, Jiang H, Zhang Y, Jiang C, Ma X. Design, Synthesis and Pharmacological Evaluation of Three Novel Dehydroabietyl Piperazine Dithiocarbamate Ruthenium (II) Polypyridyl Complexes as Potential Antitumor Agents: DNA Damage, Cell Cycle Arrest and Apoptosis Induction. Molecules. 2021; 26(5):1453. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051453
Chicago/Turabian StyleWang, Haoran, Jianhua Wei, Hong Jiang, Ye Zhang, Caina Jiang, and Xianli Ma. 2021. "Design, Synthesis and Pharmacological Evaluation of Three Novel Dehydroabietyl Piperazine Dithiocarbamate Ruthenium (II) Polypyridyl Complexes as Potential Antitumor Agents: DNA Damage, Cell Cycle Arrest and Apoptosis Induction" Molecules 26, no. 5: 1453. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051453