4.1. Chemistry
All required chemicals, solvents, and reagents were purchased from Sigma-Aldrich (Arklow, Ireland) and were of reagent grade. Reaction progress was monitored on pre-coated thin layer chromatographic aluminum sheets (silica gel 60 F254, Merck, Carrigtwohill, Ireland), and TLC visualization was done using a UV lamp. Fourier transform infrared spectra were carried out with neat film coated samples on diamond using a Nicolet
TM iS
TM 10 FT-IR spectrophotometer (Thermo Fisher, Dublin, Ireland). Significant absorption peak (νmax) values are given in cm
−1.
1H- and
13C-NMR spectra were recorded on an Avance 400 spectrometer (Bruker, Rheinstetten, Germany) at 400 MHz and 100 MHz, respectively, in CDCl
3 and CD
3OD, using tetramethylsilane (TMS) as the internal standard (Spectra available in
Supplementary Materials). Chemical shift values are given on the δ (ppm) scale, with signals described as follows: s (singlet), d (doublet), dd (double doublet), t (triplet), q (quartet), br. (broad signal), m (multiplet), and coupling constants (
J) expressed in Hz. Mass spectral analyses were recorded using a LCT Premiere XE (ESI-TOF MS) instrument (Waters, Dublin, Ireland). All calculated exact mono isotopic mass distributions were calibrated against internal reference standards.
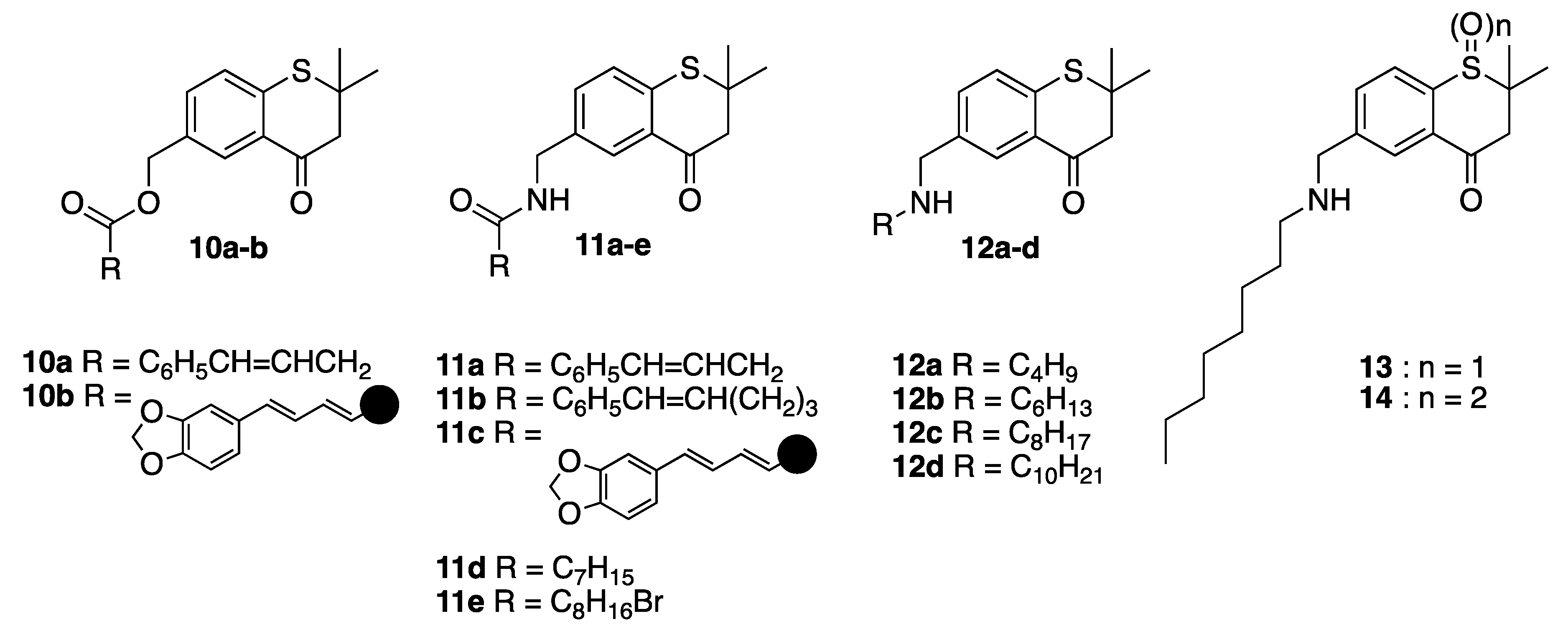
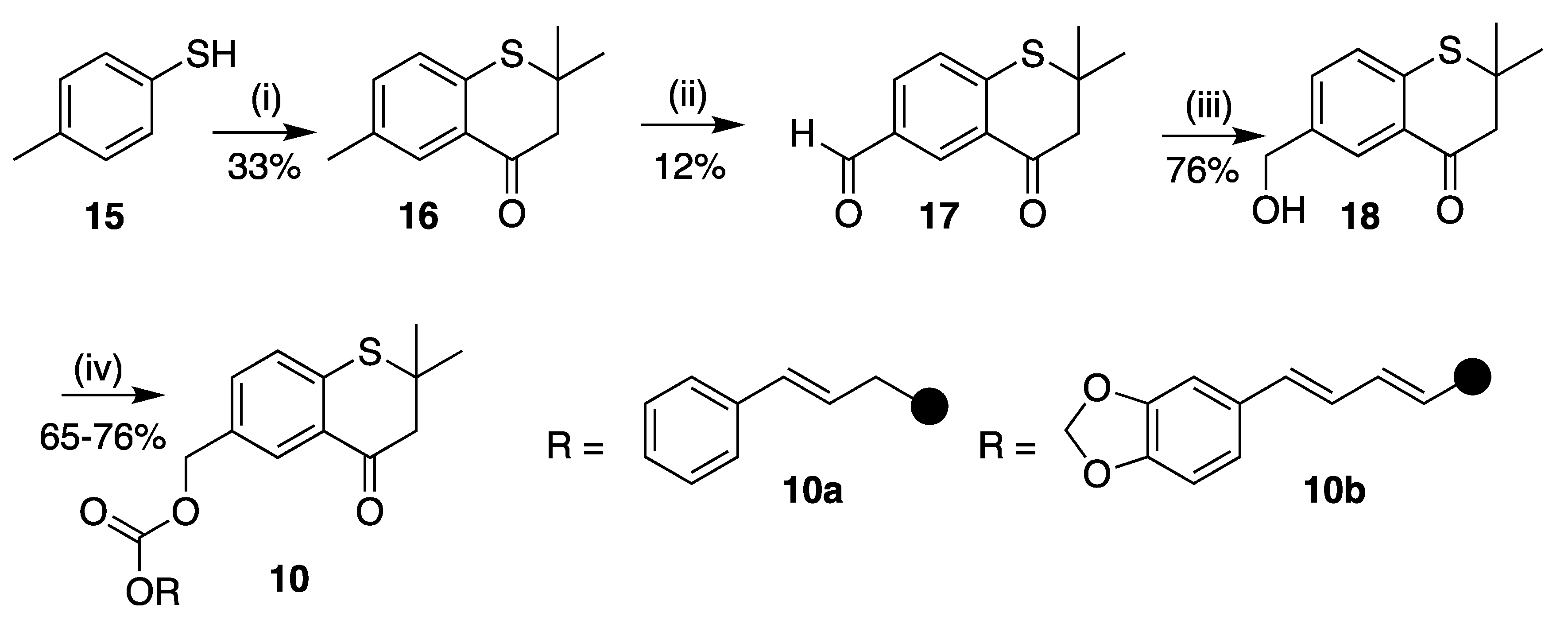
2,2,6-Trimethylthiochroman-4-one (16)
This compound was prepared and characterized as previously described [
17]. Yield: 33%.
2,2-Dimethyl-4-oxothiochromane-6-carbaldehyde (17)
To a solution of 16 (0.53 g, 2.57 mmol) in acetonitrile (15 mL) was added a solution of potassium persulfate (1.39 g, 5.14 mmol) in water (15 mL), and copper sulfate (0.13 g, 0.52 mmol). The resulting solution was stirred at 75–80 °C for 1 h and then cooled. The cold mixture was diluted with saturated sodium bicarbonate solution and extracted with diethyl ether (3 × 50 mL). The diethyl ether extracts were concentrated in vacuo and the residue purified by flash chromatography (pet. ether/EtOAc 10:1) to give 17 as a pale oil that solidified on standing (70 mg, 12%). 1H-NMR (CDCl3) δH 1.43 (6H, s, C(CH3)2), 2.86 (2H, s, CH2), 7.30 (1H, d, J = 8.2 Hz, ArH), 7.84 (1H, d, J = 7.9 Hz, ArH), 8.47 (1H, s, H5), 9.90 (1H, s, CHO). 13C-NMR δC 28.6 ((CH3)2), 45.3 (C(CH3)2), 53.2 (CH2), 128.4 (ArCH), 129.6 (ArC), 131.6 (ArCH), 132.1 (ArCH), 133.1 (ArC), 149.5 (ArC), 190.7 (CHO), 193.8 (C=O).
6-(Hydroxymethyl)-2,2-dimethylthiochroman-4-one (18)
To 17 (70 mg, 0.032 mmol) in DMF (1 mL) was added distilled water (50 mL) and freshly cut slices of D. carota (10 g). The resulting mixture was stirred vigorously at room temperature for 72 h. The reaction was filtered, and the filtrate washed with ethyl acetate (50 mL). The water/ethyl acetate mixture was separated, and the ethyl acetate extract dried over Na2SO4. The crude orange oil was purified by flash column chromatography (pet. ether/EtOAc 7:1) to afford alcohol 18, (54 mg, 76%). 1H-NMR (CDCl3) δH 1.47 (6H, s, CH3), 2.88 (2H, s, CH2), 4.69 (2H, s, CH2OH), 5.30 (1H, s, OH), 7.24 (1H, d, ArH), 7.46 (1H, dd, J = 8.3, 1.7 Hz, ArH), 8.08 (1H, s, H5).
(2,2-Dimethyl-4-oxothiochroman-6-yl)methyl (E)-4-phenylbut-3-enoate (10a)
To a solution of trans-styrylacetic acid (15 mg, 0.09 mmol) in dichloromethane (5 mL) was added EDC-HCl (30 mg, 0.16 mmol) and 4-(dimethylamino)pyridine (DMAP) (2 mg). To this solution was added 18 (20 mg, 0.09 mmol). The reaction was stirred for 72 h at room temperature. The residual solvent was removed in vacuo, and the crude residue purified by flash column chromatography (pet. ether/EtOAc 10:1) to afford 10a as a pale, straw-coloured oil (25 mg, 76%). IR (neat) νmax 964, 1147, 1190, 1678, 1733, 2924 cm−1. 1H- NMR (CDCl3) δH 1.47 (6H, s, (CH3)2), 2.88 (2H, s, CH2CO), 3.30 (2H, d, J = 7.1 Hz, CH2COO), 5.12 (2H, s, OCH2), 6.31 (1H, m, CH=C), 6.50 (1H, m, CH=C), 7.21–7.43 (7H, m, 7 × ArH), 8.11 (1H, d, J = 1.5 Hz, H5). 13C NMR δC 28.6 (2C), 38.3, 44.8, 53.7, 65.8, 121.4, 126.3 (2C), 127.6, 128.0, 128.5, 128.6 (2C), 129.7, 132.5, 133.6, 133.7, 136.8, 141.7, 171.4, 194.7. HRMS (M + NH4)+ 384.1611, C22H26NO3S requires 384.1628.
(2,2-Dimethyl-4-oxothiochroman-6-yl)methyl (2E,4E)-5-(benzo[d][1,3]dioxol-5-yl)penta-2,4-dienoate (10b)
Compound 18 (25 mg, 0.11 mmol) was reacted as for 10a with piperic acid (24 mg, 0.11 mmol) to afford 10b as a straw-coloured oil (31 mg, 65%). IR (neat) νmax 994, 1035, 1126, 1232, 1250, 1445, 1603, 1677, 1705, 2924 cm−1. 1H-NMR (CDCl3) δH 1.47 (6H, s, (CH3)2), 2.88 (2H, s, CH2CO), 5.17 (2H, s, ArCH2O), 5.98 (3H, m, OCH2O & CH=C), 6.70 (1H, m, C=CH), 6.78–6.64 (2H, m, 2 × C=CH), 6.91 (1H, d, J = 8.3 Hz, C=CH), 6.99 (1H, s, C=CH), 7.24 (1H, d, J = 8.1 Hz, C=CH), 7.42–7.48 (2H, m, 2 × C=CH), 8.13 (1H, br. s, H5). 13C-NMR δC 28.6 (2C), 44.8, 53.8, 65.4, 101.4, 105.9, 108.6, 119.7, 123.1, 124.4, 127.9, 128.5, 129.7, 130.5, 133.0, 133.6, 140.7, 141.5, 145.6, 148.3, 148.7, 166.9, 194.7. HRMS (M + NH4)+ 440.1570, C24H26NO5S requires 440.1526.
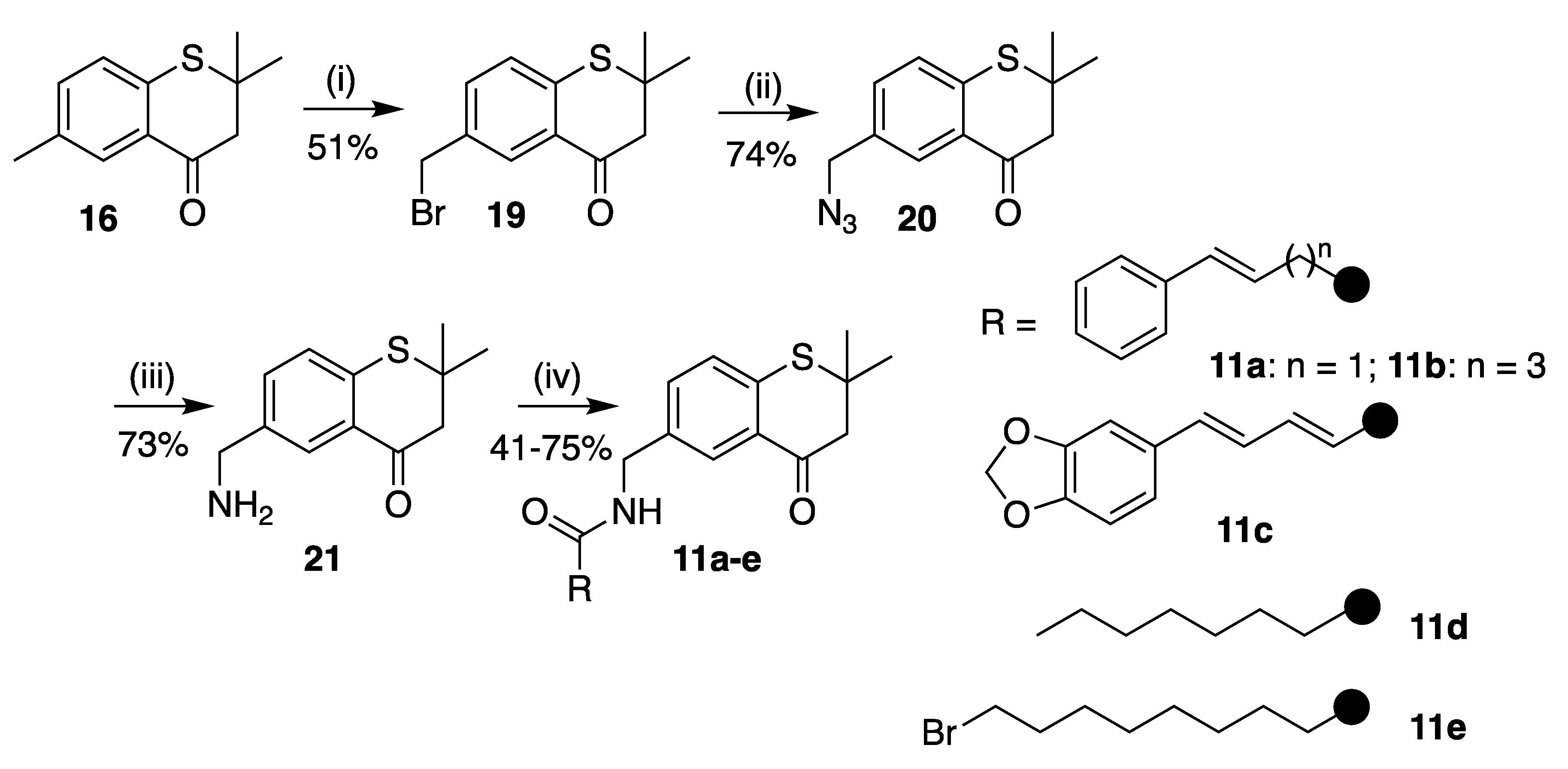
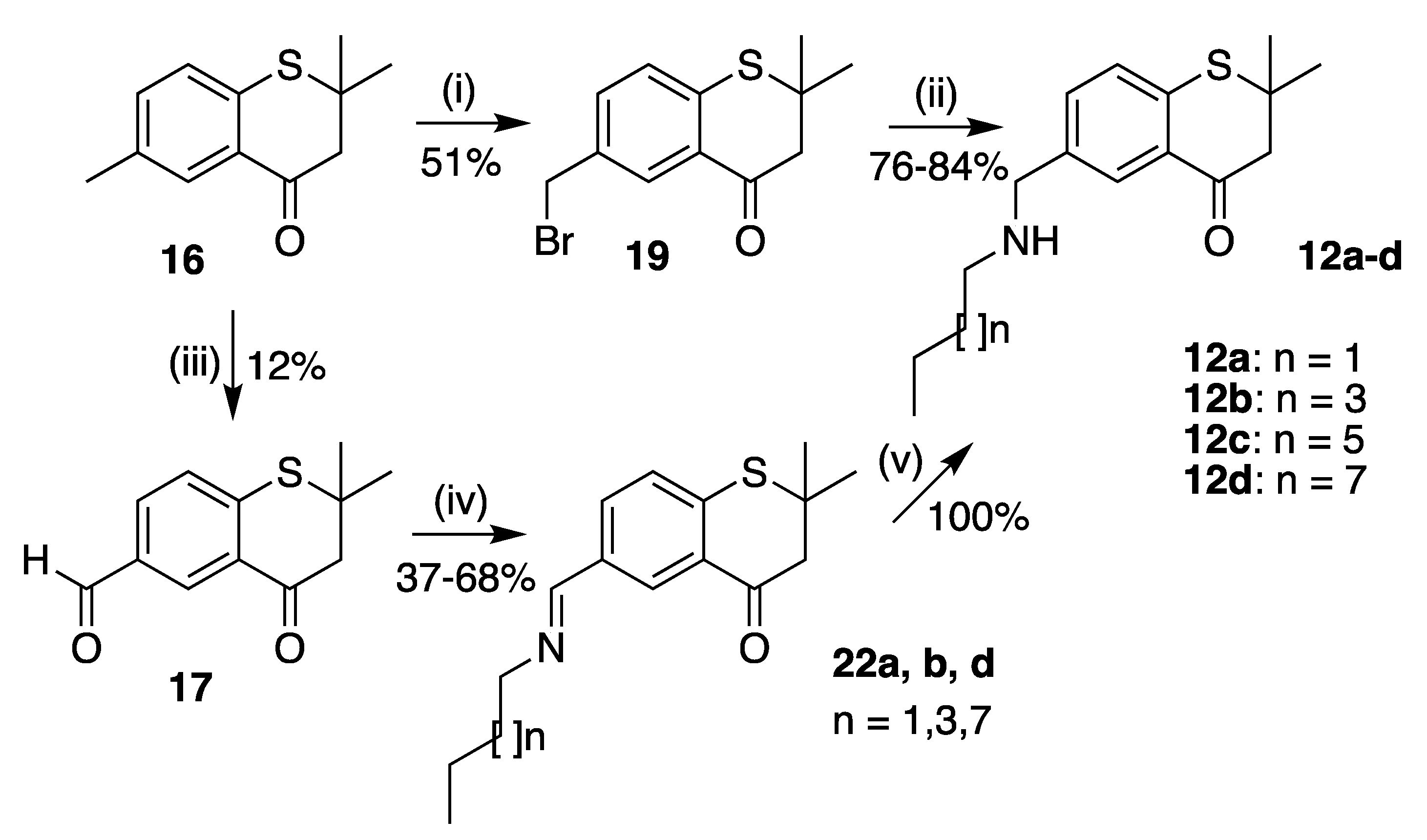
6-(Bromomethyl)-2,2-dimethylthiochroman-4-one (19)
Compound 16 (1.55 g (7.5 mmol) was dissolved in cyclohexane (25 mL). To this stirred solution was added N-bromosuccinimide (2.66 g, 14.9 mmol) and a catalytic quantity of dibenzoylperoxide, and the reaction mixture was stirred at 100 °C for 6 h. Upon completion, the solvent was removed in vacuo, and the residue purified by flash chromatography (pet. ether/EtOAc 10:1) to give 19 as a golden oil (1.072 g, 51%), which solidified on standing. IR νmax (neat) 1676 cm−1; 1H-NMR (CDCl3) δH 1.47 (6H, s, (CH3)2, 2.87 (2H, s, CH2), 4.48 (2H, s, ArCH2), 7.22 (1H, d, J = 8.2 Hz, H8), 7.45 (1H, dd, J = 8.2, 2.1 Hz, H7), 8.10 (1H, d, J = 2.2 Hz, H5); 13C-NMR δC 28.6 ((CH3)2), 32.7 (CH2), 44.9(C(CH3)2), 53.6 (CH2), 128.3 (ArCH), 128.9 (ArCH), 129.7 (ArC), 134.2 (ArCH), 134.4 (ArC), 141.9 (ArC), 194.5 (C=O).
6-(Azidomethyl)-2,2-dimethylthiochroman-4-one (20)
To a solution of 19 (0.81 g, 2.84 mmol) in DMF (6 mL) was added sodium azide (2 g, 30.8 mmol). The resultant slurry was heated on an oil bath for 4 h at 50 °C, and then partitioned between water and ethyl acetate. The aqueous layer was washed twice with ethyl acetate (2 × 20 mL portions) and the combined organic layers dried over sodium sulfate and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (pet. ether/EtOAc 10:1) to yield the azide (0.52 g, 74%) as a bright yellow oil. 1H- NMR (CDCl3) δH 1.48 (6H, s, (CH3)2, 2.89 (2H, s, CH2CO), 4.34 (2H, s, CH2N3), 7.27 (1H, d, J = 8 Hz, H8), 7.38 (1H, dd, J = 8, 2 Hz, H7), 8.04 (1H, d, J = 1.7 Hz, H5); 13C-NMR δC 28.6 ((CH3)2), 44.8 (C2), 53.7 (CH2), 54.1 (CH2), 128.3 (ArCH), 128.3 (ArCH), 129.7 (ArC), 132.1 (ArC), 133.3 (ArCH), 141.7 (ArC), 194.6 (C=O).
6-(Aminomethyl)-2,2-dimethylthiochroman-4-one (21)
To 20 (57 mg, 0.23 mmol) in EtOH/H2O (3:1, 5 mL) was added Zn (20 mg, 0.31 mmol) and NH4Cl (25 mg, 0.47 mmol). The mixture was stirred vigorously at room temperature for 24 h followed by reflux for 6 h. Ethyl acetate (20 mL) and NaOH (1 M) (1 mL) were added. The mixture was filtered, and the filtrate was washed with brine, dried over anhydrous sodium sulfate. Removal of solvent under reduced pressure afforded a yellow oil. 1H-NMR (CDCl3) δH 1.47 (6H, s, (CH3)2, 2.88 (2H, s, CH2CO), 3.86 (2H, s, CH2NH2), 7.21 (1H, d, J = 8.1 Hz, H8), 7.40 (1H, dd, J = 8.1, 1.8 Hz, H7), 8.04 (1H, d, J = 1.3 Hz, H5); 13C- NMR δC 28.6 ((CH3)2), 44.7 (C2), 45.7 (CH2), 53.9 (CH2), 127.0 (ArCH), 127.9 (ArCH), 129.6 (ArC), 132.9 (ArCH), 133.5 (ArC), 139.7 (ArC), 195.1 (C=O). As an alternative method, to a mixture of 20 (462 mg, 1.87 mmol) in THF (10 mL) and H2O (1 mL) was added PPh3 (0.86 g, 3.29 mmol). The reaction mixture was stirred at RT for 12 h. The mixture was acidified to pH = 1 with 1N HCl and extracted with EtOAc (100 mL). The aqueous layer was separated and the pH adjusted to pH 10 with 1N NaOH, and extracted with DCM (3 × 100 mL). The residual solvent was removed in vacuo to afford 302 mg (73%) of 21.
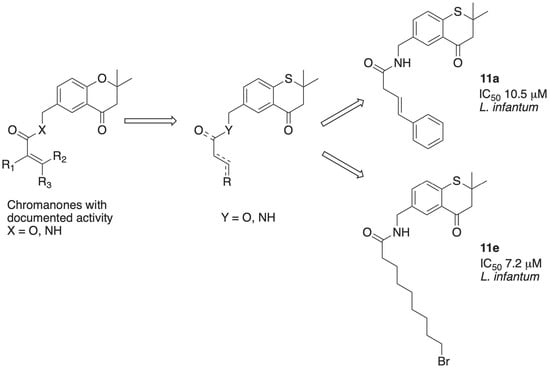
(E)-N-((2,2-Dimethyl-4-oxothiochroman-6-yl)methyl)-4-phenylbut-3-enamide (11a)
To a solution of trans-styrylacetic acid (41 mg, 0.25 mmol) in dichloromethane (5 mL) at 0 °C under a N2 atmosphere was added EDC-HCl (100 mg, 0.52 mmol) and triethylamine (0.09 mL, 0.65 mmol). After 30 min, 21 (50 mg, 0.23 mmol) was added to this solution. The reaction was allowed to reach room temperature and stirred for 24 h. The residual solvent was removed in vacuo, the residue extracted with saturated NaHCO3/DCM, and the crude organic residue purified by flash column chromatography (pet. ether/EtOAc 5:1) to afford amide 11a as a pale oil (56 mg, 67%). IR (neat) νmax 965, 1186, 1212, 1304, 1468, 1538, 1675, 2923 cm−1; 1H-NMR (CDCl3) δH 1.46 (6H, s, (CH3)2, 2.86 (2H, s, CH2CO), 3.22 (2H, d, J = 7.1 Hz, CH2C=C), 4.43 (2H, d, J = 5.9 Hz, CH2N), 5.97 (1H, br., NH), 6.31 (1H, m, CH2CH=CH), 6.55 (1H, m, CH2CH=CH), 7.19–7.39 (7H, m, 7 × ArH), 7.98 (1H, d, J = 1.7 Hz, H5); 13C-NMR δC 28.6 ((CH3)2), 40.8 (CH2), 43.0 (CH2), 44.7 (C2), 53.8 (CH2), 122.0, 126.4 (2C), 127.6, 127.9, 128.2, 128.7 (2C), 133.4, 134.9 (ArC), 135.1, 140.7 (ArC), 170.7 (NHC=O), 194.8 (C=O) (2C signals obscured). HRMS (M + Na)+ 388.1361, C22H23NO2NaS requires 388.1347.
(2,2-Dimethyl-4-oxothiochroman-6-yl)methyl-6-phenylhex-5-enoate (E:Z 3:1) (11b)
Compound 21 (87 mg (0.39 mmol) was reacted as described for 11a with 6-phenylhex-5-enoic acid (75 mg, 0.39 mmol) to afford 11b as a pale straw-coloured oil that on standing became a semi-solid gum (113 mg, 73%). IR (neat) νmax 964, 1091, 1213, 1304, 1466, 1648, 1676, 2852, 2922, 2955 cm−1; 1H-NMR (CDCl3) δH 1.46 (6H, s, (CH3)2, 1.82–1.90 (2H, m, CH2), 2.19–2.40 (4H, m, NHCOCH2 & C=CHCH2), 2.86 (2H, s, CH2CO), 4.30 & 4.41 (2H, 2 × d, J = 5.7 Hz, ArCH2N), 5.53 & 5.81 (1H, br., NH), 6.18 (1H, m, CH=CHAr), 6.37 & 6.46 (1H, 2 × d, J = 15.9 & 11.7 Hz, CH=CHAr), 7.15–7.36 (7H, m, 7 × ArH), 7.93 & 7.97 (1H, 2 × s, H5); 13C-NMR δC 25.1 (CH2), 28.6 ((CH3)2), 32.4 (CH2), 35.9 (CH2), 42.9 (CH2), 44.7 (C2), 53.8 (CH2), 126.0 (2C), 127.0, 127.5, 128.1, 128.2, 128.5 (2C), 129.7, 130.8, 133.4, 135.2 (ArC), 137.5 (ArC), 140.6 (ArC), 172.7 (NHC=O), 194.8 (C=O), (signals for E isomer reported, 1 quaternary signal obscured); HRMS (M + Na)+ 416.1645, C24H27NO2NaS requires 416.1660.
(2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)-N-((2,2-dimethyl-4-oxothiochroman-6-yl)methyl)penta-2,4-dienamide (11c)
Compound 21 (80 mg, 0.36 mmol) was reacted as for 11a with piperic acid (79 mg, 0.36 mmol) to afford 11c as a pale yellow solid (63 mg, 41%). IR (neat) νmax 982, 1036, 1190, 1250, 1443, 1536, 1614, 1644, 1679, 3278 cm−1; 1H-NMR (CDCl3-) δH 1.45 (6H, s, (CH3)2, 2.85 (2H, s, CH2), 4.51 (2H, d, J = 5.9 Hz, CH2N), 5.94 (1H, d, J = 14.9 Hz, COCH=C), 5.98 (2H, s, OCH2O), 6.04 (1H, br. t, NH), 6.65 & 6.68 (1H, 2 × d, J = 10.6 Hz, CH=CH), 6.78 (2H, m, 2 × CH=C), 6.89 (1H, d, J = 8.1 Hz, CH=C), 6.97 (1H, br. s, CH=C), 7.20 (1H, d, J = 8.1 Hz, CH=C), 7.39 (2H, m, 2 × CH=C), 8.00 (1H, br. s, CH=C); 13C-NMR δC 28.6 ((CH3)2), 43.0 (CH2), 44.7 (C(CH3)2), 53.8 (CH2), 101.3 (OCH2O), 105.8 (CH=C), 108.5 (CH=C), 122.5 (CH=C), 122.8 (CH=C), 124.5 (CH=C), 127.5 (CH=C), 128.1 (CH=C), 129.6 (ArC), 130.8 (ArC), 133.5 (CH=C), 135.2 (ArC), 139.4 (CH=C), 140.6 (ArC), 141.8 (ArCH), 148.2 (ArC), 148.3 (ArC), 166.2 (NC=O), 195.0 (C=O); HRMS (M + H)+ 422.1432, C24H24NO4S requires 422.1426.
N-((2,2-Dimethyl-4-oxothiochroman-6-yl)methyl)octanamide (11d)
Compound 21 (87 mg, 0.39 mmol) was reacted as for 11a with octanoic acid (56 mg, 0.39 mmol) to afford 11d as a pale straw-coloured oil that on standing became a semi-solid gum (102 mg, 75%). IR (neat) νmax 1193, 1306, 1407, 1462, 1530, 1639, 1680, 2852, 2923, 3297 cm−1; 1H-NMR (CDCl3, 400 MHz) δH 0.87 (3H, t, CH2CH3), 1.28 (m, 8H, 4 × CH2), 1.46 (6H, (CH3)2), 1.65 (2H, m, CH2), 2.22 (2H, m, CH2CONH), 2.86 (2H, s, CH2CO), 4.41 (2H, d, J = 5.9 Hz, CH2N), 5.86 (1H, br., NH), 7.20 (1H, d, J = 8.1 Hz, H8), 7.36 (1H, dd, J = 8.1, 2.0 Hz, H7), 7.97 (1H, J = 1.5 Hz, H5); 13C-NMR δC 14.1 (CH3), 22.6 (CH2), 25.7 (CH2), 28.6 ((CH3)2), 29.0 (CH2), 29.3 (CH2), 31.7 (CH2), 36.8 (CH2), 42.8 (CH2), 44.7 (C(CH3)2), 53.8 (CH2), 127.4 (ArCH), 128.1 (ArCH), 129.6 (ArC), 133.4 (ArCH), 135.3 (ArC), 140.6 (ArC), 173.2 (NC=O), 194.9 (C=O); HRMS (M + Na)+ 370.1831, C20H29NO2NaS requires 370.1817.
9-Bromo-N-((2,2-dimethyl-4-oxothiochroman-6-yl)methyl)nonanamide (11e)
Compound 21 (100 mg, 0.45 mmol) was reacted as for 11a with 9-bromononanoic acid (100 mg, 0.42 mmol) to afford 11e as a pale straw-coloured oil that on standing became a semi-solid gum (123 mg, 62%). IR (neat) νmax 1215, 1300, 1463, 1539, 1647, 1675, 2855, 2924 cm−1; 1H-NMR (CDCl3) δH 1.25–1.86 (m, 12H, 6 × CH2), 1.46 (6H, s, (CH3)2, 2.22 (2H, m, NHCOCH2), 2.86 (2H, s, CH2CO), 3.41 (2H, m, CH2Br), 4.42 (2H, d, J = 5.9 Hz, CH2N), 5.82 (1H, br., NH), 7.21 (1H, d, J = 8.2 Hz, H8), 7.36 (1H, dd, J = 8.2, 2.2 Hz, H7), 7.97 (1H, dd, J = 2, 0.2 Hz, H5); 13C-NMR δC 25.6 (CH2), 28.1 (CH2), 28.5 (CH2), 28.6 ((CH3)2), 29.1 (CH2), 29.2 (CH2), 32.8 (CH2), 34.0 (CH2), 36.7 (CH2), 42.8 (CH2), 44.7 (C(CH3)2), 53.8 (CH2), 127.4 (ArCH), 128.1 (ArCH), 129.7 (ArC), 133.4 (ArCH), 135.3 (ArC), 140.6 (ArC), 173.0 (NC=O), 194.9 (C=O); HRMS (M + Na)+ 462.1078, C21H30NO2NaSBr requires 462.1078.
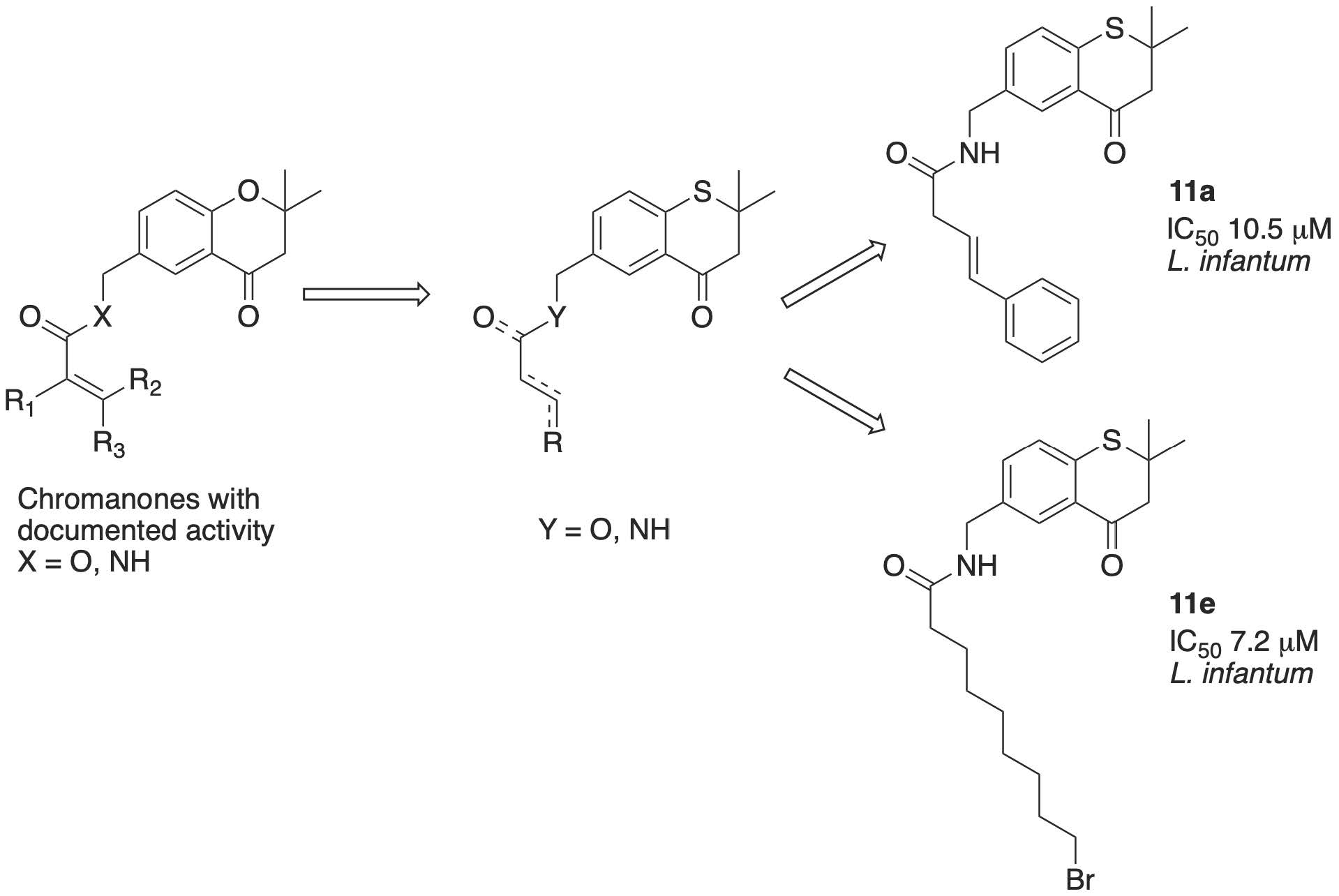
6-((Butylamino)methyl)-2,2-dimethylthiochroman-4-one (12a)
Compound 19 (100 mg, 0.35 mmol) was dissolved in acetonitrile (25 mL) under aerobic conditions. To this stirred solution was added excess n-butylamine, and the reaction mixture was stirred at 100 °C for 2 h. Upon completion, the solvent was removed in vacuo, and the residue purified by flash chromatography (pet. ether/EtOAc 2:1) to give 12a as a pale brown oil (74 mg, 76%), without contamination with any tertiary amine. IR νmax (neat) 1249, 1464, 1601, 1679 cm−1; 1H-NMR (CDCl3) δH 0.91 (3H, t, J = 7.3 Hz, CH3), 1.34 (2H, m, (CH2), 1.46 (6H, s, (CH3)2, 1.48 (2H, m, CH2), 2.61 (2H, t, J = 7.2 Hz, (NCH2CH2), 2.87 (2H, s, CH2), 3.76 (2H, s, ArCH2), 7.20 (1H, d, J = 8.1 Hz, H8), 7.42 (1H, dd, J = 8.1, 1.6 Hz, H7), 8.03 (1H, br. d, H5); 13C-NMR δC 14.0 (CH3), 20.5 (CH2), 28.6 ((CH3)2), 32.2 (CH2), 44.6 (C(CH3)2), 49.1 (CH2), 53.3 (CH2), 53.9 (CH2), 127.8 (ArCH), 128.1 (ArCH), 129.5 (ArC), 133.8 (ArCH), 137.3 (ArC), 139.8 (ArC), 195.1 (C=O); HRMS (M + H)+ 278.1565, C16H24NOS requires 278.1579.
6-((Hexylamino)methyl)-2,2-dimethylthiochroman-4-one (12b)
Compound 19 (100 mg, 0.35 mmol) was reacted as for 12a with n-hexylamine to afford 12b as a pale brown oil (82 mg, 77%). IR νmax (neat) 1251, 1454, 1528, 1683, 3336 cm−1; 1H- NMR (CDCl3) δH 0.88 (3H, t, J = 6.7 Hz, CH3), 1.28 (6H, m, (CH2)3), 1.46 (6H, s, (CH3)2, 1.48 (2H, m, CH2), 2.60 (2H, t, J = 7.3 Hz, (NCH2CH2), 2.87 (2H, s, CH2), 3.76 (2H, s, ArCH2), 7.20 (1H, d, J = 8.2 Hz, H8), 7.42 (1H, dd, J = 8.1, 2.1 Hz, H7), 8.02 (1H, d, J = 1.3 Hz, H5); 13C-NMR δC 14.1 (CH3), 22.6 (CH2), 27.0 (CH2), 28.6 ((CH3)2), 30.0 (CH2), 31.8 (CH2), 44.6 (C(CH3)2), 49.5 (CH2), 53.3 (CH2), 53.9 (CH2), 127.7 (ArCH), 128.1 (ArCH), 129.5 (ArC), 133.8 (ArCH), 137.3 (ArC), 139.8 (ArC), 195.1 (C=O); HRMS (M+H)+ 306.1899, C18H28NOS requires 306.1892.
2,2-Dimethyl-6-((octylamino)methyl)thiochroman-4-one (12c)
Compound 19 (100 mg, 0.35 mmol) was reacted as for 12a with n-octylamine (45 mg, 0.35 mmol) to afford 12c as a pale brown oil (98 mg, 84%). IR νmax (neat) 1303, 1465, 1600, 1678, 2922 cm−1; 1H-NMR (CDCl3) δH 0.87 (3H, t, J = 6.8 Hz, CH3), 1.22–1.32 (10H, m, (CH2)5), 1.46 (6H, s, (CH3)2, 1.48 (2H, m, CH2), 2.60 (2H, t, J = 7.2 Hz, (NCH2CH2), 2.87 (2H, s, CH2), 3.76 (2H, s, ArCH2), 7.20 (1H, d, J = 8.2 Hz, H8), 7.42 (1H, dd, J = 8.1, 1.8 Hz, H7), 8.02 (1H, d, J = 1.5 Hz, H5); 13C-NMR δC 14.1 (CH3), 22.7 (CH2), 27.4 (CH2), 28.6 ((CH3)2), 29.3 (CH2), 29.5 (CH2), 30.1 (CH2), 31.8 (CH2), 44.6 (C(CH3)2), 49.5 (CH2), 53.3 (CH2), 53.9 (CH2), 127.8 (ArCH), 128.1 (ArCH), 129.5 (ArC), 133.8 (ArCH), 137.3 (ArC), 139.8 (ArC), 195.1 (C=O); HRMS (M+H)+ 334.2195, C20H32NOS requires 334.2205.
2,2-Dimethyl-6-((decylamino)methyl)thiochroman-4-one (12d)
Compound 19 (100 mg, 0.35 mmol) was reacted as for 12a with n-decylamine (55 mg, 0.35 mmol) to afford 12d as a pale golden oil (100 mg, 79%). IR νmax (neat) 1189, 1301, 1387, 1468, 1510, 1675, 2852, 2922 cm−1; 1H-NMR (CDCl3-) δH 0.88 (3H, t, J = 6.8 Hz, CH3), 1.20–1.33 (14H, m, (CH2)7), 1.47 (6H, s, (CH3)2, 1.49 (2H, m, CH2), 2.61 (2H, t, J = 7.2 Hz, (NCH2CH2), 2.87 (2H, s, CH2), 3.77 (2H, s, ArCH2), 7.20 (1H, d, J = 8.1 Hz, H8), 7.43 (1H, dd, J = 8.2, 1.6 Hz, H7), 8.02 (1H, d, J = 1.2 Hz, H5); 13C-NMR δC 14.2 (CH3), 22.7 (CH2), 27.3 (CH2), 28.6 ((CH3)2), 29.3 (CH2), 29.57 (CH2), 29.59 (CH2), 29.62 (CH2), 30.0 (CH2), 31.9 (CH2), 44.7 (C(CH3)2), 49.4 (CH2), 53.2 (CH2), 53.9 (CH2), 127.8 (ArCH), 128.2 (ArCH), 129.5 (ArC), 133.9 (ArCH), 137.1 (ArC), 139.9 (ArC), 195.1 (C=O); HRMS (M+H)+ 362.2519, C22H36NOS requires 362.2518.
6-((Butylimino)methyl)-2,2-dimethylthiochroman-4-one (22a)
To a solution of 17 (100 mg, 0.45 mmol) in tetrahydrofuran (10 mL) was added n-butylamine (33 mg, 0.45 mmol) and sodium triacetoxyborohydride (144 mg, 0.68 mmol). To the stirred suspension was added acetic acid (20 μL, 0.35 mmol). The reaction was stirred under a N2 atmosphere at room temperature overnight. The reaction mixture was quenched by adding aqueous saturated NaHCO3, and the product was extracted with EtOAc (3 × 30 mL). The EtOAc extract was dried (Na2SO4), and the solvent was evaporated to give the crude imine as a clear oil (84 mg, 67%). 1H-NMR (CDCl3-) δH 0.86 (3H, t, J = 7.4 Hz, CH2CH3), 1.29 (2H, m, CH2), 1.40 (6H, s, C(CH3)2), 1.58 (2H, m, CH2), 2.81 (2H, s, CH2), 3.53 (2H, t, J = 7 Hz, NCH2), 7.19 (1H, d, J = 8.3 Hz, H8), 7.84 (1H, dd, J = 8.3, 1.4 Hz, H7), 8.18 (2H, m, H5 & CH=N). 13C-NMR δC 13.9 (CH3), 20.4 (CH2), 28.6 ((CH3)2), 32.9 (CH2), 44.9 (C(CH3)2), 53.6 (CH2), 61.3 (CH2), 128.0 (ArCH), 129.3 (ArCH), 129.5 (ArC), 131.4 (ArCH), 133.2 (ArC), 144.1 (ArC), 159.4 (N=CH), 194.5 (C=O).
6-((Hexylimino)methyl)-2,2-dimethylthiochroman-4-one (22b)
Compound 17 (98 mg, 0.44 mmol) was reacted as described for 22a with n-hexylamine (45 mg, 0.44 mmol) to afford 22b as a clear oil (92 mg, 68%). 1H-NMR (CDCl3) δH 0.81 (3H, t, CH2CH3), 1.23–1.29 (6H, m, (CH2)3), 1.41 (6H, s, C(CH3)2), 1.61 (2H, m, CH2), 2.82 (2H, s, CH2), 3.52 (2H, t, J = 7 Hz, NCH2), 7.20 (1H, d, J = 8.3 Hz, H8), 7.85 (1H, dd, J = 8.3, 2.1 Hz, H7), 8.20 (2H, m, H5 & CH=N). 13C-NMR δC 13.1 (CH3), 21.6 (CH2), 26.0 (CH2), 27.6 ((CH3)2), 29.8 (CH2), 30.6 (CH2), 43.9 (C(CH3)2), 52.6 (CH2), 60.7 (CH2), 126.8 (ArCH), 128.3 (ArCH), 128.5 (ArC), 130.3 (ArCH), 132.2 (ArC), 143.1 (ArC), 158.3 (N=CH), 193.4 (C=O).
6-((Decylimino)methyl)-2,2-dimethylthiochroman-4-one (22d)
Compound 17 (87 mg, 0.39 mmol) was reacted as for 22a with n-decylamine (62 mg, 0.39 mmol) to afford 22d as a yellow oil (52 mg, 37%). 1H-NMR (CDCl3) δH 0.78 (3H, t, CH2CH3), 1.20 (10H, m, 5 × CH2), 1.38 (6H, s, C(CH3)2), 1.59 (2H, m, CH2), 2.79 (2H, s, CH2), 3.50 (2H, t, J = 7 Hz, NCH2), 7.16 (1H, d, J = 8.3, H8), 7.82 (1H, d, J = 8.3 Hz, H7), 8.16 (2H, m, H5 & CH=N). 13C-NMR δC 14.1 (CH3), 22.7, 25.6, 27.3, 28.5 ((CH3)2), 29.3, 29.4, 29.6, 30.9, 31.9, 44.9 (C(CH3)2), 53.6 (CH2), 61.7 (CH2), 127.9 (ArCH), 129.3 (ArCH), 129.5 (ArC), 131.4 (ArCH), 133.2 (ArC), 144.0 (ArC), 159.2 (N=CH), 194.3 (C=O).
Reduction of 22a–d with one equivalent of sodium borohydride in methanol for one hour at 0 °C to room temperature afforded 12a–d in quantitative yield.
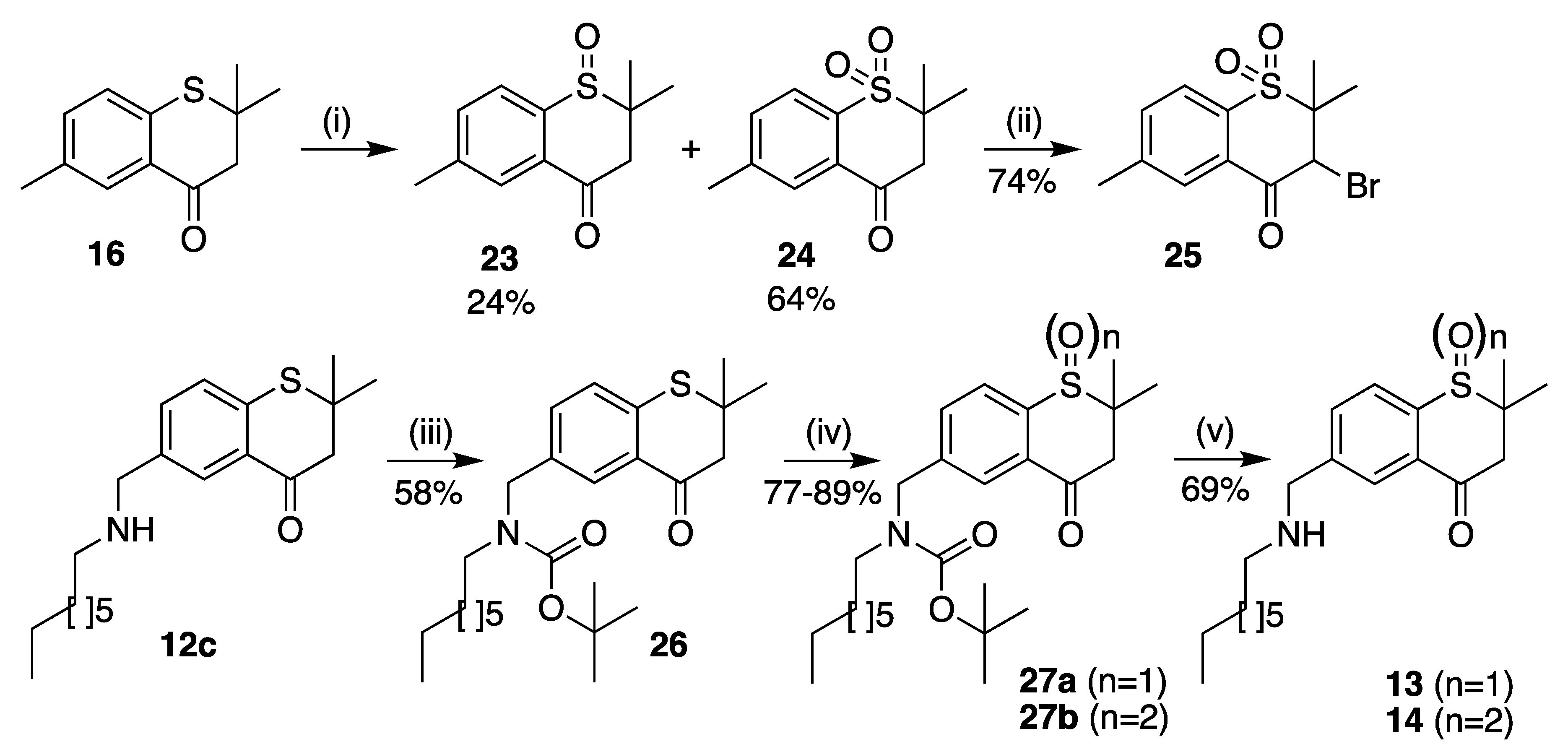
2,2,6-Trimethylthiochroman-4-one 1-oxide (23) and 2,2,6-trimethylthiochroman-4-one 1,1-dioxide (24)
To a suspension of 16 (100 mg, 0.48 mmol) in glacial acetic acid (10 mL) was added sodium perborate·4H2O (75 mg, 0.49 mmol), in portions. After stirring at 55 °C for 4 h, the reaction was poured into ice/water (100 mL). Ethyl acetate was added and the aqueous layer extracted (3 × 50 mL). The organic layers were combined, the solvent removed in vacuo and the residue purified by flash chromatography (pet. ether/EtOAc 8:1) to give two products. 1. Sulfone 24, a clear oil (69 mg, 64%); 1H-NMR (CDCl3) δH 1.53 (6H, s, (CH3)2, 2.49 (3H, s, (CH3), 3.24 (2H, s, CH2), 7.64 (1H, d, J = 8.1 Hz, ArH), 7.91 (1H, s, H5), 7.96 (1H, d, J = 8.1 Hz, ArH); 13C-NMR δC 21.1 ((CH3)2), 21.6 (CH3), 50.6 (CH2), 58.6 (C(CH3)2), 125.0 (ArCH), 128.4 (ArCH), 130.4 (ArC), 135.8 (ArCH), 136.2 (ArC), 144.2 (ArC), 191.0 (C=O). 2. Sulfoxide 23, straw coloured oil (28 mg, 24%); 1H-NMR (CDCl3) δH 1.35 & 1.41 (6H, 2 × s, (CH3)2, 2.47 (3H, s, (CH3), 2.73 (1H, d, J = 17.5 Hz, 1H of CH2), 3.24 (1H, d, J = 17.5 Hz, 1H of CH2), 7.60 (1H, d, J = 7.9 Hz, ArH), 7.78 (1H, d, J = 8.1 Hz, ArH), 7.95 (1H, s, H5); 13C- NMR δC 20.0 (CH3), 21.4 (CH3), 24.0 (CH3), 45.4 (CH2), 56.2 (C(CH3)2), 128.7 (ArCH), 129.0 (ArC), 129.6 (ArCH), 135.6 (ArCH), 140.6 (ArC), 142.4 (ArC), 192.7 (C=O).
3-Bromo-2,2,6-trimethylthiochroman-4-one 1,1-dioxide (25)
Compound 24 (100 mg, 0.45 mmol) was dissolved in cyclohexane/CH3CN (1:1) (25 mL). To this stirred solution was added N-bromosuccinimide (120 mg, 0.67 mmol) and a catalytic quantity of dibenzoylperoxide, and the reaction mixture was stirred at 100 °C for 6 h. Upon completion, the solvent was removed in vacuo, and the residue purified by flash chromatography (pet. ether/EtOAc 10:1) to give an orange oil (100 mg, 74%). 1H-NMR (CDCl3) δH 1.44 & 1.75 (6H, 2 × s, (CH3)2, 2.52 (3H, s, (CH3), 5.82 (1H, s, CH), 7.70 (1H, d, J = 8.1 Hz, ArH), 7.95 (1H, d, J = 7.9 Hz, ArH), 8.00 (1H, s, H5); 13C-NMR δC 18.8 (CH3), 20.2 (CH3), 21.7 (CH3), 62.0 (CH), 64.8 (C(CH3)2), 125.0 (ArCH), 127.7 (ArC), 129.8 (ArCH), 135.1 (ArC), 136.4 (ArCH), 144.9 (ArC), 184.8 (C=O).
tert-Butyl ((2,2-dimethyl-4-oxothiochroman-6-yl)methyl)(octyl)carbamate (26)
To a solution of 12c (200 mg, 0.60 mmol) in DCM (10 mL) at 0 °C was added di-tert-butyl dicarbonate (130 mg, 0.60 mmol) and triethylamine (0.08 mL, 0.60 mmol). The reaction was stirred overnight, allowing it to reach room temperature. The solvent was evaporated and the residue purified by flash chromatography (pet. ether/EtOAc 8:1) to give 26 as a yellow oil (150 mg, 58%). 1H-NMR (CDCl3, 400 MHz) δH 0.87 (3H, t, J = 6.6 Hz, CH3), 1.25 (10H, m, (CH2)5), 1.45–1.50 (17H, m, 5 × CH3 and CH2), 2.87 (2H, s, CH2CO), 3.16 (2H, m, NCH2CH2), 4.39 (2H, m, NCH2Ar), 7.20 (1H, dd, J = 8.1, 0.4 Hz, ArH), 7.32 (1H, br. d, ArH), 7.97 (1H, br. d, ArH).
2,2-Dimethyl-6-((octylamino)methyl)thiochroman-4-one 1-oxide (13)
To compound 26 (27 mg, 0.06 mmol) in methanol (1 mL) was added 35% H2O2 (11.3 mg solution, 0.1 mmol) and Montmorillonite K10 (20 mg). The resulting mixture was stirred at room temperature overnight, after which the clay was removed by filtration. The solvent was evaporated to yield BOC-sulfoxide 27a (25 mg, 89%). 1H-NMR (CDCl3,-) δH 0.88 (3H, t, J = 6.9 Hz, CH3), 1.26 (10H, m, (CH2)5), 1.41–1.54 (17H, m, 5 × CH3 and CH2), 2.79 (1H of CH2CO), 3.22 (3H, m, 1H of CH2CO and NCH2CH2), 4.52 (2H, m, NCH2Ar), 7.68 (1H, br., ArH), 7.90 (1H, br., ArH), 8.01 (1H, br. d, ArH). To a solution of 27a in DCM (2mL) at 0 °C was added trifluoroacetic acid (2 mL). The reaction was stirred overnight, after which time the reaction was washed with saturated sodium bicarbonate solution and extracted with DCM (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, evaporated and purified by flash column chromatography (pet. ether/EtOAc 3:1) to yield the deprotected sulfoxide 13. 1H-NMR (CDCl3-) δH 0.80 (3H, t, J = 6.7 Hz, CH3), 1.19 (12H, m, (CH2)6), 1.48 (6H, s, (CH3)2, 2.54 (2H, t, J = 7.2 Hz, NCH2CH2), 3.17 (2H, s, CH2CO), 3.85 (2H, s, NCH2Ar), 7.78 (1H, d, J = 8.1 Hz, ArH), 7.95 (1H, d, J = 7.8 Hz, ArH), 7.98 (1H, s, ArH); 13C-NMR δC 14.1 (CH3), 21.1 ((CH3)2), 22.7, 27.3, 29.3, 29.5, 29.9, 31.8, 49.4, 50.6, 53.0 (9 × CH2), 58.7 (C(CH3)2), 125.2 (ArCH), 127.5 (ArCH), 130.5 (ArC), 134.7 (ArCH), 137.4 (ArC), 146.5 (ArC), 190.8 (C=O).
2,2-Dimethyl-6-((octylamino)methyl)thiochroman-4-one 1,1-dioxide (14)
To a suspension of 26 (40 mg, 0.09 mmol) in glacial acetic acid (10 mL) was added sodium perborate·4H2O (30 mg, 0.2 mmol), in portions. After stirring at 55 °C for 4 h, the reaction was poured into ice/water (100 mL). Ethyl acetate was added and the aqueous layer extracted (3 × 50 mL). The organic layers were combined, the solvent removed in vacuo and the residue purified by flash chromatography (pet. ether/EtOAc 8:1) to give the BOC-sulfone 27b (33 mg, 77%). 1H-NMR (CDCl3) δH 0.87 (3H, t, J = 6.8 Hz, CH3), 1.26 (10H, m, (CH2)5), 1.37–1.55 (17H, m, 5 × CH3 and CH2), 3.14–3.25 (4H, m, CH2CO and NCH2CH2), 4.52 (2H, m, NCH2Ar), 7.71 (1H, br., ArH), 7.95 (1H, br., ArH), 8.03 (1H, d, J = 8.1 Hz, ArH). To a solution of 27b in DCM (2 mL) at 0 °C was added trifluoroacetic acid (2 mL). The reaction was stirred overnight, after which time the reaction was washed with saturated sodium bicarbonate solution and extracted with DCM (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, evaporated and purified by flash column chromatography (pet. ether/EtOAc 3:1) to yield the deprotected sulfone 14 (18 mg, 69%). 1H-NMR (CDCl3) δH 0.87 (3H, t, J = 6.6 Hz, CH3), 1.26 (12H, m, (CH2)6), 1.53 (6H, s, (CH3)2, 2.64 (2H, t, J = 7.3 Hz, NCH2CH2), 3.24 (2H, s, CH2CO), 3.95 (2H, s, NCH2Ar), 7.87 (1H, d, J = 8.2 Hz, ArH), 8.03 (1H, d, J = 7.9 Hz, ArH), 8.06 (1H, s, ArH); 13C- NMR δC 14.1 (CH3), 21.1 ((CH3)2), 22.7, 27.2, 29.2, 29.4, 29.5, 31.8, 49.2, 50.6, 52.7 (9 × CH2), 58.7 (C(CH3)2), 125.2 (ArCH), 127.7 (ArCH), 130.6 (ArC), 134.9 (ArCH), 137.7 (ArC), 145.5 (ArC), 190.8 (C=O).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}