
From Oncogenic Signaling Pathways to Single-Cell Sequencing of Immune Cells: Changing the Landscape of Cancer Immunotherapy

 ,
,  ,
,  ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tumorigenesis and Signaling Pathways
2.1. Cell Proliferation
2.2. Cell Survival
2.3. Cell Metabolism
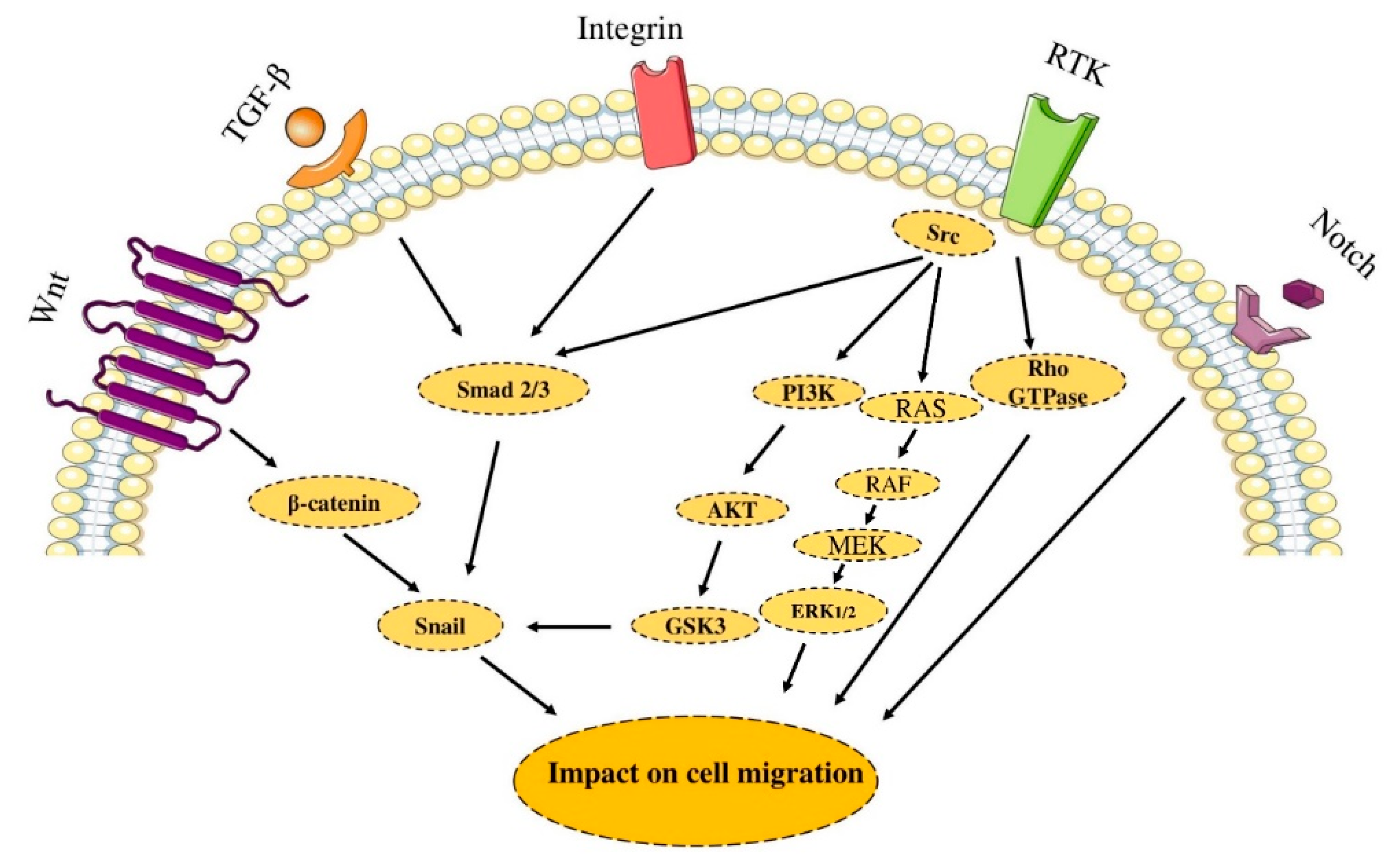
2.4. Cell Migration
2.5. Cell Polarity
2.6. Cell Differentiation
3. Cancer and Extracellular Matrix (ECM)
3.1. Cell Surface Proteoglycans
3.1.1. The Classes of Cell Surface Proteoglycans
3.1.2. CD44-Specific Signaling
3.1.3. Syndecans and Cancer
4. Angiogenesis
5. Inflammation
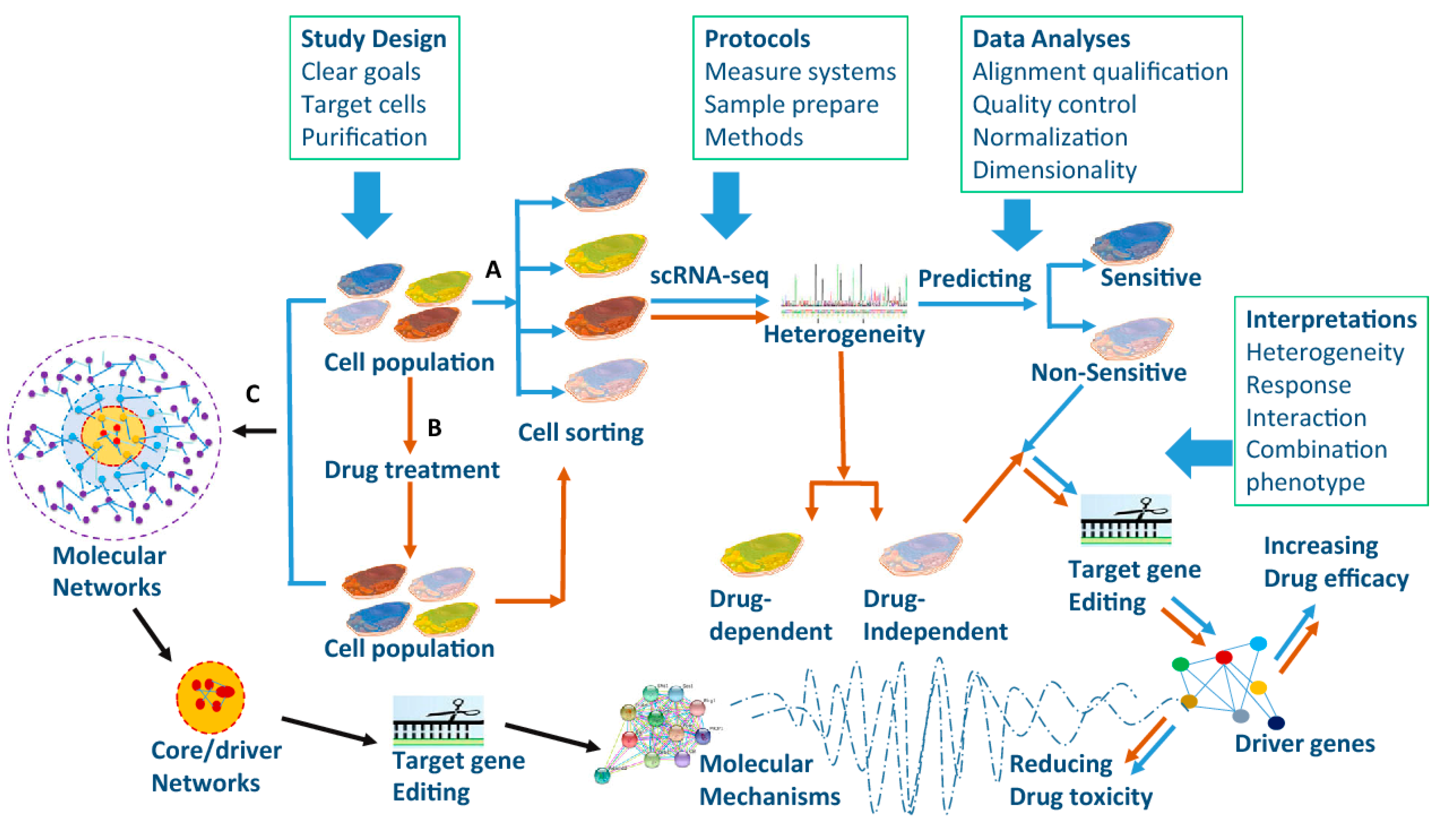
6. Single-Cell Sequencing
6.1. Dissecting Key Cellular and Molecular Functions in Cancers
6.2. Molecular Mechanisms of Drug Resistance
6.3. Immunotherapy and Single-Cell Sequencing: Overcoming the Barriers?
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Solimini, N.L.; Luo, J.; Elledge, S.J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 130, 986–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, R. The Biology of Cancer; Garland Science: New York City, NY, USA, 2013. [Google Scholar]
- Navin, N.; Hicks, J. Future medical applications of single-cell sequencing in cancer. Genome Med. 2011, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Kang, B.; Zhang, Z. Understanding tumor ecosystems by single-cell sequencing: Promises and limitations. Genome Biol. 2018, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kamaruzman, N.I.; Aziz, N.A.; Poh, C.L.; Chowdhury, E.H. Oncogenic Signaling in Tumorigenesis and Applications of siRNA Nanotherapeutics in Breast Cancer. Cancers 2019, 11, 632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, A.; Licchetta, A.; Scarpa, A.; Azzariti, A.; Brunetti, A.E.; Simone, G.; Nardulli, P.; Santini, D.; Aieta, M.; Delcuratolo, S. Carcinogenesis of pancreatic adenocarcinoma: Precursor lesions. Int. J. Mol. Sci. 2013, 14, 19731–19762. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Lotfipour, F.; Hallaj-Nezhadi, S.; Valizadeh, H.; Dastmalchi, S.; Baradaran, B.; Jalali, M.B.; Dobakhti, F.; Sciences, P. Preparation of chitosan-plasmid DNA nanoparticles encoding interleukin-12 and their expression in CT-26 colon carcinoma cells. J. Pharm. Pharm. Sci. 2011, 14, 181–195. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Shirjang, S.; Baradaran, B. HMGI-C suppressing induces P53/caspase9 axis to regulate apoptosis in breast adenocarcinoma cells. Cell Cycle 2016, 15, 2585–2592. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.-H.; Huang, C.-H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris IV, J.P.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R. p53 represses the mevalonate pathway to mediate tumor suppression. Cell 2019, 176, 564–580.e519. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, S.; Azzariti, A.; Silvestris, N.; Porcelli, L.; Russo, A.; Paradiso, A. p53 as the main traffic controller of the cell signaling network. Front. Biosci 2010, 15, 1172–1190. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Glass, C.K. Signaling by nuclear receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a016709. [Google Scholar] [CrossRef] [Green Version]
- Silvestris, N.; Tommasi, S.; Petriella, D.; Santini, D.; Fistola, E.; Russo, A.; Numico, G.; Tonini, G.; Maiello, E.; Colucci, G. The dark side of the moon: The PI3K/PTEN/AKT pathway in colorectal carcinoma. Oncology 2009, 77, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [Green Version]
- Günthert, U.; Hofmann, M.; Rudy, W.; Reber, S.; Zöller, M.; Hauβmann, I.; Matzku, S.; Wenzel, A.; Ponta, H.; Herrlich, P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell 1991, 65, 13–24. [Google Scholar] [CrossRef]
- Shadbad, M.A.; Hajiasgharzadeh, K.; Baradaran, B. Cross-talk between myeloid-derived suppressor cells and Mucin1 in breast cancer vaccination: On the verge of a breakthrough. Life Sci. 2020, 258, 118128. [Google Scholar] [CrossRef]
- Shadbad, M.A.; Hajiasgharzadeh, K.; Derakhshani, A.; Silvestris, N.; Baghbanzadeh, A.; Racanelli, V.; Baradaran, B. From Melanoma Development to RNA-Modified Dendritic Cell Vaccines: Highlighting the Lessons from the Past. Front. Immunol. 2021, 12, 331. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832.e823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colak, S.; ten Dijke, P. Targeting TGF-β signaling in cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Derakhshani, A.; Silvestris, N.; Hemmat, N.; Asadzadeh, Z.; Abdoli Shadbad, M.; Nourbakhsh, N.S.; Mobasheri, L.; Vahedi, P.; Shahmirzaie, M.; Brunetti, O. Targeting TGF-β-Mediated SMAD Signaling pathway via novel recombinant cytotoxin II: A potent protein from naja naja oxiana venom in Melanoma. Molecules 2020, 25, 5148. [Google Scholar] [CrossRef] [PubMed]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e282. [Google Scholar] [CrossRef] [Green Version]
- Pedram, M.; Heidari, M.; Keikhaei, B.; Azizi Malamiri, R.; Poopak, B.; Fekri, K. Impact of N-myc amplification on median survival in children with neuroblastoma. J. Compr. Ped. 2012, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Sansone, R.; Strigini, P.; Badiali, M.; Dominici, C.; Fontana, V.; Iolascon, A.; De Bernardi, B.; Tonini, G.P. Age-dependent prognostic significance of N-myc amplification in neuroblastoma: The Italian experience. Cancer Genet. Cytogenet. 1991, 54, 253–257. [Google Scholar] [CrossRef]
- Yu, S.; Sun, L.; Jiao, Y.; Lee, L.T.O. The role of G protein-coupled receptor kinases in cancer. Int. J. Biol. Sci. 2018, 14, 189. [Google Scholar] [CrossRef] [Green Version]
- Pedrini, B.; Tsai, C.-J.; Capitani, G.; Padeste, C.; Hunter, M.S.; Zatsepin, N.A.; Barty, A.; Benner, W.H.; Boutet, S.; Feld, G.K. 7 Å resolution in protein two-dimensional-crystal X-ray diffraction at Linac Coherent Light Source. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130500. [Google Scholar] [CrossRef] [PubMed]
- Schöneberg, T.; Schulz, A.; Biebermann, H.; Hermsdorf, T.; Römpler, H.; Sangkuhl, K. Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol. Ther. 2004, 104, 173–206. [Google Scholar] [CrossRef]
- Cohen, S.M.; Ellwein, L.B. Cell proliferation in carcinogenesis. Science 1990, 249, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.P. Notch, apoptosis and cancer. Adv. Exp. Med. Biol. 2012, 727, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Li, P.; Lu, F.; Liu, N.; Dai, J.; Ye, J.; Qu, X.; Sun, X.; Ma, D.; Park, J. Notch1 is required for hypoxia-induced proliferation, invasion and chemoresistance of T-cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 2013, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aburjania, Z.; Jang, S.; Whitt, J.; Jaskula-Stzul, R.; Chen, H.; Rose, J.B. The Role of Notch3 in Cancer. Oncologist 2018, 23, 900–911. [Google Scholar] [CrossRef] [Green Version]
- Alqudah, M.A.; Agarwal, S.; Al-Keilani, M.S.; Sibenaller, Z.A.; Ryken, T.C.; Assem, M. NOTCH3 is a prognostic factor that promotes glioma cell proliferation, migration and invasion via activation of CCND1 and EGFR. PLoS ONE 2013, 8, e77299. [Google Scholar] [CrossRef]
- Richardson, C.J.; Schalm, S.S.; Blenis, J. PI3-kinase and TOR: PIKTORing cell growth. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2004; pp. 147–159. [Google Scholar]
- Diehl, J.A.; Zindy, F.; Sherr, C.J. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997, 11, 957–972. [Google Scholar] [CrossRef] [Green Version]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [Green Version]
- Rössig, L.; Jadidi, A.S.; Urbich, C.; Badorff, C.; Zeiher, A.M.; Dimmeler, S. Akt-dependent phosphorylation of p21Cip1 regulates PCNA binding and proliferation of endothelial cells. Mol. Cell. Biol. 2001, 21, 5644–5657. [Google Scholar] [CrossRef] [Green Version]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef] [Green Version]
- Burgering, B.M.; Medema, R.H. Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J. Leukoc. Biol. 2003, 73, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Hou, Y.; Yin, X.; Bao, L.; Tang, A.; Song, L.; Li, F.; Tsang, S.; Wu, K.; Wu, H. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell 2012, 148, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of β-catenin by AKT promotes β-catenin transcriptional activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.A. The jak/stat pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, K.F.; Hariharan, I.K. The hippo pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duronio, R.J.; Xiong, Y. Signaling pathways that control cell proliferation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008904. [Google Scholar] [CrossRef] [PubMed]
- Capaccione, K.M.; Pine, S.R. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 2013, 34, 1420–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.-M.; Tergaonkar, V. NFκB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis 2009, 14, 348–363. [Google Scholar] [CrossRef]
- O’Reilly, L.A.; Kruse, E.A.; Puthalakath, H.; Kelly, P.N.; Kaufmann, T.; Huang, D.C.; Strasser, A. MEK/ERK-mediated phosphorylation of Bim is required to ensure survival of T and B lymphocytes during mitogenic stimulation. J. Immunol. (Baltim. Md. 1950) 2009, 183, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Traub, B.; Shi, J.; Huber, N.; Schreiner, S.; Chen, G.; Zhou, S.; Henne-Bruns, D.; Knippschild, U.; Kornmann, M. c-Jun N-terminal kinase 2 suppresses pancreatic cancer growth and invasion and is opposed by c-Jun N-terminal kinase 1. Cancer Gene Ther. 2021, 1–14. [Google Scholar] [CrossRef]
- Pan, C.-W.; Liu, H.; Zhao, Y.; Qian, C.; Wang, L.; Qi, J. JNK2 downregulation promotes tumorigenesis and chemoresistance by decreasing p53 stability in bladder cancer. Oncotarget 2016, 7, 35119. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plas, D.R.; Thompson, C.B. Akt-dependent transformation: There is more to growth than just surviving. Oncogene 2005, 24, 7435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606. [Google Scholar] [CrossRef] [PubMed]
- Barthel, A.; Okino, S.T.; Liao, J.; Nakatani, K.; Li, J.; Whitlock, J.P.; Roth, R.A. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J. Biol. Chem. 1999, 274, 20281–20286. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.J.; Gunter, L.B.; Carayannopoulos, M.O. Akt2 modulates glucose availability and downstream apoptotic pathways during development. J. Biol. Chem. 2010, 285, 17673–17680. [Google Scholar] [CrossRef] [Green Version]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, A.; Kawanishi, I.; Eguchi, S.; Yu, E.H.; Eguchi, S.; Oshiro, N.; Yoshino, K.-i.; Kikkawa, U.; Yonezawa, K. Association of CAD, a multifunctional protein involved in pyrimidine synthesis, with mLST8, a component of the mTOR complexes. J. Biomed. Sci. 2013, 20, 24. [Google Scholar] [CrossRef] [Green Version]
- Gingras, A.-C.; Kennedy, S.G.; O’Leary, M.A.; Sonenberg, N.; Hay, N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt (PKB) signaling pathway. Genes Dev. 1998, 12, 502–513. [Google Scholar] [CrossRef]
- Bakan, I.; Laplante, M. Connecting mTORC1 signaling to SREBP-1 activation. Curr. Opin. Lipidol. 2012, 23, 226–234. [Google Scholar] [CrossRef]
- Jeon, T.-I.; Osborne, T.F. SREBPs: Metabolic integrators in physiology and metabolism. Trends Endocrinol. Metab. 2012, 23, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Hlavin Bell, E.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014, 20, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. AACR 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-de-Cossio-Diaz, J.; Vazquez, A. Limits of aerobic metabolism in cancer cells. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb Perspect Med. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.-J.; Zhang, Y.; You, M.J. Hexokinase 2-mediated Warburg effect is required for PTEN-and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Moon, R.R.-B.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia10, A.; Bissell11, M.J.; Osborne12, T.F.; Tian, B.; Lowe, S.W.; Silva, J.M. Mutant p53 Disrupts Mammary Acinar Morphogenesis via the Mevalonate Pathway. Cell 2012, 148, 244–258. [Google Scholar]
- Vicente-Manzanares, M.; Horwitz, A.R. Cell migration: An overview. In Cell Migration; Springer: Cambridge, MA, USA, 2011; pp. 1–24. [Google Scholar]
- Devreotes, P.; Horwitz, A.R. Signaling networks that regulate cell migration. Cold Spring Harb. Perspect. Biol. 2015, 7, a005959. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.R.; Toker, A. Akt isoform-specific signaling in breast cancer: Uncovering an anti-migratory role for palladin. Cell Adhes. Migr. 2011, 5, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Birchmeier, W.; Birchmeier, C. Epithelial-mesenchymal transitions in development and tumor progression. In Epithelial-Mesenchymal Interactions in Cancer; Springer: Cambridge, MA, USA, 1995; pp. 1–15. [Google Scholar]
- Larue, L.; Bellacosa, A. Epithelial–mesenchymal transition in development and cancer: Role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005, 24, 7443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doble, B.W.; Woodgett, J.R. Role of glycogen synthase kinase-3 in cell fate and epithelial-mesenchymal transitions. Cells Tissues Organs 2007, 185, 73–84. [Google Scholar] [CrossRef]
- Hussey, G.S.; Chaudhury, A.; Dawson, A.E.; Lindner, D.J.; Knudsen, C.R.; Wilce, M.C.; Merrick, W.C.; Howe, P.H. Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Mol. Cell 2011, 41, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsum, I.A.; Martin, C.; Humbert, P.O. Scribble regulates an EMT polarity pathway through modulation of MAPK-ERK signaling to mediate junction formation. J. Cell Sci. 2013, 126, 3990–3999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasaka, K.; Seiki, T.; Yamashita, A.; Massimi, P.; Subbaiah, V.K.; Thomas, M.; Kranjec, C.; Kawana, K.; Nakagawa, S.; Yano, T. A novel interaction between hScrib and PP1γ downregulates ERK signaling and suppresses oncogene-induced cell transformation. PLoS ONE 2013, 8, e53752. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Wu, H.; Xu, H.; Han, N.; Chu, Q.; Yu, S.; Chen, Y.; Wu, K. Meta-analysis reveals the correlation of Notch signaling with non-small cell lung cancer progression and prognosis. Sci. Rep. 2015, 5, 10338. [Google Scholar] [CrossRef] [Green Version]
- Dow, L.; Elsum, I.; King, C.; Kinross, K.; Richardson, H.; Humbert, P. Loss of human Scribble cooperates with H-Ras to promote cell invasion through deregulation of MAPK signalling. Oncogene 2008, 27, 5988. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, S.; Collier, D.A.; Sham, P.C.; Ball, D.; Aitchison, K.; Vallada, H.; Kerwin, R.; Smith, I.; Gill, M. Allelic association between a Ser-9-Gly polymorphism in the dopamine D3 receptor gene and schizophrenia. Hum. Genet. 1996, 97, 714–719. [Google Scholar] [CrossRef]
- Wu, M.; Pastor-Pareja, J.C.; Xu, T. Interaction between Ras V12 and scribbled clones induces tumour growth and invasion. Nature 2010, 463, 545. [Google Scholar] [CrossRef] [Green Version]
- Iden, S.; van Riel, W.E.; Schäfer, R.; Song, J.-Y.; Hirose, T.; Ohno, S.; Collard, J.G. Tumor type-dependent function of the par3 polarity protein in skin tumorigenesis. Cancer Cell 2012, 22, 389–403. [Google Scholar] [CrossRef] [Green Version]
- McCaffrey, L.M.; Montalbano, J.; Mihai, C.; Macara, I.G. Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell 2012, 22, 601–614. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Krishnamurthy, K.; Allred, D.C.; Muthuswamy, S.K. Loss of Par3 promotes breast cancer metastasis by compromising cell–cell cohesion. Nat. Cell Biol. 2013, 15, 189. [Google Scholar] [CrossRef] [Green Version]
- Siahmansouri, H.; Somi, M.H.; Babaloo, Z.; Baradaran, B.; Jadidi-Niaragh, F.; Atyabi, F.; Mohammadi, H.; Ahmadi, M.; Yousefi, M. Pharmacology. Effects of HMGA 2 si RNA and doxorubicin dual delivery by chitosan nanoparticles on cytotoxicity and gene expression of HT-29 colorectal cancer cell line. J. Pharm. Pharmacol. 2016, 68, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Maity, A. PI3K/AKT/mTOR pathway in angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusse, R. Wnt signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011163. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R. Notch signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingham, P.W. Hedgehog signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; Ansell, S.; Haug, J.; Grote, D.; Kimlinger, T.; Stenson, M.; Timm, M.; Wellik, L.; Halling, T.; Rajkumar, S.V. MRK003, a γ-secretase inhibitor exhibits promising in vitro pre-clinical activity in multiple myeloma and non-Hodgkin’s lymphoma. Leukemia 2012, 26, 340. [Google Scholar] [CrossRef] [Green Version]
- Xiong, G.-F.; Xu, R. Function of cancer cell-derived extracellular matrix in tumor progression. J. Cancer Metastasis Treat. 2016, 2, 357–364. [Google Scholar] [CrossRef]
- Schober, M.; Jesenofsky, R.; Faissner, R.; Weidenauer, C.; Hagmann, W.; Michl, P.; Heuchel, R.L.; Haas, S.L.; Löhr, J. Desmoplasia and chemoresistance in pancreatic cancer. Cancers 2014, 6, 2137–2154. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.-J. The role of the extracellular matrix in cancer stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Keely, P.J. Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J. Mammary Gland Biol. Neoplasia 2011, 16, 205. [Google Scholar] [CrossRef] [PubMed]
- Bolós, V.; Gasent, J.M.; Lopez-Tarruella, S.; Grande, E. The dual kinase complex FAK-Src as a promising therapeutic target in cancer. Oncotargets Ther. 2010, 3, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Guan, J.-L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.-C.; Qu, X.-J.; Gao, Z.-H. Integrins: Players in cancer progression and targets in cancer therapy. Anti-Cancer Drugs 2014, 25, 1107–1121. [Google Scholar] [CrossRef] [PubMed]
- Cantor, D.; Cheruku, H.; Nice, E.; Baker, M. Integrin αvβ6 sets the stage for colorectal cancer metastasis. Cancer Metastasis Rev. 2015, 34, 715–734. [Google Scholar] [CrossRef] [PubMed]
- Östman, A.; Augsten, M. Cancer-associated fibroblasts and tumor growth–bystanders turning into key players. Curr. Opin. Genet. Dev. 2009, 19, 67–73. [Google Scholar] [CrossRef]
- Pietras, K.; Östman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Eke, I.; Cordes, N. Focal adhesion signaling and therapy resistance in cancer. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2015; Volume 31, pp. 65–75. [Google Scholar]
- Xu, D.; Esko, J.D. Demystifying heparan sulfate–protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef]
- Milner, C.; Tongsoongnoen, W.; Rugg, M.; Day, A. The molecular basis of inter-α-inhibitor heavy chain transfer on to hyaluronan. Biochem. Soc. Trans. 2007, 35, 672–676. [Google Scholar] [CrossRef]
- Nikitovic, D.; Tzardi, M.; Berdiaki, A.; Tsatsakis, A.; Tzanakakis, G.N. Cancer microenvironment and inflammation: Role of hyaluronan. Front. Immunol. 2015, 6, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filmus, J.; Capurro, M.; Rast, J. Glypicans. Genome Biol. 2008, 9, 224. [Google Scholar] [CrossRef]
- Xian, X.; Gopal, S.; Couchman, J.R. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res. 2010, 339, 31. [Google Scholar] [CrossRef] [PubMed]
- Lowy, C.M.; Oskarsson, T. Tenascin C in metastasis: A view from the invasive front. Cell Adhes. Migr. 2015, 9, 112–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurbuz, I.; Chiquet-Ehrismann, R. CCN4/WISP1 (WNT1 inducible signaling pathway protein 1): A focus on its role in cancer. Int. J. Biochem. Cell Biol. 2015, 62, 142–146. [Google Scholar] [CrossRef] [Green Version]
- Vahidian, F.; Safarzadeh, E.; Mohammadi, A.; Najjary, S.; Mansoori, B.; Majidi, J.; Babaloo, Z.; Aghanejad, A.; Shadbad, M.A.; Mokhtarzadeh, A. siRNA-mediated silencing of CD44 delivered by Jet Pei enhanced Doxorubicin chemo sensitivity and altered miRNA expression in human breast cancer cell line (MDA-MB468). Mol. Biol. Rep. 2020, 47, 9541–9551. [Google Scholar] [CrossRef]
- Orian-Rousseau, V. CD44, a therapeutic target for metastasising tumours. Eur. J. Cancer 2010, 46, 1271–1277. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Bikle, D. Selective hyaluronan–CD44 signaling promotes miRNA-21 expression and interacts with vitamin D function during cutaneous squamous cell carcinomas progression following UV irradiation. Front. Immunol. 2015, 6, 224. [Google Scholar] [CrossRef] [Green Version]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef] [Green Version]
- Keener, A.B. Single-cell sequencing edges into clinical trials. Nat. Med. 2019, 25, 1322. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Song, S.; Hochster, H.S.; Steinberg, I.B. Cancer stem cells: The promise and the potential. In Seminars in Oncology; WB Saunders: Cambridge, MA, USA, 2015; Volume 42, pp. S3–S17. [Google Scholar]
- Sanderson, R.; Couchman, J. Targeting syndecan shedding in cancer. Extracell. Matrix Pathobiol. Signal. (Karamanos NkEd.) 2012, 802–812. [Google Scholar] [CrossRef]
- Yoneda, A.; Lendorf, M.E.; Couchman, J.R.; Multhaupt, H.A. Breast and ovarian cancers: A survey and possible roles for the cell surface heparan sulfate proteoglycans. J. Histochem. Cytochem. 2012, 60, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Ramani, V.C.; Purushothaman, A.; Stewart, M.D.; Thompson, C.A.; Vlodavsky, I.; Au, J.L.S.; Sanderson, R.D. The heparanase/syndecan-1 axis in cancer: Mechanisms and therapies. FEBS J. 2013, 280, 2294–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, C.M.; Reichsman, F.; Hinkes, M.T.; Lincecum, J.; Becker, K.A.; Cumberledge, S.; Bernfield, M. Syndecan-1 is required for Wnt-1-induced mammary tumorigenesis in mice. Nat. Genet. 2000, 25, 329. [Google Scholar] [CrossRef] [PubMed]
- Simone, V.; Brunetti, O.; Lupo, L.; Testini, M.; Maiorano, E.; Simone, M.; Longo, V.; Rolfo, C.; Peeters, M.; Scarpa, A. Targeting angiogenesis in biliary tract cancers: An open option. Int. J. Mol. Sci. 2017, 18, 418. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.; Brunetti, O.; Gnoni, A.; Cascinu, S.; Gasparini, G.; Lorusso, V.; Ribatti, D.; Silvestris, N. Angiogenesis in pancreatic ductal adenocarcinoma: A controversial issue. Oncotarget 2016, 7, 58649. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia. Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Xu, B.; An, Z.; Wang, Z.; Li, Y.; Wei, L.; Wei, D. Evolutionary analysis of TSP-1 gene in Plateau zokor (MyospalaxBaileyi) and its expression pattern under hypoxia. Cell. Mol. Biol. (Noisy-Le-GrandFr.) 2019, 65, 48–57. [Google Scholar] [CrossRef]
- Zaslavsky, A.; Chen, C.; Grillo, J.; Baek, K.H.; Holmgren, L.; Yoon, S.S.; Folkman, J.; Ryeom, S. Regional control of tumor growth. Mol. Cancer Res.: Mcr 2010, 8, 1198–1206. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W.J.C. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumsteg, A.; Christofori, G. Corrupt policemen: Inflammatory cells promote tumor angiogenesis. Curr. Opin. Oncol. 2009, 21, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Ferrara, N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010, 21, 21–26. [Google Scholar] [CrossRef]
- Patenaude, A.; Parker, J.; Karsan, A.J. Involvement of endothelial progenitor cells in tumor vascularization. Microvasc. Res. 2010, 79, 217–223. [Google Scholar] [CrossRef]
- Ziegelhoeffer, T.; Fernandez, B.; Kostin, S.; Heil, M.; Voswinckel, R.; Helisch, A.; Schaper, W. Bone marrow-derived cells do not incorporate into the adult growing vasculature. Circ. Res. 2004, 94, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Yoder, M.C.; Ingram, D.A. Endothelial progenitor cell: Ongoing controversy for defining these cells and their role in neoangiogenesis in the murine system. Curr. Opin. Hematol. 2009, 16, 269–273. [Google Scholar] [CrossRef]
- Leone, P.; Di Lernia, G.; Solimando, A.G.; Cicco, S.; Saltarella, I.; Lamanuzzi, A.; Ria, R.; Frassanito, M.A.; Ponzoni, M.; Ditonno, P.; et al. Bone marrow endothelial cells sustain a tumor-specific CD8+ T cell subset with suppressive function in myeloma patients. Oncoimmunology 2019, 8, e1486949. [Google Scholar] [CrossRef] [Green Version]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: Implications for immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Österreicher, C.H.; Hung, K.E. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254. [Google Scholar] [CrossRef] [Green Version]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e2105. [Google Scholar] [CrossRef]
- Thomas, M.U.; Messex, J.K.; Dang, T.; Abdulkadir, S.A.; Jorcyk, C.L.; Liou, G.Y. Macrophages expedite cell proliferation of prostate intraepithelial neoplasia through their downstream target ERK. FEBS J. 2020. [Google Scholar] [CrossRef]
- Shi, J.-S.; Zhou, L.-S.; Han, Y.; Zhu, A.-J.; Sun, X.-J.; Yang, Y.-J. Expression of tumor necrosis factor and its receptor in gallstone and gallbladder carcinoma tissue. Hepatobiliary Pancreat. Dis. Int. Hbpd. Int. 2004, 3, 448–452. [Google Scholar]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? 1. Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, B.B.; Langefeld, C.; Olivier, M.; Cox, L.A. Integrated omics: Tools, advances and future approaches. J. Mol. Endocrinol. 2019, 62, R21–R45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solimando, A.G.; Da Vià, M.C.; Cicco, S.; Leone, P.; Di Lernia, G.; Giannico, D.; Desantis, V.; Frassanito, M.A.; Morizio, A.; Delgado Tascon, J.; et al. High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. J. Clin. Med. 2019, 8, 997. [Google Scholar] [CrossRef] [Green Version]
- Kuenzi, B.M.; Ideker, T. A census of pathway maps in cancer systems biology. Nat. Rev. Cancer 2020. [Google Scholar] [CrossRef]
- Sambath, J.; Patel, K.; Limaye, S.; Kumar, P. Single-Cell Multiomics: Dissecting Cancer. In Statistical Modelling and Machine Learning Principles for Bioinformatics Techniques, Tools, and Applications; Srinivasa, K.G., Siddesh, G.M., Manisekhar, S.R., Eds.; Springer Singapore: Singapore, 2020; pp. 289–317. [Google Scholar] [CrossRef]
- Wang, W.; Gao, D.; Wang, X. Can single-cell RNA sequencing crack the mystery of cells? Cell Biol. Toxicol. 2018, 34, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.Y. Identification of ERBB pathway-activated cells in triple-negative breast cancer. Genom. Inform. 2019, 17, e3. [Google Scholar] [CrossRef]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Hu, J.; Zhang, J.; Guo, F.; Zhou, M.; Zhang, G.; Yu, F.; Su, J. scTPA: A web tool for single-cell transcriptome analysis of pathway activation signatures. Bioinformatics 2020, 36, 4217–4219. [Google Scholar] [CrossRef]
- Lim, B.; Lin, Y.; Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer cell 2020, 37, 456–470. [Google Scholar] [CrossRef]
- Ho, Y.-J.; Anaparthy, N.; Molik, D.; Mathew, G.; Aicher, T.; Patel, A.; Hicks, J.; Hammell, M.G. Single-cell RNA-seq analysis identifies markers of resistance to targeted BRAF inhibitors in melanoma cell populations. Genome Res. 2018, 28, 1353–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.J.S. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell 2018, 173, 879–893.e813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseinkhani, N.; Derakhshani, A.; Kooshkaki, O.; Abdoli Shadbad, M.; Hajiasgharzadeh, K.; Baghbanzadeh, A.; Safarpour, H.; Mokhtarzadeh, A.; Brunetti, O.; Yue, S.C. Immune Checkpoints and CAR-T Cells: The Pioneers in Future Cancer Therapies? Int. J. Mol. Sci. 2020, 21, 8305. [Google Scholar] [CrossRef] [PubMed]
- Luoma, A.M.; Suo, S.; Williams, H.L.; Sharova, T.; Sullivan, K.; Manos, M.; Bowling, P.; Hodi, F.S.; Rahma, O.; Sullivan, R.J. Molecular pathways of colon inflammation induced by cancer immunotherapy. Cell 2020, 182, 655–671.e622. [Google Scholar] [CrossRef] [PubMed]
- Borcherding, N.; Vishwakarma, A.; Voigt, A.P.; Bellizzi, A.; Kaplan, J.; Nepple, K.; Salem, A.K.; Jenkins, R.W.; Zakharia, Y.; Zhang, W. Mapping the immune environment in clear cell renal carcinoma by single-cell genomics. Commun. Biol. 2021, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 2018, 175, 998–1013.e1020. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.Y.; Kwek, S.S.; Raju, S.S.; Li, T.; McCarthy, E.; Chow, E.; Aran, D.; Ilano, A.; Pai, C.-C.S.; Rancan, C. Intratumoral CD4+ T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell 2020, 181, 1612–1625.e1613. [Google Scholar] [CrossRef]
- Durante, M.A.; Rodriguez, D.A.; Kurtenbach, S.; Kuznetsov, J.N.; Sanchez, M.I.; Decatur, C.L.; Snyder, H.; Feun, L.G.; Livingstone, A.S.; Harbour, J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Heppt, M.V.; Steeb, T.; Schlager, J.G.; Rosumeck, S.; Dressler, C.; Ruzicka, T.; Nast, A.; Berking, C. Immune checkpoint blockade for unresectable or metastatic uveal melanoma: A systematic review. Cancer Treat. Rev. 2017, 60, 44–52. [Google Scholar] [CrossRef]
- Clarke, J.; Panwar, B.; Madrigal, A.; Singh, D.; Gujar, R.; Wood, O.; Chee, S.J.; Eschweiler, S.; King, E.V.; Awad, A.S. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J. Exp. Med. 2019, 216, 2128–2149. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Derakhshani, A.; Rostami, Z.; Safarpour, H.; Shadbad, M.A.; Nourbakhsh, N.S.; Argentiero, A.; Taefehshokr, S.; Tabrizi, N.J.; Kooshkaki, O.; Astamal, R.V.; et al. From Oncogenic Signaling Pathways to Single-Cell Sequencing of Immune Cells: Changing the Landscape of Cancer Immunotherapy. Molecules 2021, 26, 2278. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082278
Derakhshani A, Rostami Z, Safarpour H, Shadbad MA, Nourbakhsh NS, Argentiero A, Taefehshokr S, Tabrizi NJ, Kooshkaki O, Astamal RV, et al. From Oncogenic Signaling Pathways to Single-Cell Sequencing of Immune Cells: Changing the Landscape of Cancer Immunotherapy. Molecules. 2021; 26(8):2278. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082278
Chicago/Turabian StyleDerakhshani, Afshin, Zeinab Rostami, Hossein Safarpour, Mahdi Abdoli Shadbad, Niloufar Sadat Nourbakhsh, Antonella Argentiero, Sina Taefehshokr, Neda Jalili Tabrizi, Omid Kooshkaki, Reza Vaezi Astamal, and et al. 2021. "From Oncogenic Signaling Pathways to Single-Cell Sequencing of Immune Cells: Changing the Landscape of Cancer Immunotherapy" Molecules 26, no. 8: 2278. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082278